

Summary
Hemostasis and fibrinolysis, the biological processes that maintain proper blood flow, are the consequence of a complex series of cascading enzymatic reactions. Serine proteases involved in these processes are regulated by feedback loops, local cofactor molecules, and serine protease inhibitors (serpins). The delicate balance between proteolytic and inhibitory reactions in hemostasis and fibrinolysis, described by the coagulation, protein C and fibrinolytic pathways, can be disrupted, resulting in the pathological conditions of thrombosis or abnormal bleeding. Medicine capitalizes on the importance of serpins, using therapeutics to manipulate the serpin-protease reactions for the treatment and prevention of thrombosis and hemorrhage. Therefore, investigation of serpins, their cofactors, and their structure-function relationships is imperative for the development of state-of-the-art pharmaceuticals for the selective fine-tuning of hemostasis and fibrinolysis. This review describes key serpins important in the regulation of these pathways: antithrombin, heparin cofactor II, protein Z-dependent protease inhibitor, α1-protease inhibitor, protein C inhibitor, α2-antiplasmin and plasminogen activator inhibitor-1. We focus on the biological function, the important structural elements, their known non-hemostatic roles, the pathologies related to deficiencies or dysfunction, and the therapeutic roles of specific serpins.
Keywords: α1-protease inhibitor, α2-antiplasmin, antithrombin, fibrinolysis, hemostasis, heparin, heparin cofactor II, plasminogen activator inhibitor-1, protein C inhibitor, protein Z-dependent protease inhibitor, serpins, thrombosis
Introduction
Blood flow is maintained by the proper balance of hemostasis and fibrinolysis, an interdependent network of physiological processes and succession of proteolytic reactions. Hemostasis, the physiological cessation of bleeding, involves the interaction of vasoconstriction, platelet aggregation and coagulation. The end result of coagulation is the deposition of cross-linked fibrin polymers to form blood clots. Both the protein C and the fibrinolytic pathways are activated by the coagulation pathway and serve to restrict excessive clot formation or thrombosis. The enzymatic reactions that propel these pathways are dominated by serine proteases and are subject to control by serpins and their local cofactors. Dysfunction, deficiencies or over-expression of serpins can cause either abnormal bleeding or thrombosis. Investigations into the structure and related activities of serpins, their target proteases and cofactors have provided valuable information regarding both serpin-related disease states and potential mechanisms by which medicine can manipulate serpin-protease interactions for the treatment and prevention of thrombosis and bleeding.
Hemostasis
Coagulation pathway
The factors of the coagulation pathway generally circulate in an inactive state until they are activated through proteolysis by an upstream factor. While the end goal of coagulation is fibrin polymerization, the most crucial feature of the coagulation pathway is the generation of thrombin (Fig. 1). Thrombin is responsible for cleaving fibrinogen to fibrin, activating factor (F) XIII to FXIIIa (which cross-links fibrin), activating platelets, and positively feeding back into the cycle by activating upstream factors [1].
Fig. 1.
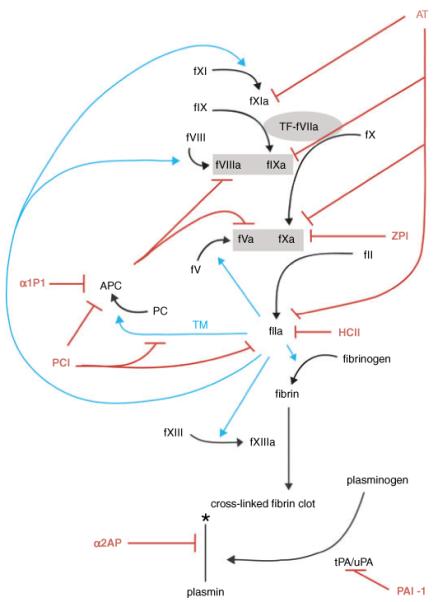
Serpin regulation of coagulation, protein C and fibrinolytic pathways. Serpins and inhibitory functions are shown in red, thrombin activity is shown in cyan. Prothrombinase and tenase complexes are shown in gray boxes. Coagulation is initiated by the exposure of tissue factor to factor VIIa shown in a gray oval. The symbol * indicates degradation. Necessary cofactors, Ca++, phospholipids, proteins S and Z, vitronectin and GAGs are not shown to maintain the simplicity of the schematic.
Thrombin generation is initiated when damage to a vessel wall exposes the blood to tissue factor (TF) in the subendothelium [2]. TF is also expressed by activated platelets and leukocytes [3]. Therefore, coagulation can also be initiated by inflammation. TF forms a complex with FVIIa and activates FX. Together, FVa and FXa form the prothrombinase complex, which then cleaves a small amount of prothrombin (FII) to thrombin (FIIa). This small amount of thrombin activates platelets, FV, FVIII and FXI, feeding back into the cycle to increase thrombin formation. Factor IXa, previously activated by either TF-VIIa or by FXIa on the platelet surface, and FVIIIa in the presence of calcium, complex on the platelet surface to form the platelet tenase complex. Platelet tenase activates more FX, which with FVa, generates a ‘thrombin burst’ (Fig. 1). It is this burst of thrombin rather than the initial thrombin activation that is crucial for the formation of a stable hemostatic plug [2].
In addition to its role in hemostasis, thrombin regulates many proinflammatory processes including leukocyte adhesion molecule expression on the endothelium, platelet activation, leukocyte chemotaxis and endothelial cell production of prothrombotic factors [4]. Thrombin is also a potent growth factor, initiating endothelial, fibroblast and smooth muscle cell proliferation and up-regulating other cytokines and growth factors [5]. These activities have been attributed to proteolytic cleavage of insulin-like growth factor binding proteins [6] and protease activated receptors -1, -3 and -4 (PAR-1, -3, -4) [7] on cell surfaces, and account for thrombin’s central role in atherosclerotic lesion formation [8].
Coagulation is regulated predominantly by antithrombin (AT) [9], tissue factor pathway inhibitor (TFPI) [10], the protein C pathway [11] and to a lesser extent heparin cofactor II (HCII) [12] and protein Z-dependent protease inhibitor (ZPI) [13]. Protein C inhibitor (PCI) and plasminogen activator inhibitor-1 (PAI-1) may also contribute by inhibiting thrombin [14,15]. TFPI is not a member of the serpin family and so will not be discussed in this paper.
Protein C pathway
The protein C pathway works in hemostasis to control thrombin formation in the area surrounding the clot [16]. The zymogen protein C (PC) is localized to the endothelium by endothelial cell protein C receptor (EPCR) [17]. Thrombin, generated via the coagulation pathway, is localized to the endothelium by binding to the integral membrane protein, thrombomodulin (TM). TM occupies exosite I on thrombin, which is needed for fibrinogen binding and cleavage, thus reducing thrombin’s procoagulant activities [18]. However, on the endothelial cell surface TM bound thrombin is able to cleave PC to activated protein C (APC), a serine protease [19]. In the presence of protein S, APC inactivates FVa and FVIIIa [20] (Fig. 1). This limits further thrombin generation on the clot periphery where the endothelium is not damaged [21].
The protein C pathway is also associated with non-hemostatic functions. APC has been shown to be an anti-inflammatory protein [22,23] and modulates gene expression [24]. It also enhances vascular permeability by signaling through both PAR-1 and sphingosine 1-phosphate receptor-1 [25]. In focal ischemic stroke animal models, APC treatment restored blood flow, and reduced infarct volume and inflammation [26]. These neuroprotective effects of APC were shown to be mediated through EPCR, PAR-1 [27] and PAR-3 [28]. In the PROWESS Study, patients diagnosed with severe sepsis were treated with recombinant human APC, resulting in a mortality reduction of 19.4% [29].
The proteolytic activity of APC is regulated predominantly by protein C inhibitor (PCI) [9]. Additionally, plasminogen activator inhibitor-1 (PAI-1) [30] and α1-protease inhibitor (α1PI) [31] have been shown to inhibit APC, although their role in hemostasis is not well understood.
