

Abstract
DNA binding activity of p53 is crucial for its tumor suppressor function. Our recent studies have shown that four molecules of the DNA binding domain of human p53 (p53DBD) bind the response elements with high cooperativity and bend the DNA. By using A-tract phasing experiments, we find significant differences between the bending and twisting of DNA by p53DBD and by full-length human wild-type (wt) p53. Our data show that four subunits of p53DBD bend the DNA by 32–36°, whereas wt p53 bends it by 51–57°. The directionality of bending is consistent with major groove bends at the two pentamer junctions in the consensus DNA response element. More sophisticated phasing analyses also demonstrate that p53DBD and wt p53 overtwist the DNA response element by ≈35° and ≈70°, respectively. These results are in accord with molecular modeling studies of the tetrameric complex. Within the constraints imposed by the protein subunits, the DNA can assume a range of conformations resulting from correlated changes in bend and twist angles such that the p53–DNA tetrameric complex is stabilized by DNA overtwisting and bending toward the major groove at the CATG tetramers. This bending is consistent with the inherent sequence-dependent anisotropy of the duplex. Overall, the four p53 moieties are placed laterally in a staggered array on the external side of the DNA loop and have numerous interprotein interactions that increase the stability and cooperativity of binding. The novel architecture of the p53 tetrameric complex has important functional implications including possible p53 interactions with chromatin.
Keywords: wild-type p53, p53 DNA binding domain, DNA–protein complex, A-tract phasing analysis, bent DNA
The tumor suppressor protein p53 plays a central role in the regulation of cellular growth and is a potent transcription factor that is activated in response to a variety of DNA damaging agents, leading to cell cycle arrest at the G1/S phase checkpoint or to induction of apoptosis (1, 2). Disruption of this pathway occurs in a wide variety of human cancers and correlates with the development of the tumorogenic mutants that are defective in DNA binding and consequently cannot activate transcription. Thus, it is becoming clear that sequence-specific DNA binding and transactivation are the key activities that control the biological functions of p53 (1, 3).
Wild-type (wt) p53 binds as a tetramer (4–7) to over 100 different naturally occurring response elements, of which approximately 60 show functionality. The human genome has been estimated to contain approximately 200–300 such sites (8). Response elements differ in the details of their specific base sequence, but all contain two tandem decamers, each a pentameric inverted repeat. Most decamers follow the consensus sequence pattern (4) PuPuPuC(A/t)|(T/a)GPyPyPy, where Pu and Py are purines and pyrimidines, respectively, and the vertical bar denotes the center of pseudodyad symmetry. These decamers may be separated by as many as 21 bp without complete loss of p53 binding affinity (9), but functional sites (defined as the ability to activate a nearby reporter gene) have short intervening spacers (8). Of special importance to understanding the pleiotropic character of p53 binding and hence its function is the sequence of the tetramers that span the pseudodyad in each half-site. These are most commonly CA|TG but can also be CA|AG:CT|TG (8), all of which are known to exhibit a large flexibility for bending or kinking into the major groove (10–12). Because many architectural proteins utilize CA:TG dimers (13–15), it is natural to suspect that these dimers may play an important role in specific DNA recognition by transcription factors, especially those with the functional multiplicity of p53.
To date, only one crystallographic structure of a p53 nucleoprotein complex has been reported (16). This structure clearly showed the direct interactions between a p53 DNA binding domain (p53DBD) and the target DNA but could not address the role of the tetrameric complex in specific DNA recognition. We therefore performed ligase-mediated cyclization (17) and chemical probe analysis (18) combined with molecular modeling (18, 19), the latter to develop a tetrameric model consistent with the demonstration of DNA bending by ligase-mediated cyclization. Surprisingly, it was found that the p53DBD self-assembles as a tetrameric complex when bound to a full (20-bp) p53 response element (although it exists as a monomer in solution) and that binding occurs with high cooperativity (17, 20). Subsequent studies showed that the binding affinity of the tetrameric p53DBD complex was correlated with DNA bending and that response elements with greatest flexibility at the pentameric junctions, e.g., one or more CA|TG elements, actually bound with the highest affinity (21). However, none of these earlier experiments yield either the directionality of DNA bending or the local changes in DNA twisting associated with p53 binding. The latter is of special importance because stereochemical analysis (19) suggested that the DNA twisting increases on complex formation and that local supercoiling in genomic DNA might therefore play a role in the regulation of specific sequence recognition. Finally, none of the above studies examined the binding of wt p53.