Fibrinolysis
Fibrinolytic pathway
Fibrinolysis is the physiological breakdown of fibrin to limit and resolve blood clots [32]. Fibrin is degraded primarily by the serine protease, plasmin, which circulates as a zymogen, plasminogen. In an auto-regulatory manner, fibrin serves as both the cofactor for the activation of plasminogen and the substrate for plasmin (Fig. 1). In the presence of fibrin, tissue plasminogen activator (tPA) cleaves plasminogen to plasmin, which proteolyzes the fibrin. Because it is a necessary cofactor for the reaction, the degradation of fibrin limits further activation of plasminogen [33-35]. The serine protease, tPA, is synthesized and released by endothelial cells [32]. In addition to binding fibrin, tPA binds Annexin II (AnII) and other receptors on endothelial cell and platelet surfaces [36]. Thus, plasmin generation and fibrinolysis are restricted to the site of thrombus formation.
In addition to its role in fibrinolysis, plasmin has other physiological functions as evidenced by its ability to degrade components of the extracellular matrix [37] and activate matrix metalloproteases 2 and 9 [38,39]. Plasminogen can also be converted to plasmin by the serine protease, urokinase plasminogen activator (uPA) [37]. Urokinase-catalyzed events are localized on the cell surface through the uPA receptor (uPAR). Complex formation and subsequent reactions are thought to be more important during pericellular proteolysis, cell adhesion and migration than they are for vascular fibrinolysis [32]. These additional functions contribute to the role of the fibrinolytic pathway in cancer [37,40,41].
Fibrinolysis is controlled predominantly by α2-antiplasmin (α2AP) [42], PAI-1 [33,43] and thrombin activatable fibrinolysis inhibitor (TAFI) [44]. PCI can inhibit tPA and uPA [45,46], but its role in fibrinolysis is unclear. TAFI is not a member of the serpin family and so will not be discussed in this paper.
Serpin overview
Serpins
Serpins are a superfamily of proteins classified into 16 clades (A-P). The systematic name of each serpin is, SERPINXy where X is the clade and y is the number within the clade [47]. Serpins have been identified in the genomes of organisms representing all of the branches of life (Bacteria, Archaea, Eukarya and Viruses), and the genome of humans contains c. 36 serpins [48]. While serpins are named for their ability to inhibit serine proteases (of the chymotrypsin family) (Table 1), some are capable of cross-class inhibition of proteases from the subtilisin, papain and caspase families. In addition, some serpins utterly lack protease inhibitory activity and serve other roles, such as hormone transporters, molecular chaperones or catalysts for DNA condensation. Serpins are typically composed of c. 400 amino acids, but can have large N-, C-terminal or internal insertion loops [47]. Serpins can also be post-translationally modified by glycosylation, sulfation, phosphorylation and oxidation to alter their function. In spite of a low overall primary sequence identity for the family, serpins share a highly conserved three-dimensional fold comprised of a bundle of 9 α-helices (A-I) and a β-sandwich composed of three β-sheets (A-C) (Fig. 2A). It is useful to view a serpin in the ‘classic orientation’ to illustrate the important structural features (Fig. 2A, left panel). In this view the main β-sheet A is facing and the reactive site loop (RSL) is on top. The RSL is typically composed of 20 amino acids running from P17 at the N-terminus (at the C-terminal end of strand 5A) to P3′ at the C-terminal end (using the nomenclature of Schechter and Berger, where residues are numbered from the scissile P1-P1′ bond). In the normal native state of a serpin, β-sheet A is composed of five strands and the RSL (bridging the C-terminus of strand 5A to the N-terminus of strand 1C) is exposed. This state is, however, not the most stable. An astounding increase in thermodynamic stability (best estimate - 32 kcal mol-1) [49] can be achieved through the incorporation of the RSL into β-sheet A, triggered either through extension of strand 1C (to form the so-called ‘latent’ state), or through proteolytic nicking anywhere near the scissile bond (the cleaved state). The metastability of the native serpin is critical for its unusual mechanism of protease inhibition [50].
Table 1.
Second order rate constants of protease inhibition by serpins in the presence and absence of cofactors*
| Serpin | Systematic name | Target protease | Cofactor | Second order rate k2 (m-1 s-1) | Citation |
|---|---|---|---|---|---|
| AT | SERPINC1 | Thrombin | - | 7.5 × 103, 1 × 104 | [168], [169] |
| UFH | 2 × 107, 4.7 × 107 | [168],[170] | |||
| LMWH | 5.3 × 106 | [170] | |||
| Pentasaccharide | 2 × 104 | [169] | |||
| FXa | - | 2.5 × 103, 6 × 103 | [171] | ||
| UFH | 5 × 106, 6.6 × 106 | [169], [170] | |||
| LMWH | 1.3 × 106 | [170] | |||
| Pentasaccharide | 7.5 × 105 | [169] | |||
| FIXa | - | 1.3 × 102, 5 × 102 | [169], [172] | ||
| UFH | 8 × 106, 1.75 × 106 | [169], [172] | |||
| LMWH | 3.7 × 105 | [172] | |||
| Pentasaccharide | 3 × 104 | [169] | |||
| HCII | SERPIND1 | Thrombin | - | 6 × 102 | [168] |
| UFH | 5 × 106 | [168] | |||
| LMWH | ∼5 × 106 | [95] | |||
| Dermatan sulfate | 1 × 107 | [168] | |||
| Hexasaccharide | 2 × 104 | [173] | |||
| ZPI | SERPINA10 | FXa | - , Ca++, PL | 2.3 × 103 | [174] |
| Protein Z, Ca++, PL | 6.1 × 105 | [174] | |||
| FIXa | - | 2 × 105 | [101] | ||
| UFH | 4 × 105 | [101] | |||
| PCI | SERPINA5 | Thrombin | - | 1.7 × 104 | [57] |
| UFH | ∼2 × 105 | [57] | |||
| Thrombomodulin | 2.4 × 106 | [57] | |||
| APC | - | 3 × 102 | [175] | ||
| UFH | 5 × 104 | [175] | |||
| UFH, Ca++ | 2.9 × 105 | [176] | |||
| tPA (2-chain) | - | 8 × 102 | [45] | ||
| UFH | 3 × 104 | [45] | |||
| α1PI | SERPINA1 | Thrombin | - | 4.8 × 101 | [177] |
| APC | - | 4 × 101 | [178] | ||
| α1PIPittsburgh | Thrombin | - | 4.8 × 105 | [179] | |
| APC | - | 7 × 104 | [179] | ||
| α2AP | SERPINF2 | Plasmin | - | 2 × 107 | [42,180] |
| Plasminogen activator inhibitor-1 | SERPINE1 | Thrombin | - | 7.9 × 102 | [15] |
| UFH | 1.6 × 105 | [141] | |||
| Vitronectin | 1.9 × 105 | [141] | |||
| APC | - | 5.7 × 102 | [30] | ||
| Vitronectin | 1.8 × 105 | [30] | |||
| tPA (1- , 2-chain) | - | 4 × 107, 1.5 × 108 | [181] |
The rate constants indicated here are from selected references and may vary slightly under different experimental conditions. AT, antithrombin; APC, activated protein C; HCII, heparin cofactor II; ZPI, Z-dependent protease inhibitor; PCI, protein C inhibitor; α1-PI, α1-protease inhibitor; t-PA, tissue plasminogen activator; UHF, unfractionated heparin; LMWH, low-molecular-weight heparin; PL, phospholipids.
Fig. 2.