In the present work, we have used A-tract phasing analysis (22) to determine both the directionality and magnitude of DNA bending in the p53DBD– and wt p53–DNA complexes. In this technique, a protein-induced DNA bend is helically phased with one or more poly(A) tracts whose bending magnitude and directionality are known (23). By examining the gel mobility as a function of variable spacer length between these elements, an absolute determination of both bending magnitude and direction is possible. To determine twisting changes, we employed more sophisticated phasing constructs that include an additional A tract (24). Results show substantial differences in both DNA bending and twisting between the p53DBD and wt p53 complexes. Bending directionality confirms earlier model predictions for the p53DBD complex (18, 19). Based on the new results, we further develop this model, including the N- and C-terminal fragments, in addition to the DBD subunits, which allows new insights into possible p53 structure-function relationships.
METHODS
Plasmid Construction and Protein Purification.
A human p53 cDNA clone encoding amino acid residues 96–308 was amplified by PCR using p53-specific primers 5′-ATATCATATGGTCCCTTCCCAGAAAACCTA-3′ and 5′-ATATGGATCCTCACAGTGCTCGCTTAGTGCTC-3′. The amplified product was cloned in the pet12a expression vector (Novagen), and the core DNA binding domain was overproduced in Escherichia coli BL21(DE3) and purified as described (17). The full human wt p53 with 27-aa N-terminal tag was expressed in baculovirus and purified by Ni+2 affinity column chromatography as described (25). Purified proteins were analysed by SDS/PAGE and were stained by Coomassie blue. All p53s were >90% pure.
Design of the p53 Binding Site.
The principal considerations dictating the design of the response element used here were (i) to ensure that each monomer interacts with the DNA pentamer as in the crystal (16), (ii) to maintain a flexibility element CATG at the pentamer junctions in each half-site (8), and (iii) to maintain dyad symmetry in the sequence to facilitate molecular modeling (19). The binding site used here was composed of a fourfold repeat of the consensus pentamer, organized into half-sites AGGCA|TGCCT. This selection guarantees that the trimers GGC:GCC are the same as in the cocrystal structure (16) where the p53DBD subunit is bound to the pentamer GGGCA forming hydrogen bonds with the italicized three central base pairs.
Phasing Analyses for DNA Bending and Twisting.
The phasing probes for the DNA bending analysis were constructed by partial annealing of a fixed length oligonucleotide having a p53 consensus sequence to a series of oligonucleotides having three phased A-tract sequences (the three-segment constructs in Fig. 1). The annealed oligonucleotides were extended by Klenow fragments, digested by EcoRI and HindIII, and cloned at the EcoRI–HindIII-digested plasmid Litmus 39. The plasmids were sequenced to confirm the sizes and sequences of the inserts. The sequences are organized as follows: 5′–BsrGI–EcoRI–(p53 site: AGGCATGCCT|AGGCATGCCT)–variable spacer–(A-tract DNA: CGGGCAAAAAACGGGCAAAA|AACGGCAAAAAACGGGC)–HindIII–ApaI–3′. The vertical bars denote the center of the p53 binding site and the center of the A-tract DNA curvature. The lengths of the fragments between the vertical bars vary between 30 and 50 bp (S3 in Fig. 1A). The variable spacer changes from zero for S3 = 30 bp to 20 bp for S3 = 50 bp. The Twist angles calculated for the S3 fragments based on Kabsch et al. (26) values are the closest to 360° × n for S3 = 42 bp (Twist = 360° × n + 3°) .
Figure 1.

(A) Three- and four-segment DNA constructs (22, 24) used for the A-tract phasing analyses of DNA bending and twisting in the complex with p53 tetramer. The spacer S3 is variable, whereas the segments S1, S2, and S4 have fixed lengths. (B) Scheme for calculation of the DNA bend angle in the three-segment construct in A. P stands for the p53-bound end of DNA and A is for the A-tract end. C denotes the center of the horizontal fragment, so that s = S3/2, dp = S2, and da = S4.
The four-segment constructs for analyzing the DNA twisting were made by digesting the bending plasmids with EcoRI and MluI and subcloning an oligonucleotide with three phased A-tracts such that the distance between the p53 bend center and center of A-tracts remained 41 bp (S2 in Fig. 1A). All of the recombinant plasmids were digested with BsrGI, labeled with [32P]dATP and Klenow fragment, and digested with ApaI. The labeled probes were further purified on 10% native PAGE to eliminate the free label.