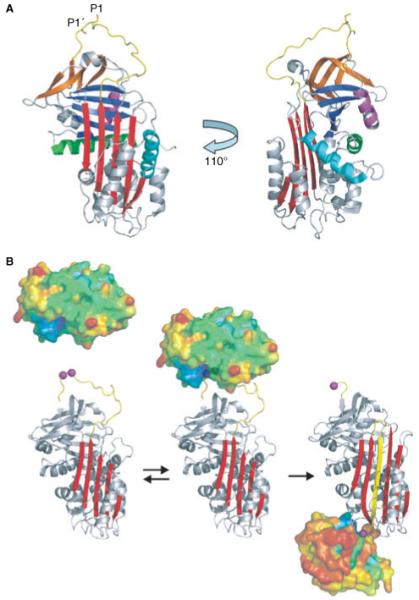
Serpin structure and mechanism of protease inhibition. (A) The shared serpin fold is illustrated by the structure of the prototypical native serpin α1PI. The ‘classic’ orientation shown on the left places the RSL (yellow) on top and the main β-sheet A (red) to the front. Sheets B and C are blue and orange, respectively, and helices A, D and H are colored green, cyan and magenta. The accessibility of the RSL is illustrated by rotating the molecule by 110° to the left along the long axis. It shows how the P1-P1′ (rods) scissile bond is exposed for proteolytic attack. Also clearer in this orientation are helices D and H, which are the heparin binding helices. (B) The serpin mechanism of protease inhibition is minimally expressed as a two-step process. In the first step, native serpin (ribbon with the P1 and P1′ residues as magenta balls) interacts reversibly with a protease (surface representation, colored according to temperature factors from blue to red) to form the Michaelis complex (middle). After formation of the acyl-enzyme intermediate the protease is flung to the opposite pole of the serpin and its catalytic architecture is destroyed, and consequently there is a loss of ordered structure (notice the smaller size and increase in temperature factors).
The serpin mechanism of protease inhibition
The serpin mechanism of protease inhibition has been worked out over the last 20 years through a series of biochemical, fluorescence and structural studies. A minimalist kinetic scheme is composed of two steps: the formation of the encounter complex (also known as the Michaelis complex) where the sequence of the RSL is recognized by the protease as a substrate; and the formation of a final covalent complex where the protease is trapped in an inactive state (Fig. 2B). The rates of formation and dissociation of the reversible Michaelis complex, along with colocalization in tissues, determines the specificity of the serpin-protease interaction [51,52]. While the obligate RSL-active site contacts contribute significantly to the formation of the Michaelis complexes, exosite interactions may also be involved. As with actual substrates of serine proteases, this step is followed by the nucleophilic attack of the peptide bond between the P1-P1′ residues by the catalytic Ser195 of the protease. This ultimately results in the formation of a covalent ester bond between the P1 residue and Ser195 of the protease (acyl-enzyme intermediate), and then separation of the P’ residues from the active site of the protease. At this stage the serpin rapidly adopts its lowest energy conformation through the incorporation of the N-terminal portion of the RSL into β-sheet A. The tethered protease is thus flung from the top to the bottom of the serpin (c. 70Å), and the resulting pulling force exerted on the catalytic loop results in a conformational distortion of the protease [53]. The acyl-enzyme intermediate is thus trapped, with deacylation prevented, largely because of the destruction of the oxyanion hole. Two structures of final complexes have been solved by X-ray crystallography [54,55], with one showing an additional distortion of c. 37% of the protease structure [55]. This mechanism is particularly well suited to tightly regulated processes such as hemostasis and fibrinolysis because inhibition is irreversible, and the conformational changes in the serpin and the protease alter cofactor interactions. An example of the physiologic relevance of the conformational change in the protease component of the complex is the complete destruction of thrombin’s exosite I in complex with serpins [56]. Thus, when PCI inhibits thrombin bound to thrombomodulin the interaction with thrombomodulin is broken, allowing the serpin-protease complex to diffuse away so that another thrombin molecule can bind [57].
Cofactor interactions
Because serpin specificity is determined largely by the rate of formation of the Michaelis complex, cofactors that bind to serpins (and sometimes the protease) can radically alter specificity [51]. Table 1 presents serpin second order rates of protease inhibition in the presence and absence of relevant cofactors. The best understood cofactor for serpins is the glycosaminoglycan (GAG), heparin. It binds to and activates most of the serpins involved in hemostasis and thrombosis [58]. Acceleration of protease inhibition is generally conferred through a template effect where the protease and the serpin bind to the same heparin chain. The hypothesis is that this co-occupation will limit the diffusional freedom from three to one dimension to increase the likelihood (rate) of encounter. In addition, heparin also provides a bridge between the serpin and the protease to help stabilize the Michaelis complex. However, heparin and other GAGs are also capable in some cases of altering the conformation of the serpin to permit more rapid complexation with proteases. The best-characterized examples are AT and HCII, whose activation by heparin is the basis of its therapeutic anticoagulant effect. In the next sections we describe each of the serpins involved in hemostasis and fibrinolysis, their targets, the role of cofactors and available structural data.
Serpins in hemostasis and fibrinolysis
Antithrombin: SERPINC1
Antithrombin is a 58 kDa, 432 amino acid glycoprotein [59], synthesized in the liver, circulating at approximately 150 μg mL-1 with a half-life of c. 3 days [60]. It is the most important physiological inhibitor of the coagulation pathway [61]. As its name implies, AT inhibits thrombin. In addition, AT is capable of inhibiting all of the other proteolytic coagulation factors (e.g. FIXa, Xa and XIa). The predominance of its anticoagulant activity, however, is focused on the regulation of FXa, FIXa and thrombin. Measurement of thrombin-AT (TAT) complex is used as a marker of hemostatic activation and helps diagnose thrombotic events [62]. Thrombin bound to fibrin, clot-bound thrombin, is protected from inhibition by AT [63]. This may explain the occurrence of rethrombosis after fibrinolytic therapy as clot-bound thrombin is released from the dissolving hemostatic plug [64].
The anticoagulant activity of AT is dependent on its cofactor, heparin. Consisting of variably sulfated repeating disaccharide units, heparin can have a molecular weight ranging from 3 to 40 kDa [65-67]. A unique pentasaccharide sequence in heparin is responsible for the high affinity binding to AT [68]. In vivo, forms of heparin relevant to AT include heparan sulfate, found on the endothelium, and heparin released from endothelium-associated mast cell granules. The interaction of AT with heparan sulfate on the endothelium and subendothelium localizes AT activity to the vessel wall and maintains its normal, non-thrombogenic nature [60]. AT is expressed as both an α-form and a β-form. α-AT represents 90% of AT and is glycosylated at all four positions. While comprising only 10% of AT, β-AT, which is not glycosylated at one position (N135), has a higher affinity for heparin and is thought to exert an overall larger anticoagulant effect [69].
Heparin uses two distinct mechanisms to accelerate protease inhibition by AT. AT undergoes a well-characterized conformational change upon heparin binding, which expels the N-terminus of the RSL from β-sheet A (Fig. 3). This ‘liberation’ of the RSL is sufficient to confer the majority of the acceleration of FIXa and FXa inhibition, but thrombin inhibition is not appreciably affected. Recently, the structures of the AT-heparin-protease Michaelis complexes have been solved [54,70] revealing the interactions behind the allosteric and template mechanisms (Fig. 3B).
Fig. 3.
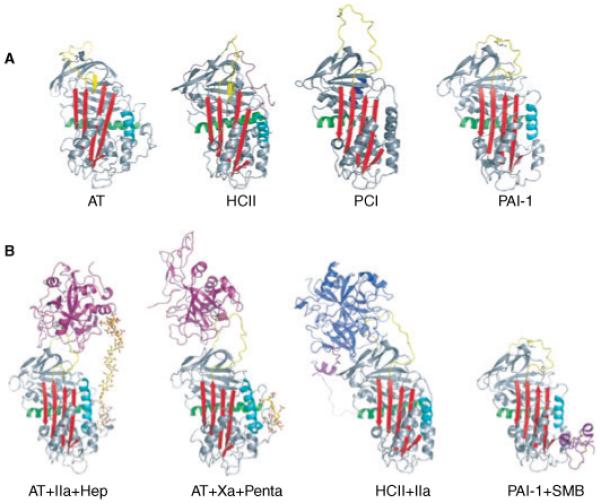
Native and complexed serpin structures. (A) The native structures of important hemostatic and fibrinolytic serpins are shown as ribbon diagrams, colored essentially as in Fig. 2. The monomeric structure of antithrombin is shown in the left panel, and is similar to that of heparin cofactor II (HCII) with the partial insertion of the N-terminal portion of the reactive site loop (RSL). A modeled position for the N-terminal tail of HCII is shown in magenta although its true position is not known. For AT, HCII and plasminogen activator inhibitor-1 the heparin binding helix (helix D) is shown in cyan, but for protein C inhibitor (PCI) heparin binds to helix H (blue). The increased size and flexibility of the RSL of PCI is also evident from this depiction. (B) Some important serpin complexes are shown. Using S195A proteases it was possible to obtain the structures of the AT Michaelis complexes with thrombin (magenta) and FXa (magenta) with their activating synthetic heparins (SR123781 and fondaparinux, rods). Similarly, the HCII-thrombin (blue) complex was also solved. The somatomedin (SMB) domain of VN (magenta) binds to s1A and helix E to prevent the latent transition through expansion of sheet A.