The labeled probes (5 × 103 cpm, 50 ng) were mixed with poly(dI-dC) (200 ng) and incubated with the binding protein (p53DBD peptide or wt p53) in DNA binding buffer (50 mM Bistris propane⋅HCl, pH 6.8/1 mM DTT/100 mM sodium chloride) at 4°C for 30 min and analyzed on 12% polyacrylamide gel. The binding reaction with wt p53 was carried out by incubating 2 ng of the probe, 30 ng of 30-bp nonspecific competitor DNA, and 50 ng of p53 in 50 mM Tris⋅Cl/100 mM NaCl/1 mM DTT at 4°C for 30 min and analyzed on 12% polyacrylamide gel. The concentration of the protein was adjusted so that it forms an approximately 50:50 complex with the probe.
Intrinsic DNA bend standards were prepared by partial annealing of two oligonucleotides each having three phased A-tracts. The lengths of the oligonucleotides were adjusted such that the distances between two bend centers varied between 37 and 45 bp. The standard probes were run on the same gel for comparison (Fig. 2 A and B).
Figure 2.


Phasing gel analysis of DNA bending. p53DBD (A) and wt p53 (B) are bound to the three-segment constructs with spacers S3 (Fig. 1 A and B) with the lengths (in bp) indicated. Lane M shows mobilities of the markers. Labels C stand for the p53 complexes and F stands for free DNA. A-tract DNAs serve as “standard probes” to determine the adjusting parameter k (see Methods). (C and D) Relative mobility plots for the p53DBD-bound DNA (○ in C), for the wt p53–DNA complex (● in C), and free DNA (D). To calculate the relative gel mobilities, the free DNA mobility as a function of the spacer length was approximated by using a linear function. The observed mobilities (both for free DNA and for the complexes) were divided by these “ideal” values corresponding to a length-dependent retardation of DNA without intrinsic bends. The values so obtained were normalized separately by the maximum mobilities in each case so that the maximum relative mobility is always 1.0.
Calculation of the DNA Bend Angle.
To estimate the p53-induced DNA bend angle, we used a scheme similar to that developed by Kerppola and Curran (27). According to their formalism, the DNA trajectory is approximated by a V shape, i.e., the two DNA bends, one in the center of the A tract and the other in the center of the protein binding site, are assumed to be closely spaced. In the present work, however, the DNA spacer between the two bend centers, S3 = 30–50 bp, is comparable in length to the two DNA “arms”, S2 = 50 bp and S4 = 55 bp (Fig. 1A). Therefore, the formalism (27) requires modification.
We consider the two bends separately and assume that DNA forms a C-shaped trajectory (Fig. 1B). The angle α = (180° − ∠PCA) is used as a measure of the overall DNA bend. For the cis configuration, α = γa + γp, and for the trans configuration, α = γa − γp, in which P refers to protein and A to the A tracts. Thus, our expression equivalent to the formalism of Kerppola and Curran (27) becomes
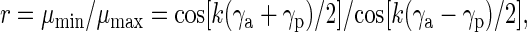 |
1 |
where k ≈ 1 is a parameter adjusting for specific electrophoresis conditions. In turn, the angles γp and γa are related to the protein-induced bend, βp, and the A tracts’ intrinsic bend angle, βa, by
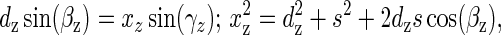 |
2 |
where subscript “z” means either “a” or “p”. The parameters da, dp, xa, xp, and s are shown in Fig. 1B. To calculate the protein-induced bend angle, βp, the angle βa is fixed at the selected value of 54° or 60°, corresponding to the A6-tract bend angle of 18° or 20° (28), and the angle βp is varied from 0° to 90° with an increment of 1°. The angles γa, γp and the corresponding ratio r = μmin/μmax are calculated from Eqs. 2 and 1, respectively. The value of βp when the two ratios (theoretical and experimental) are equal, is selected as the estimate of the protein-induced bend angle βp.
The coefficient k is obtained from the data for the standard probes containing two sets of A tracts (Fig. 2 A and B). In this case, the βp value is known (βp = βa), and Eq. 1 is used to find the value of k corresponding to the ratio r = μmin/μmax = 0.69 observed under our experimental conditions. Based on our formalism, we estimate the values of the coefficient k = 1.04 and 1.17 for βa = 60° and 54°, respectively, whereas the Kerppola and Curran formalism (27) with our data gives corresponding values k = 0.77 and 0.85. This suggests that our “C-shape model” provides a more realistic approximation to the observed DNA gel mobility.
RESULTS AND DISCUSSION
Directionality of DNA Bending in the p53–DNA Complexes.
The phasing analysis data for the p53DBD and wt p53 bound to the three-segment DNA constructs are shown in Fig. 2 A and B, with their relative mobility plots in Fig. 2C. The p53–DNA complexes demonstrate the cosine mobility pattern characteristic of protein-induced DNA bending (22, 27). The probes with 35- and 45-bp spacers show maximum mobility on the gel, whereas the probes with 30-, 40-, and 50-bp spacers move the slowest. The former correspond to the trans DNA configuration, and the latter to the cis DNA shown in Fig. 1. In other words, the cis DNA is formed when the centers of the two bends (in the p53–DNA complex and in the A tract) are separated by an integral number of helical turns. Therefore, the overall p53-DNA bend is directed in the same way as that in the A tract, into the minor groove (22, 23).
To establish the actual directionality of the local p53-induced bending, it is necessary to specify the centers of bending. Because the p53 response element consists of two decameric repeats with the flexible CA|TG pentameric junctions, the DNA bends are presumably located at these flexible junctions (13–15). In this case, we conclude that the local DNA bends occur in the opposite direction, into the major grooves, because the CA|TG junctions are separated by 5 bp from the center of the p53 binding site.
Both wt p53 and p53DBD reveal the same directionality of DNA bending. It is clear that although the wt p53–DNA complexes show a reduced mobility on the gel, probably because of their higher molecular weight (Fig. 2B), the overall pattern of gel retardation remains the same (Fig. 2C). The DNA bend angles are calculated to be 32–36° in the case of p53DBD–DNA complex and 51–57° for the wt p53–DNA complex (Table 1).
Table 1.
Gel mobility ratio and p53-induced DNA bending and twisting
| p53 | Ratio, r | Bend1, ° | Bend2, ° | ΔTwist, ° |
|---|---|---|---|---|
| p53DBD | 0.81 | 32.0 | 36.0 | ≈35 |
| wt p53 | 0.71 | 50.5 | 56.5 | ≈70 |
Ratio, r, is the value of relative mobility shown in Fig. 2C for the spacer of 40 bp (minimum mobility of the complex divided by maximum mobility). The averages for three independent measurements are given, with the standard errors not exceeding 0.02. The bend angles are calculated following Eq. 2 assuming that the DNA bending per A6 tract is 18° (bend1) or 20° (bend2) (see ref. 28). The changes in DNA twisting, ΔTwist, are deduced from Figs. 3 C and D.
The free DNA fragments also demonstrate the cosine-like profile of the gel mobility as a function of the spacer size (Fig. 2D). Thus, in addition to the A tracts, free DNA is intrinsically curved in some other sites or there would be no periodicity in the gel retardation. The directionality of the free DNA bending is close to that in the p53–DNA complexes, although its magnitude is much smaller. This suggests that the free p53 binding site can flex in the direction close to that observed in the p53–DNA complex: presumably, into the major groove in the CATG tetramers.
Increase in DNA Twisting On p53 Binding.
The mobility minima for the p53 complexes are shifted to a shorter spacer, i.e., to 40 bp instead of 42 bp for the free DNA (Fig. 2 C and D). This shift probably indicates overtwisting of the DNA in the complex compared with free DNA. To test this hypothesis, we analyzed the four-segment twisting constructs (Fig. 3). It is important for comparing the free DNA with the complex that the segment S2 was 41 bp in this case, which ensures that S1 and S3 are in the cis orientation in both cases (Fig. 1).
Figure 3.

Phasing gel analysis of DNA twisting. Gel mobilities of the p53DBD–DNA complex (A and C) and wt p53–DNA complex (B and D) are compared with the free four-segment DNA constructs (Fig. 1A). M, C and F are as in Fig. 2. The average mobility plots in C and D are given for three independent experiments (the individual measuments vary by no more than 0.4 cm for the complexes and 0.6 cm for free DNA).