In addition to its anticoagulant activity, AT has been shown to have anti-inflammatory and anti-angiogenic functions. These properties are independent of AT’s inhibitory activity. AT regulates inflammation by signaling through heparan sulfate on endothelial and leukocyte cell surfaces [71]. Latent and cleaved AT exert anti-angiogenic effects [72] by binding cell surface heparan sulfate. This blocks fibroblast growth factor-2 and vascular endothelial cell growth factor from forming pro-angiogenic ternary signaling complexes with their protein receptors and the heparin sulfate co-receptors [73].
Antithrombin in disease
Inherited and acquired AT deficiency predisposes individuals to different degrees of thrombotic disease. The severity of thromophilia can be exacerbated by other risk factors for thrombosis. Inherited AT is classified as type I or type II. Type I deficiencies, which generally confer a higher thrombotic risk, are caused by genetic mutations that impair the synthesis and secretion of AT. Type II deficiencies are caused by genetic mutations that functionally impair AT. Variations in the degree of thrombophilia in inherited AT deficiencies can be attributed to homozygosity vs. heterozygosity and where the mutation lies in the AT structure [74]. An up-to-date database of AT mutations can be found online at http://www1.imperial.ac.uk/medicine/about/divisions/is/haemo/coag/antithrombin [75]. Some research suggests that certain mutations predispose AT to convert to its latent form, which preferentially dimerizes with native β-AT. Dimerization reduces the presence of highly active AT monomers, thus increasing thrombogenicity [76]. Concern has been raised that therapeutic preparations of AT-concentrates (contain > 10% latent AT) might have thrombotic effects. However, a recent study demonstrated that the addition of latent AT alone does not decrease the activity of AT in plasma [77]. Other mutated forms of AT do not show impaired activity or decreased AT levels in the standard hospital laboratory assays despite associated thrombophilia. In particular, the AT Cambridge II (A384S) variant was undetected by some protocols, but estimated to be the most frequent cause of AT deficiency in Caucasian populations [78]. These results suggest a need for alternative methods for detection of AT deficiencies [79].
Antithrombin-related treatments for coagulation disorders
Therapeutic unfractionated heparin (UFH), derived from porcine mucosa, is one of the most commonly used anticoagulant agents administered for treatment and prophylaxis of thrombotic events. Additionally, UFH is used to coat blood collection tubes and surgical devices to prevent clotting on their surfaces. UFH’s primary mechanism of action is to accelerate AT’s inhibition of thrombin, FXa and FIXa. UFH also accelerates thrombin inhibition by other circulating serpins. It has a very short half-life and optimal dosing of heparin is notoriously difficult to achieve, therefore requiring frequent monitoring [80]. Still in trials, an orally available form of heparin, sodium N-(8-[2-hydroxybenzoyl] amino) caprylate bound heparin or SNAC-heparin, has dose-dependent anti-thrombotic effects, and has an efficacy comparable with low-molecular weight heparin in reducing venous thrombosis in patients undergoing hip replacement surgery [81]. Contra-intuitively, heparin can cause a dangerous thrombotic condition called heparin-induced thrombocytopenia (HIT). In this autoimmune reaction antibodies develop against platelets [82]. Currently in development stages, synthetic oligosaccharide heparin mimetics show thrombin and FXa inhibition comparable with UFH without inducing HIT and with far fewer side effects [83].
Low-molecular-weight heparin (LMWH) is a fractionated preparation of heparin fragments between 1 and 10 kDa with an enriched population of high affinity pentasaccharide sequences. Because of the smaller average size, LMWH acts predominantly by inducing conformational changes in AT, the mechanism which activates FXa. It has a longer half-life than UFH and does not need coagulation monitoring. LMWH also has significantly reduced risk of HIT. Multiple LMWH variants are available or in clinical trials [84].
Fondiparinux and idraparinux are synthetic pentasaccharide sequences derived from heparin, which activate AT to inhibit FXa specifically. Because of this the single target, the side effect of over-anticoagulation, bleeding, is reduced. Both have longer half-lives than LMWH. Additionally, neither preparation causes HIT [84,85].
Thrombotic events because of AT deficiency are treated with AT purified from human plasma. A covalent AT-heparin complex is currently under preliminary investigations for possible use as a novel anticoagulant because unlike AT or heparin alone, it is able to inhibit clot-bound thrombin [86].
Heparin cofactor II: SERPIND1
HCII is a 66.5 kDa, 480 amino acid glycoprotein synthesized in the liver circulating at c. 80 μg mL-1 with a half-life of 2-3 days. HCII inhibits thrombin in the presence of many polyanionic molecules including the GAGs heparin and dermatan sulfate [12]. A unique hexasaccharide sequence within dermatan sulfate has been determined to be responsible for its high affinity binding to HCII [87]. Dermatan sulfate does not accelerate any other serpin activity. HCII has a unique N-terminal extension of c. 80 residues that contains two acidic regions, critical for its GAG-associated anti-thrombin activity [88]. HCII inhibits thrombin and clot-bound thrombin, but not other coagulation proteases [12]. Evidence suggests that HCII contributes 20-30% to thrombin inhibition in coagulation. Neither humans nor mice deficient in HCII exhibit thrombophilia under normal conditions [89]. However, HCII homozygous deficient mice form occlusive thrombi faster than wild-type mice after photochemical vascular endothelial cell injury to the carotid artery [90]. Recent data suggest that the primary physiological function of HCII is to inhibit thrombin’s non-hemostatic roles such as in the development of atherosclerosis. Elevated levels of HCII are shown to protect against atherosclerosis and restenosis [91-93].
The structure of native HCII was solved in 2002 and revealed a surprising resemblance to native AT [88], with the N-terminus of the RSL inserted into β-sheet A (Fig. 3). As HCII also shares a similar heparin binding site along helix D [9,94], it was proposed that HCII underwent a similar conformational change upon heparin binding [88]. Recently, it was shown that the smallest heparin length capable of tight binding to HCII was 14 monosaccharide unit chains, and that the majority of the acceleration effect was due to this allosteric change in HCII conformation [95]. Disappointingly, however, the native structure could not resolve the position of the N-terminal extension. Several mutagenesis studies concluded that the acidic tail binds to the basic heparin binding site in native HCII [94], but it is still unclear how the tail interacts with the body of HCII in the native state. From the structure of HCII bound to S195A thrombin [88] it was clear how the tail confers specificity to thrombin. The tail binds to exosite I of thrombin in a manner similar to hirudin, primarily through hydrophobic contacts. The tail was found sandwiched between thrombin and the body of HCII, essentially providing a shared exosite (Fig. 3B). A complex allosteric mechanism has been proposed based on these structures, supporting biochemical studies and analogy to AT [52,58].
HCII-related treatments for coagulation disorders
As alternatives to heparin-based treatments, dermatan sulfate derivatives and other polyanionic molecules that act to accelerate HCII’s antithrombotic activity are being investigated. They are of particular interest for use in HIT, AT deficiency and for the inhibition of clot-bound thrombin. Two types of fractionated dermatan sulfate enriched for the hexasaccharide sequence (Intimatan™ and Desmin™) have been tested in humans [96,97]. Intimatan is beginning Phase I trials. Other HCII agonists in laboratory investigations include over-sulfated dermatan sulfate [98], fucosylated chondroitin sulfate [99] and fucoidan [100].
Protein Z-dependent protease inhibitor: SERPINA10
ZPI is a 72 kDa, 444 amino acid glycoprotein, synthesized in the liver, circulating at c. 1.5 μg mL-1. In the presence of protein Z, phospholipids and calcium, ZPI rapidly inhibits FXa. In the absence of cofactors, ZPI also inhibits FXIa and can be accelerated 2-fold by heparin. It is thought that the major physiological function of ZPI is to attenuate the coagulation response prior to the formation of the prothrombinase complex [101]. In humans, mutations in ZPI are associated with increased risk of venous thrombosis[102-104]. Additionally, reductions in protein Z plasma concentrations result in an aggravated thromboembolic risk in humans and mice with factor V Leiden (FV Leiden) [105].
Protein C inhibitor: SERPINA5
PCI is a 57 kDa, 387 amino acid glycoprotein [106], synthesized in the liver, circulating at c. 5 μg mL-1. It is also found in other bodily fluids, including urine, saliva, amniotic fluid, milk, tears and seminal fluid [107]. PCI is a heparin-binding serpin that inhibits many proteases, including APC [108], IIa, IIa bound to TM [57], tPA and uPA [109]. PCI may have contrary anticoagulant and procoagulant functions depending on the target protease and the presence of specific cofactors. In the presence of heparin, PCI is anticoagulant, inhibiting the proteolytic cleavage of fibrinogen by thrombin. However, in the presence of TM, PCI is procoagulant, inhibiting the activation of PC by thrombin [9].
The structures of RSL-cleaved PCI [106] and of native PCI (PDB #2HI9 and #2OL2) (Fig. 3A) have now been solved, revealing a typical serpin structure with some notable differences. The RSL of PCI is unusually long and flexible, accounting for its broad protease specificity, and its heparin-binding site is found along the highly basic helix H. The effect of heparin on PCI activity can either be to accelerate protease inhibition (e.g. APC) [110] or to abrogate it (tissue kallikrein) [111]. The position of the heparin binding site close to the protease docking site may help explain this property.
Protein C inhibitor in disease
PCI is not synthesized by the liver in mice, and thus PCI is unlikely to play a role in hemostasis or fibrinolysis in the mouse [112]. Transgenic mice that over-express human PCI (hPCI) provide evidence of PCI’s ability to inhibit thrombin, APC and tPA. These mice do not exhibit symptoms of pulmonary hypertension induced by monocrotaline. TAT complexes are reduced compared with their wild-type counterparts, suggesting that PCI competes with AT to inhibit thrombin. Additionally, a decrease in free tPA and subsequent reduction in fibrinolysis is seen. Finally, when APC was administered for endotoxemia, hPCI-expressing transgenic mice demonstrated a reduction in the anticoagulant and anti-inflammatory effects of the treatment [113]. Male homozygous PCI-knockout mice were infertile because of abnormal spermatogenesis caused by loss of the Sertoli cell barrier [114] because of unregulated proteolytic activity.
In humans, APC-PCI complex is indicative of atherosclerosis and aortic aneurysms [115], an early indicator of myocardial infarction [116] and predicts poor patient outcome after aortic surgery [117]. Additionally, APC-PCI complex is increased (4-fold) in patients with FV Leiden who have suffered a previous venous thrombosis [118]. PCI alone has been shown to be elevated in survivors of acute coronary events [119].
α1-Protease inhibitor: SERPINA1
α1PI, historically known as α1-antitrypsin, is a 51 kDa, 394 amino acid glycoprotein, synthesized in the liver, circulating at c. 1.3 mg mL-1 with a half-life of 4.5 days (structure shown in Fig. 2). Its physiological target is neutrophil elastase [120]; however, it has also been shown to inhibit APC in a heparin-independent manner [31]. In pediatric ischemic stroke patients, α1PI levels were significantly increased independent of other prothrombotic factors. Some authors suggest this pathology is due to APC inhibition [121].
α1PI is not thought to contribute significantly to coagulation. However, a variant of the protein (α1PIPittsburgh) with a reactive site mutation (M358R) can cause a fatal bleeding disorder [122]. The Met to Arg polymorphism creates a potent inhibitor of several coagulation serine proteases, especially thrombin and APC, that is not dependent on heparin or other cofactors [123]. Therapies utilizing recombinant α1PIPittsburgh were considered but abandoned because of side-effects of promiscuous protease inhibition [124,125].
α2-Antiplasmin: SERPINF2
α2AP is a 63 kDa, 452 amino acid glycoprotein, synthesized in the liver, circulating at c. 70 μg mL-1 with a half-life of 2.6 days [42,126,127]. α2AP is the primary physiological inhibitor of plasmin, but has also been reported to inhibit other enzymes such as trypsin, elastase and APC. Homozygous deficiency of α2AP results in uncontrolled fibrinolysis and subsequent severe hemorrhagic tendencies [126,128]. While α2AP has all of the key structural features of the serpin family, it uniquely has both N- and C-terminal extensions of 42 and 55 residues, respectively [42,129]. In thrombus formation, the N-terminal region of α2AP is cross-linked to fibrin by FXIIIa, and the C-terminal Lys binds to the Lys-binding site of plasmin. The rate of fibrinolysis is proportional to cross-linked α2AP.
Plasminogen activator-1: SERPINE1
PAI-1 is a 50 kDa, 379 amino acid glycoprotein, synthesized in endothelial cells, platelets and other mesenchymal cells surrounding the vasculature [130-132]. This serpin is relatively unstable, with a half-life of 1-2 h in circulation [133]. However, PAI-1 is found bound to the extracellular matrix protein, vitronectin (VN) [134,135]. The PAI-1-VN complex has an enhanced half-life of 4-6 h [133]. PAI-1 regulates both tPA and uPA and is considered the main physiological inhibitor of plasminogen activation [32,33,43,136]. As platelets are activated following vessel injury, they release PAI-1 to protect the developing thrombus from premature fibrinolysis. Later in the coagulation process, tPA and plasminogen/plasmin are bound to fibrin within the thrombus, which protects tPA from inhibition by PAI-1, resulting in plasmin generation and fibrinolysis [32,33,43,136].
Several structures of PAI-1 have been solved, but, because of its rapid conversion to the latent form, all structures of native PAI-1 are of a stabilized quadruple mutant [137-139]. Although the structure shows a native state similar to α1PI, not AT and HCII, some mutagenesis studies suggest an equilibrium for wild-type PAI-1 where the native state is in equilibrium between α1PI-like and AT-like states [140]. The structure of the stabilized mutant bound to the somatomedin domain of VN revealed the mechanism of stabilization of the native state through a blocking of the expansion of β-sheet A [135] (Fig. 3B).
PAI-1 can also inhibit APC [30] and thrombin [15,141] in the presence of VN and/or heparin. It is not known to what extent these activities contribute to coagulation. Previously, it has been shown that APC cleaves PAI-1, inactivating the serpin [142,143]. Recently, it has been shown that PAI-1 inhibits APC and the rate of inhibition increases in the presence of VN c. three hundredfold [30].
Plasminogen activator-1 in disease
Studies show that PAI-1 levels are sensitive to many different pathophysiological factors and increased synthesis of PAI-1 contributes to numerous cardiovascular disease states. In metabolic syndrome, both glucose and insulin increase PAI-1 synthesis in vascular endothelial and smooth muscle cells [144]. Controlling hyperglycemia in type 2 diabetes results in a decrease in PAI-1 levels. One of the clinical benefits of ‘statins’ may be due to their decrease of PAI-1 expression and simultaneous increase of tPA expression, altering the balance of the fibrinolytic pathway [145,146]. Circadian clock proteins, including CLOCK, BMAL and CRY, regulate PAI-1 gene expression, which may explain the increased risk of adverse cardiovascular events in the morning [147]. Inhibition of nitric oxide synthase induces PAI-1 expression, which contributes to the development of perivascular fibrosis [148]. Increased PAI-1 levels are associated with coronary artery disease and myocardial infarction. However, studies examining the association of cardiovascular disease with a polymorphism within the PAI-1 promoter region (4 G/5 G) which increases the expression of PAI-1 are controversial [149]. Stents with rapamycin and paclitaxel are used in interventional cardiology because of the antiproliferative effects of these drugs [150,151]. These stents have been shown to be associated with an increased risk of thrombosis and it is speculated that this may be due to an up-regulation of PAI-1 by rapamycin and paclitaxel [152]. There is also strong evidence for a role of PAI-1 in cancer metastasis independent of its protease inhibitory activity [41,153,154].