The free DNA gel mobility is characterized by easily detectable maxima and minima, which are located exactly where they should be according to the DNA twisting calculations based on the scheme of Kabsch et al. (26). These calculations predict that the two A tracts are in a cis orientation (C shape, Fig. 1A) when the spacer S3 is 32 bp or 42 bp, and in trans orientation (S shape) when S3 is 37 bp (data not shown). Accordingly, in Fig. 3 C and D, the free DNA mobility is at a minimum for S3 = 32 bp and 42 bp and a maximum for S3 = 37 bp. So, the gel mobilities of the four-segment constructs with two sets of A tracts are consistent with simple “S–C–S transition” related to DNA twisting, and thus can be used for measurements of the twist angle changes induced by p53 binding.
It is clear that the DNA mobility as a function of the spacer S3 length has a cosine wave appearance for both free DNA and the p53 complex, the former being shifted to the right compared with the latter (Fig. 3 C and D). For the p53DBD–DNA complex this shift is 1 bp, and for the wt p53 the shift is 2 bp (within the precision of these measurements). In terms of the DNA twist angle, these shifts indicate ≈35° and ≈70° increase in the DNA twisting for p53DBD and wt p53, respectively.
The four-segment constructs for both free DNA and the complexes show similar amplitudes in their periodic mobility profiles (Fig. 3). This has the advantage of effectively increasing the sensitivity of the method for measuring and comparing DNA twisting. By contrast, the three-segment constructs had a much smaller amplitude for free DNA compared with the p53 complex. Thus, the combination is exceptionally powerful: the three-segment constructs are ideal for measuring the directionality and magnitude of the DNA bending, whereas the four-segment constructs permit a realistic estimate of changes in DNA twisting on binding to protein.
Molecular Modeling of the DNA Bending and Twisting in the p53 Complex.
The tetrameric p53–DNA complex was modeled by using the coordinates from the x-ray structure of a single p53DBD bound to the DNA obtained by Pavletich and coworkers (16). The details of the computations are described elsewhere (19). We present below the principal results and compare these with the phasing gel data.
The p53 response element studied here is formed by four pentamers.
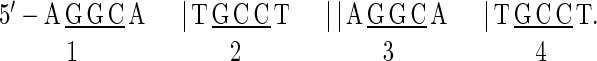 |
The base pairs in the underlined central trimers in each pentamer are hydrogen bonded with p53DBD in the cocrystal monomer structure (16), and unlikely to be distorted in the tetrameric complex. To retain the symmetry of the complex, the DNA duplex must flex along the dyad axes at the pentamer junctions indicated by vertical bars, i.e., in the Roll direction. Our data suggest that the decamer junction indicated by the double bar remains less distorted than the flexible junctions CA|TG (19). Thus, the number of the independent degrees of freedom is reduced.
The energy landscape of the complex is shown in Fig. 4, where the energy of the p53DBD tetramer is presented as a function of two angles, Bend and Twist, measured between the centers of pentamers 1 and 2 (the angles for pentamers 3 and 4 are identical because of the symmetry of the complex). Two main results follow from this energy profile: (i) DNA bending in the positive direction, i.e., into the major groove of CA|TG, is much preferred; and (ii) bending and twisting are strongly correlated, so the positive bend is accompanied by an increase in DNA twisting (19), entirely consistent with the gel data (Table 1).
Figure 4.

Energy profile and contour map (Bend, Twist) for the interprotein interactions in the p53DBD tetramer bound to DNA (29). The “global” Bend and Twist angles describe the relative orientation of the adjacent pentamers in the p53 response element (see text). The positive sign of Bend indicates the major groove bending in the CA|TG tetramer. The area in the contour map with the prohibitive interaction energy of >20 kcal/mol is cross-hatched. The areas with the energy <−15 kcal/mol are shaded. The crosses correspond to conformations of free DNA in solution (F), in the complex with p53DBD (DBD), and in the complex with wt p53 (WT), as found by phasing gel analyses. They denote the following (Bend, Twist) parameters: F (0°, 170°), DBD (17°, 179°), and wt (27°, 187.5°), as deduced from Table 1.
The DNA bending is likely localized mostly at the two CA|TG junctions (18, 19). Therefore, to establish equivalency with the computations, the measured total bend must be divided by two (Bend/2 = 17° for p53DBD and 27° for wt p53); these values are noted by crosses in Fig. 4. For simplicity, it is also presumed that the increase in the Twist angle is localized within the 20-bp response element interacting directly with the protein (16, 18) and the DNA twisting is uniformly distributed between the pentamers. Therefore, each of the two “center-to-center” fragments, i.e., from the center of pentamer 1 to 2, and from 3 to 4, absorbs ≈25% of the total overtwisting (ΔTwist/4 = 35°/4 ≈ 9° for p53DBD and 70°/4 = 17.5° for wt p53; see Fig. 4). The absolute value of Twist = 170° for free DNA in solution (cross F in Fig. 4) was calculated by using the dimeric twist angles of Kabsch et al. (26).