Effectors of plasminogen activator inhibitor-1 activity and synthesis
Physiological levels of PAI-1 provide crucial regulation of fibrinolysis, yet excess levels contribute to disease. Numerous factors have been found that up-regulate PAI-1 expression and secretion, including inflammatory cytokines, angiotensin II, aldosterone, transforming growth factor-β, and very low density lipoproteins [43,155,156]. Monoclonal antibodies have been prepared against PAI-1, which express inhibitory activity by: (i) preventing the formation of the encounter complex between PAI-1 and tPA/uPA, (ii) increasing PAI’s susceptibility to cleavage by target proteases, and (iii) promoting the tendency of PAI-1 to become latent and inactive [157]. Sequence-specific catalytic DNA enzyme, short-interfering RNA structures and antisense technology have all been used to down-regulate PAI-1 levels [158]. Negatively charged organochemical compounds have been found to bind to a hydrophobic site on PAI-1 and induce polymerization and inactivation. Finally, several small molecules (XR5118, ZK4044 and PAI-039) have been developed to inhibit PAI-1 activity by either reduction of accessibility to the RSL, by promoting a latent-like state or by favoring a substrate-like conformation [159-167].
Closing statement
In this State of the Art manuscript, we have described our current knowledge of serpins that regulate hemostasis and fibrinolysis. Utilizing the ‘suicide substrate’ mechanism unique to serpins AT, HCII, ZPI, α1PI, PCI, α2AP and PAI-1 provides rapid and specific inhibition of the activated serine proteases in the coagulation, protein C and fibrinolytic pathways. These pathways are not single independent systems, but they represent a dynamic balance between procoagulant, anticoagulant, profibrinolytic and antifibrinolytic states with serpins playing multiple and sometimes conflicting roles. While we have learned a considerable amount about their physiological control, their structure, related activities and regulation by local cofactors, much is left to be understood. Of note, this includes deciphering the primary physiologic roles of HCII and PCI, resolving the crystal structures of ZPI and α2AP, and learning more about the non-hemostatic functions of AT and PAI-1. Continued research with the less-studied serpins such as ZPI and α2AP will undoubtedly provide useful information about control of hemostasis and fibrinolysis and of serpins in general. The serpins described in this paper have a multitude of functions, which under some circumstances contribute to disease, but which often can be manipulated for the benefit of medicine. Therefore, it is of paramount importance that we continue the investigations of serpins in thrombosis, hemostasis and fibrinolysis.
Acknowledgments
We gratefully acknowledge all the serpin scientists and their contributions over the last three decades, and we apologize to our respected colleagues whose important publications we were not able to cite or discuss because of space limitations. The F. C. Church Laboratory is supported in part by the National Institutes of Health (HL-32 656) and in part by the Susan G. Komen Breast Cancer Foundation (BCTR0503475 and BCTR45206). Current stipend support for J. C. Rau is through the UNC-CH Integrative Vascular Biology Program NIH grant (T32 HL69768) and the Gertrude B. Elion Mentored Medical Student Award from the Triangle Community Foundation. Current stipend support for L. M. Beaulieu is supported in part through an NRSA-NIH predoctoral fellowship (F31 NS054590) and was previously supported through the UNC-CH Integrative Vascular Biology Program NIH grant (T32 HL69768) and the Susan G. Komen Breast Cancer Foundation (BCTR0503475). The J. A. Huntington Laboratory is supported by the Medical Research Council (UK), the British Heart Foundation, and the National Institutes of Health (HL-068 629).
Footnotes
Disclosure of Conflict of Interests
The authors state that they have no conflict of interests.
References
- 1.Bode W. The structure of thrombin: a janus-headed proteinase. Semin Thromb Hemost. 2006;32(Suppl 1):16–31. doi: 10.1055/s-2006-939551. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman M, Monroe DM. Coagulation 2006: a modern view of hemostasis. Hematol Oncol Clin North Am. 2007;21:1–11. doi: 10.1016/j.hoc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7. [DOI] [PubMed] [Google Scholar]
- 4.Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation. 2006;114:1070–7. doi: 10.1161/CIRCULATIONAHA.105.574830. [DOI] [PubMed] [Google Scholar]
- 5.Harker LA, Hanson SR, Runge MS. Thrombin hypothesis of thrombus generation and vascular lesion formation. Am J Cardiol. 1995;75:12B–7B. doi: 10.1016/0002-9149(95)80004-c. [DOI] [PubMed] [Google Scholar]
- 6.Zheng B, Clarke JB, Busby WH, Duan C, Clemmons DR. Insulin-like growth factor-binding protein-5 is cleaved by physiological concentrations of thrombin. Endocrinology. 1998;139:1708–14. doi: 10.1210/endo.139.4.5945. [DOI] [PubMed] [Google Scholar]
- 7.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–82. [PubMed] [Google Scholar]
- 8.Baykal D, Schmedtje JF, Runge MS. Role of thrombin receptor in restenosis and atherosclerosis. Am J Cardiol. 1995;75:82B–7B. doi: 10.1016/0002-9149(95)80019-o. [DOI] [PubMed] [Google Scholar]
- 9.Pike RN, Buckle AM, le Bonniec BF, Church FC. Control of the coagulation system by serpins. Getting by with a little help from glycosaminoglycans. FEBS J. 2005;272:4842–51. doi: 10.1111/j.1742-4658.2005.04880.x. [DOI] [PubMed] [Google Scholar]
- 10.Sandset PM. Tissue factor pathway inhibitor (TFPI) - an update. Haemostasis. 1996;26(Suppl 4):154–65. doi: 10.1159/000217293. [DOI] [PubMed] [Google Scholar]
- 11.Dahlback B, Villoutreix BO. The anticoagulant protein C pathway. FEBS Lett. 2005;579:3310–6. doi: 10.1016/j.febslet.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Tollefsen DM. Heparin cofactor II modulates the response to vascular injury. Arterioscler Thromb Vasc Biol. 2007;27:454–60. doi: 10.1161/01.ATV.0000256471.22437.88. [DOI] [PubMed] [Google Scholar]
- 13.Broze GJ., Jr. Protein Z-dependent regulation of coagulation. Thromb Haemost. 2001;86:8–13. [PubMed] [Google Scholar]
- 14.Cooper ST, Whinna HC, Jackson TP, Boyd JM, Church FC. Intermolecular interactions between protein C inhibitor and coagulation proteases. Biochemistry. 1995;34:12991–7. doi: 10.1021/bi00040a009. [DOI] [PubMed] [Google Scholar]
- 15.van Meijer M, Smilde A, Tans G, Nesheim ME, Pannekoek H, Horrevoets AJ. The suicide substrate reaction between plasminogen activator inhibitor 1 and thrombin is regulated by the cofactors vitronectin and heparin. Blood. 1997;90:1874–82. [PubMed] [Google Scholar]
- 16.Esmon CT. The protein C pathway. Chest. 2003;3(Suppl 3):26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 17.Fukudome K, Kurosawa S, Stearns-Kurosawa DJ, He X, Rezaie AR, Esmon CT. The endothelial cell protein C receptor. Cell surface expression and direct ligand binding by the soluble receptor. J Biol Chem. 1996;271:17491–8. doi: 10.1074/jbc.271.29.17491. [DOI] [PubMed] [Google Scholar]
- 18.Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, et al. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature. 2000;404:518–25. doi: 10.1038/35006683. [DOI] [PubMed] [Google Scholar]