Under these assumptions, the point corresponding to experiment for p53DBD (the cross DBD in Fig. 4) lies at the center of a wide asymmetric area representing energetically acceptable configurations of the tetramer. (This point does not coincide precisely with the minimum in energy, but rather is shifted in the direction predicted by entropic effects.) This DBD configuration, presented in Fig. 5, is characterized by favorable interactions between both parallel and antiparallel p53 subunits. Unlike straight B-DNA, where the H1 helices from two antiparallel p53 domains overlap (18, 19), there is no H1–H1 clash in this structure. Instead, the H1–H1 contact energy has negative van der Waals and electrostatic components (19). Because of the restrictions on the system imposed in the course of simulations, the p53DBD–DNA interactions are consistent with the cocrystal structure (24) and thus preserve the sites of specific p53–DNA recognition observed in that study.
Figure 5.

Three-dimensional model for four p53DBD subunits bound to bent DNA. The H2 helices are involved in direct DNA recognition in the major groove (24). The H1–H1 interactions are operative in causing DNA bending and twisting (26, 29). The broken lines indicate that in the wt p53 tetramer bound to DNA, the N termini are located on the external side of the DNA loop, whereas the C termini fragments are on the internal side (the tetramerization and basic domains are not shown). Small arrows denote putative interactions between the proline-rich N fragments and the p53 core domains.
The experimental point for wt p53 is at the edge of the acceptable area (Fig. 4). Note, however, that the energy map is calculated for the p53DBD tetramer, not for wt p53. On the other hand, the location of the wt cross on the (Bend, Twist) map is generally consistent with the energy profile. All three crosses, F (for free DNA), DBD, and wt, lie approximately on a straight line running along the bottom of the energy “ravine” (Fig. 4), and the positive correlation between Bend and Twist described above therefore remains valid.
Direct comparison of the protein-induced DNA bend angles based on gel data (Fig. 2) is problematic because of the uncertainty caused by physical sizes of the bound proteins (29). This uncertainty is partially resolved, however, by the analysis of the “twisting” gels shown in Fig. 3. In this case, it is not important whether the protein is large or small, or whether it lies on the interior or exterior of the DNA loop. Because the measured Bend and ΔTwist angles increase concomitantly when DBD is substituted by wt p53, and because this trend is consistent with the stereochemical data (Fig. 4), we suggest that the increased Bend measured here for wt p53 reflects the real architecture of the tetrameric wt complex in solution.
CONCLUSIONS AND IMPLICATIONS OF THE MODEL
The present model for a sequence specific-tetrameric complex between p53DBD and a response element is consistent with a wide variety of biochemical evidence, including ligase-mediated cyclization (17), cyclic permutation (21), a variety of chemical probe studies (18), and the use of phasing analysis in the present work. The latter has led to the determination of both the magnitude and directionality of the DNA bend in the complex. The bending directionality agrees with our earlier prediction (18, 19), following the major groove bending at conserved CATG tetramers that has been observed in other nucleoprotein systems (13–15).
A particularly important finding in the present work is that the DNA bending and twisting angles are significantly larger in the wt p53 complex than in the complex with the p53 core domains. This strongly indicates that the p53 domains flanking the p53DBD also are involved in DNA binding.
Based on our model, we suggest that the N termini interact with the DBD, thereby affecting the DBD–DBD interactions, whereas the C termini could interact directly with the DNA. The N termini are positioned close to the adjacent DBDs, and their interactions could lead to an increased bend and twist in the DNA bound to wt p53 (Fig. 5). This interaction can also account for the variable length of spacer DNA between the p53 half-sites (8–9) because the N terminus potentially could form a bridge between separated p53 dimers. This would be an aspect of indirect recognition in the binding specificity of p53. Finally, the N termini are relatively exposed and accessible to other proteins after p53 tetramer is bound to DNA, consistent with their involvement in downstream signal transduction pathways.
The functional roles of the C termini are equally important and include moderating p53 DNA binding properties and promoting DNA looping (30, 31). In the present model, the C termini interacting electrostatically with the DNA would increase the bend simply by pulling the DNA toward the center of the bend. On the other hand, our model opens many possibilities for allosteric control of p53 binding (32, 33) implicit in the location of the N and C termini of the p53DBD on the outside and inside of the DNA loop, respectively (Fig. 5). For example, the flexible spacers in the C termini could change their orientation with respect to DNA and the entire complex. This is of special interest because the C termini undergo posttranslational modifications including both acetylation and phosphorylation, which appear to be regulated by DNA damage (34, 35).
Our model suggests that direct binding of p53 might occur to nucleosomes (19) and that p53 binding to unfolded chromatin might offer possible mechanisms for the detection of DNA damage. The external location of the p53DBD moieties on the DNA and the critical importance of major groove flexibility elements CA|TG at helically phased sites in the response elements are suggestive of such a binding because the CA:TG dimers lie on the outside of nucleosomal DNA (36, 37).
The model also offers insights into the remarkable binding specificity and selectivity of p53. Because p53 is such a pleiotropic transcription factor having many functions (including that of enhancer), the demands for binding specificity and selectivity are necessarily extraordinary. This is evidently accomplished through its tetrameric association with a repetitive binding site in which precise steric fit is extremely important. Steric fit is accommodated through both DNA bending and twisting; because these are coupled (Fig. 4), it is possible that the binding specificity of the p53 system could also be regulated by local supercoiling in promoter regions either by specific bending in larger transcriptional complexes or because of nearby architectural elements (38).
Acknowledgments
The authors thank Professor Ilga Winicov for many helpful suggestions and Dr. Mary Anderson and Professor Peter Tegtmeyer for the generous gift of purified wild-type p53 that enabled us to initiate these studies. The work was supported by research grants CA70274 and GM53517 from the National Institutes of Health (R.E.H.) and from the AIDS Targeted Anti-Viral Program of the Office of the Director of the National Institutes of Health (E.A.).
ABBREVIATIONS
- DBD
DNA binding domain
- wt
wild type
References
- 1.Levine A J. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 2.Ding H F, Fisher D E. Crit Rev Oncog. 1998;9:83–98. doi: 10.1615/critrevoncog.v9.i1.60. [DOI] [PubMed] [Google Scholar]
- 3.Meek D W. Cell Signalling. 1998;10:159–166. doi: 10.1016/s0898-6568(97)00119-8. [DOI] [PubMed] [Google Scholar]
- 4.El-Deiry W S, Kern S E, Pietenpol J A, Kinzler K W, Vogelstein B. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 5.Halazonetis T D, Kandil A N. EMBO J. 1993;12:5057–5064. doi: 10.1002/j.1460-2075.1993.tb06199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman P N, Chen X B, Bargonetti J, Prives C. Proc Natl Acad Sci USA. 1993;90:3319–3323. doi: 10.1073/pnas.90.8.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stenger J E, Tegtmeyer P, Mayr G A, Reed M, Wang Y, Wang P, Hough P V, Mastrangelo I A. EMBO J. 1994;13:6011–6020. doi: 10.1002/j.1460-2075.1994.tb06947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tokino T, Thiagalingam S, El-Deiry W S, Waldman T, Kinzler K W, Vogelstein B. Hum Mol Genet. 1994;3:1537–1542. doi: 10.1093/hmg/3.9.1537. [DOI] [PubMed] [Google Scholar]
- 9.Waterman J L F, Shenk J L, Halazonetis T D. EMBO J. 1995;14:512–519. doi: 10.1002/j.1460-2075.1995.tb07027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNamara P T, Bolshoy A, Trifonov E N, Harrington R E. J Biomol Struct Dyn. 1990;8:529–538. doi: 10.1080/07391102.1990.10507827. [DOI] [PubMed] [Google Scholar]