- 19.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci USA. 1996;93:10212–6. doi: 10.1073/pnas.93.19.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu D, Kalafatis M, Mann KG, Long GL. Comparison of activated protein C/protein S-mediated inactivation of human factor VIII and factor V. Blood. 1996;87:4708–17. [PubMed] [Google Scholar]
- 21.Esmon CT. Inflammation and thrombosis. J Thromb Haemost. 2003;1:1343–8. doi: 10.1046/j.1538-7836.2003.00261.x. [DOI] [PubMed] [Google Scholar]
- 22.Feistritzer C, Mosheimer BA, Sturn DH, Riewald M, Patsch JR, Wiedermann CJ. Endothelial protein C receptor-dependent inhibition of migration of human lymphocytes by protein C involves epidermal growth factor receptor. J Immunol. 2006;176:1019–25. doi: 10.4049/jimmunol.176.2.1019. [DOI] [PubMed] [Google Scholar]
- 23.Yuda H, Adachi Y, Taguchi O, Gabazza EC, Hataji O, Fujimoto H, et al. Activated protein C inhibits bronchial hyperresponsiveness and Th2 cytokine expression in mice. Blood. 2004;103:2196–204. doi: 10.1182/blood-2003-06-1980. [DOI] [PubMed] [Google Scholar]
- 24.Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 25.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–84. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- 26.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–72. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 27.Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, et al. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–42. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 28.Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernandez JA, et al. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–72. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 29.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 30.Rezaie AR. Vitronectin functions as a cofactor for rapid inhibition of activated protein C by plasminogen activator inhibitor-1. Implications for the mechanism of profibrinolytic action of activated protein C. J Biol Chem. 2001;276:15567–70. doi: 10.1074/jbc.C100123200. [DOI] [PubMed] [Google Scholar]
- 31.Heeb MJ, Griffin JH. Physiologic inhibition of human activated protein C by alpha 1-antitrypsin. J Biol Chem. 1988;263:11613–6. [PubMed] [Google Scholar]
- 32.Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129:307–21. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- 33.Wiman B, Collen D. Molecular mechanism of physiological fibrinolysis. Nature. 1978;272:549–50. doi: 10.1038/272549a0. [DOI] [PubMed] [Google Scholar]
- 34.Loskutoff DJ, Quigley JP. PAI-1, fibrosis, and the elusive provisional fibrin matrix. J Clin Invest. 2000;106:1441–3. doi: 10.1172/JCI11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levi M, van der Poll T, Buller HR. Bidirectional relation between inflammation and coagulation. Circulation. 2004;109:2698–704. doi: 10.1161/01.CIR.0000131660.51520.9A. [DOI] [PubMed] [Google Scholar]
- 36.Miles LA, Hawley SB, Baik N, Andronicos NM, Castellino FJ, Parmer RJ. Plasminogen receptors: the sine qua non of cell surface plasminogen activation. Front Biosci. 2005;10:1754–62. doi: 10.2741/1658. [DOI] [PubMed] [Google Scholar]
- 37.Dano K, Behrendt N, Hoyer-Hansen G, Johnsen M, Lund LR, Ploug M, et al. Plasminogen activation and cancer. Thromb Haemost. 2005;93:676–81. doi: 10.1160/TH05-01-0054. [DOI] [PubMed] [Google Scholar]
- 38.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–62. [PubMed] [Google Scholar]
- 39.Taraboletti G, D’Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol. 2002;160:673–80. doi: 10.1016/S0002-9440(10)64887-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andreasen PA, Egelund R, Peterson HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sciences. 2000;57:25–40. doi: 10.1007/s000180050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manders P, Tjan-Heijnen VC, Span PN, Grebenchtchikov N, Foekens JA, Beex LV, et al. Predictive impact of urokinase-type plasminogen activator: plasminogen activator inhibitor type-1 complex on the efficacy of adjuvant systemic therapy in primary breast cancer. Cancer Res. 2004;64:659–64. doi: 10.1158/0008-5472.can-03-1820. [DOI] [PubMed] [Google Scholar]
- 42.Coughlin PB. Antiplasmin: the forgotten serpin? FEBS J. 2005;272:4852–7. doi: 10.1111/j.1742-4658.2005.04881.x. [DOI] [PubMed] [Google Scholar]
- 43.Vaughan DE. Angiotensin, fibrinolysis, and vascular homeostasis. Am J Cardiol. 2001;87:18C–24C. doi: 10.1016/s0002-9149(01)01509-0. [DOI] [PubMed] [Google Scholar]
- 44.Mosnier LO, Bouma BN. Regulation of fibrinolysis by thrombin activatable fibrinolysis inhibitor, an unstable carboxypeptidase B that unites the pathways of coagulation and fibrinolysis. Arterioscler Thromb Vasc Biol. 2006;26:2445–53. doi: 10.1161/01.ATV.0000244680.14653.9a. [DOI] [PubMed] [Google Scholar]
- 45.Espana F, Berrettini M, Griffin JH. Purification and characterization of plasma protein C inhibitor. Thromb Res. 1989;55:369–84. doi: 10.1016/0049-3848(89)90069-8. [DOI] [PubMed] [Google Scholar]
- 46.Heeb MJ, Espana F, Geiger M, Collen D, Stump DC, Griffin JH. Immunological identity of heparin-dependent plasma and urinary protein C inhibitor and plasminogen activator inhibitor-3. J Biol Chem. 1987;262:15813–6. [PubMed] [Google Scholar]
- 47.Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, et al. An overview of the serpin superfamily. Genome Biol. 2006;7:216. doi: 10.1186/gb-2006-7-5-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, Gettins PGW, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins: evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem. 2001;276:33293–6. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- 49.Im H, Ahn HY, Yu MH. Bypassing the kinetic trap of serpin protein folding by loop extension. Protein Sci. 2000;9:1497–502. doi: 10.1110/ps.9.8.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carrell RW, Evans DL, Stein P. Mobile reactive centre of serpins and the control of thrombosis. Nature. 1991;353:576–8. doi: 10.1038/353576a0. [DOI] [PubMed] [Google Scholar]
- 51.Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002;102:4751–804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 52.Huntington JA. Shape-shifting serpins-advantages of a mobile mechanism. Biochem Sci Trends. 2006;3:427–35. doi: 10.1016/j.tibs.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 53.Olson ST, Swanson R, Day D, Verhamme I, Kvassman J, Shore JD. Resolution of Michaelis complex, acylation, and conformational change steps in the reactions of the serpin, plasminogen activator inhibitor-1, with tissue plasminogen activator and trypsin. Biochemistry. 2001;40:11742–56. doi: 10.1021/bi0107290. [DOI] [PubMed] [Google Scholar]
- 54.Dementiev A, Petitou M, Herbert JM, Gettins PG. The ternary complex of antithrombin-anhydrothrombin-heparin reveals the basis of inhibitor specificity. Nat Struct Mol Biol. 2004;11:863–7. doi: 10.1038/nsmb810. [DOI] [PubMed] [Google Scholar]
- 55.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000;407:923–6. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 56.Bock PE, Olsen ST, Bjork I. Inactivation of thrombin by antithrombin is accompanied by inactivation of regulatory exosite I. J Biol Chem. 1997;272:19837–145. doi: 10.1074/jbc.272.32.19837. [DOI] [PubMed] [Google Scholar]
- 57.Rezaie AR, Cooper ST, Church FC, Esmon CT. Protein C inhibitor is a potent inhibitor of the thrombin-thrombomodulin complex. J Biol Chem. 1995;270:25336–9. doi: 10.1074/jbc.270.43.25336. [DOI] [PubMed] [Google Scholar]
- 58.Huntington JA. Mechanisms of glycosaminoglycan activation of the serpins in hemostasis. J Thromb Haemost. 2003;1:1535–49. doi: 10.1046/j.1538-7836.2003.00305.x. [DOI] [PubMed] [Google Scholar]