- 11.Zhurkin V B, Ulyanov N B, Gorin A A, Jernigan R L. Proc Natl Acad Sci USA. 1991;88:7046–7050. doi: 10.1073/pnas.88.16.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagaich A K, Bhattacharyya D, Brahmachari S K, Bansal M. J Biol Chem. 1994;269:7824–7833. [PubMed] [Google Scholar]
- 13.Werner M H, Gronenborn A M, Clore G M. Science. 1996;271:778–784. doi: 10.1126/science.271.5250.778. [DOI] [PubMed] [Google Scholar]
- 14.Dickerson R E. Nucleic Acids Res. 1998;26:1906–1926. doi: 10.1093/nar/26.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olson W K, Gorin A A, Lu X J, Hock L M, Zhurkin V B. Proc Natl Acad Sci USA. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho Y J, Gorina S, Jeffrey P D, Pavletich N P. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 17.Balagurumoorthy P, Sakamoto H, Lewis M S, Zambrano N, Clore G M, Gronenborn A M, Appella E, Harrington R E. Proc Natl Acad Sci USA. 1995;92:8591–8595. doi: 10.1073/pnas.92.19.8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagaich A K, Zhurkin V B, Sakamoto H, Gorin A A, Clore G M, Gronenborn A M, Appella E, Harrington R E. J Biol Chem. 1997;272:14830–14841. doi: 10.1074/jbc.272.23.14830. [DOI] [PubMed] [Google Scholar]
- 19.Durell S R, Appella E, Nagaich A K, Harrington R E, Jernigan R L, Zhurkin V B. In: Structure, Motion, Interaction and Expression of Biological Macromolecules. Proceedings of the Tenth Conversation. Sarma R H, Sarma M H, editors. Vol. 2. Schenectady, NY: Adenine; 1998. pp. 277–295. [Google Scholar]
- 20.Wang Y, Schwedes J F, Parks D, Mann K, Tegtmeyer P. Mol Cell Biol. 1995;15:2157–2165. doi: 10.1128/mcb.15.4.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagaich A K, Appella E, Harrington R E. J Biol Chem. 1997;272:14842–14849. doi: 10.1074/jbc.272.23.14842. [DOI] [PubMed] [Google Scholar]
- 22.Zinkel S, Crothers D M. Nature (London) 1987;328:178–181. doi: 10.1038/328178a0. [DOI] [PubMed] [Google Scholar]
- 23.Crothers D M, Haran T E, Nadeau J G. J Biol Chem. 1990;265:7093–7096. [PubMed] [Google Scholar]
- 24.Niederweis M, Hillen W. Electrophoresis. 1993;14:693–698. doi: 10.1002/elps.11501401110. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Reed M, Wang P, Stenger J E, Mayr G, Anderson M E, Schwedes J F, Tegtmeyer P. Genes Dev. 1993;7:2575–2586. doi: 10.1101/gad.7.12b.2575. [DOI] [PubMed] [Google Scholar]
- 26.Kabsch W, Sander S, Trifonov E N. Nucleic Acids Res. 1982;10:1097–1104. doi: 10.1093/nar/10.3.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kerppola T K, Curran T. Science. 1991;254:1210–1214. doi: 10.1126/science.1957173. [DOI] [PubMed] [Google Scholar]
- 28.Crothers D M, Drak J, Kahn J D, Levene S D. Methods Enzymol. 1992;212:3–29. doi: 10.1016/0076-6879(92)12003-9. [DOI] [PubMed] [Google Scholar]
- 29.Hagerman P J. Proc Natl Acad Sci USA. 1996;93:9993–9996. doi: 10.1073/pnas.93.19.9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenger J E, Tegtmeyer P, Mayr G A, Reed M, Wang Y, Wang P, Hough P V C, Mastrangelo I A. EMBO J. 1994;13:6011–6020. doi: 10.1002/j.1460-2075.1994.tb06947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson P, Mastrangelo I, Reed M, Tegtmeyer P, Yardley G, Barrett J. Oncogene. 1998;16:283–292. doi: 10.1038/sj.onc.1201491. [DOI] [PubMed] [Google Scholar]
- 32.Halazonetis T D, Davis L J, Kandil A N. EMBO J. 1993;12:1021–1028. doi: 10.1002/j.1460-2075.1993.tb05743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefstin J A, Yamamoto K R. Nature (London) 1998;392:885–888. doi: 10.1038/31860. [DOI] [PubMed] [Google Scholar]
- 34.Gu W, Roeder R G. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 35.Sakaguchi K, Herrera J E, Saito S, Miki T, Bustin M, Vassilev A, Anderson C W, Appella E. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satchwell S, Drew H R, Travers A A. J Mol Biol. 1986;191:659–675. doi: 10.1016/0022-2836(86)90452-3. [DOI] [PubMed] [Google Scholar]
- 37.Luger K, Mader A W, Richmond R K, Sargent D F, Richmond T J. Nature (London) 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 38.Jayaraman L, Moorthy N C, Murthy K G K, Manley J L, Bustin M, Prives C. Genes Dev. 1998;12:462–472. doi: 10.1101/gad.12.4.462. [DOI] [PMC free article] [PubMed] [Google Scholar]