- 59.Johnson DJ, Langdown J, Li W, Luis SA, Baglin TP, Huntington JA. Crystal structure of monomeric native antithrombin reveals a novel reactive center loop conformation. J Biol Chem. 2006;281:35478–86. doi: 10.1074/jbc.M607204200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinsey NS, Greedy AL, Bottomley SP, Whisstock JC, Pike RN. Antithrombin: in control of coagulation. Int J Biochem Cell Biol. 2004;36:386–9. doi: 10.1016/s1357-2725(03)00244-9. [DOI] [PubMed] [Google Scholar]
- 61.Egeberg O. Inherited antithrombin III deficiency and thromboembolism. Thromb Diath Haemorrh. 1965;13:516–30. [PubMed] [Google Scholar]
- 62.Haverkate F. Levels of haemostatic factors, arteriosclerosis and cardiovascular disease. Vascul Pharmacol. 2002;39:109–12. doi: 10.1016/s1537-1891(02)00295-1. [DOI] [PubMed] [Google Scholar]
- 63.Hogg PJ, Jackson CM. Fibrin monomer protects thrombin from inactivation by heparin-antithrombin III: implications for heparin efficacy. Proc Natl Acad Sci USA. 1989;86:3619–23. doi: 10.1073/pnas.86.10.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weitz JI. Activation of blood coagulation by plaque rupture: mechanisms and prevention. Am J Cardiol. 1995;75:18B–22B. doi: 10.1016/0002-9149(95)80005-d. [DOI] [PubMed] [Google Scholar]
- 65.Weitz JI. Low-molecular-weight heparins. N Engl J Med. 1997;337:688–98. doi: 10.1056/NEJM199709043371007. [DOI] [PubMed] [Google Scholar]
- 66.Lindahl U, Kjellen L. Heparin or heparan sulfate: What is the difference? Thromb Haemost. 1991;66:44–8. [PubMed] [Google Scholar]
- 67.Rosenberg RD, Armand G, Lam L. Structure-function relationships of heparin species. Proc Natl Acad Sci USA. 1978;75:3065–9. doi: 10.1073/pnas.75.7.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Belzar KJ, Dafforn TR, Petitou M, Carrell RW, Huntington JA. The effect of reducing-end extension on pentasaccharide binding by antithrombin. JBC. 2000;275:8733–41. doi: 10.1074/jbc.275.12.8733. [DOI] [PubMed] [Google Scholar]
- 69.McCoy AJ, Pei XY, Skinner R, Abrahams JP, Carrell RW. Structure of beta-antithrombin and the effect of glycosylation on antithrombin’s heparin affinity and activity. J Mol Biol. 2003;326:823–33. doi: 10.1016/s0022-2836(02)01382-7. [DOI] [PubMed] [Google Scholar]
- 70.Johnson DJ, Li W, Adams TE, Huntington JA. Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 2006;25:2029–37. doi: 10.1038/sj.emboj.7601089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wiedermann Ch J, Romisch J. The anti-inflammatory actions of antithrombin-a review. Acta Med Austriaca. 2002;29:89–92. doi: 10.1046/j.1563-2571.2002.02012.x. [DOI] [PubMed] [Google Scholar]
- 72.O’Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science. 1999;285:1926–8. doi: 10.1126/science.285.5435.1926. [DOI] [PubMed] [Google Scholar]
- 73.Zhang W, Swanson R, Xiong Y, Richard B, Olson ST. Antiangiogenic antithrombin blocks the heparan sulfate-dependent binding of proangiogenic growth factors to their endothelial cell receptors: evidence for differential binding of antiangiogenic and anticoagulant forms of antithrombin to proangiogenic heparan sulfate domains. J Biol Chem. 2006;281:37302–10. doi: 10.1074/jbc.M604905200. [DOI] [PubMed] [Google Scholar]
- 74.van Boven HH, Lane DA. Antithrombin and its inherited deficiency states. Semin Hematol. 1997;34:188–204. [PubMed] [Google Scholar]
- 75.Lane DA, Bayston T, Olds RJ, Fitches AC, Cooper DN, Millar DS, et al. Antithrombin mutation database: 2nd (1997) update. For the Plasma Coagulation Inhibitors Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1997;77:197–211. [PubMed] [Google Scholar]
- 76.Corral J, Huntington JA, Gonzalez-Conejero R, Mushunje A, Navarro M, Marco P, et al. Mutations in the shutter region of antithrombin result in formation of disulfide-linked dimers and severe venous thrombosis. J Thromb Haemost. 2004;2:931–9. doi: 10.1111/j.1538-7836.2004.00749.x. [DOI] [PubMed] [Google Scholar]
- 77.Corral J, Rivera J, Guerrero JA, Minano A, Alberca I, Hernandez-Espinosa D, et al. Latent and polymeric antithrombin: clearance and potential thrombotic risk. Exp Biol Med. 2007;232:219–26. [PubMed] [Google Scholar]
- 78.Corral J, Hernandez-Espinosa D, Soria JM, Gonzalez-Conejero R, Ordonez A, Gonzalez-Porras JR, et al. Antithrombin Cambridge II (A384S): an underestimated genetic risk factor for venous thrombosis. Blood. 2007 doi: 10.1182/blood-2006-08-040774. in press. [DOI] [PubMed] [Google Scholar]
- 79.Kristensen SR, Rasmussen B, Pedersen S, Bathum L. Detecting antithrombin deficiency may be a difficult task - more than one test is necessary. J Thromb Haemost. 2007;5:617–8. doi: 10.1111/j.1538-7836.2007.02395.x. [DOI] [PubMed] [Google Scholar]
- 80.Bates SM, Weitz JI. The status of new anticoagulants. Br J Haematol. 2006;134:3–19. doi: 10.1111/j.1365-2141.2006.06134.x. [DOI] [PubMed] [Google Scholar]
- 81.Arbit E, Goldberg M, Gomez-Orellana I, Majuru S. Oral heparin: status review. Thromb J. 2006;4:6. doi: 10.1186/1477-9560-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Daneschvar HL, Daw H. Heparin-induced thrombocytopenia (an overview) Int J Clin Pract. 2007;61:130–7. doi: 10.1111/j.1742-1241.2006.00874.x. [DOI] [PubMed] [Google Scholar]
- 83.Petitou M, Herault JP, Bernat A, Driguez PA, Duchaussoy P, Lormeau JC, et al. Synthesis of thrombin-inhibiting heparin mimetics without side effects. Nature. 1999;398:417–22. doi: 10.1038/18877. [DOI] [PubMed] [Google Scholar]
- 84.Weitz JI. Emerging anticoagulants for the treatment of venous thromboembolism. Thromb Haemost. 2006;96:274–84. doi: 10.1160/TH06-05-0234. [DOI] [PubMed] [Google Scholar]
- 85.Hampton T. Agents to control bleeding show promise. JAMA. 2007;297:349–50. doi: 10.1001/jama.297.4.349. [DOI] [PubMed] [Google Scholar]
- 86.Patel S, Berry LR, Chan AK. Covalent antithrombin-heparin complexes. Thromb Res. doi: 10.1016/j.thromres.2006.08.003. in press. [DOI] [PubMed] [Google Scholar]
- 87.Maimone MM, Tollefsen DM. Structure of a dermatan sulfate hexasaccharide that binds to heparin cofactor II with high affinity. J Biol Chem. 1990;265:18263–71. [PubMed] [Google Scholar]
- 88.Baglin TP, Carrell RW, Church FC, Esmon CT, Huntington JA. Crystal structures of native and thrombin-complexed heparin cofactor II reveal a multistep allosteric mechanism. Proc Natl Acad Sci USA. 2002;99:11079–84. doi: 10.1073/pnas.162232399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tollefsen DM. Heparin cofactor II deficiency. Arch Pathol Lab Med. 2002;126:1394–400. doi: 10.5858/2002-126-1394-HCID. [DOI] [PubMed] [Google Scholar]
- 90.He L, Vicente CP, Westrick RJ, Eitzman DT, Tollefsen DM. Heparin cofactor II inhibits arterial thrombosis after endothelial injury. J Clin Invest. 2002;109:213–9. doi: 10.1172/JCI13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aihara K, Azuma H, Takamori N, Kanagawa Y, Akaike M, Fujimura M, et al. Heparin cofactor II is a novel protective factor against carotid atherosclerosis in elderly individuals. Circulation. 2004;109:2761–5. doi: 10.1161/01.CIR.0000129968.46095.F3. [DOI] [PubMed] [Google Scholar]
- 92.Schillinger M, Exner M, Sabeti S, Mlekusch W, Amighi J, Handler S, et al. High plasma heparin cofactor II activity protects from restenosis after femoropopliteal stenting. Thromb Haemost. 2004;92:1108–13. doi: 10.1160/TH04-05-0311. [DOI] [PubMed] [Google Scholar]
- 93.Takamori N, Azuma H, Kato M, Hashizume S, Aihara K, Akaike M, et al. High plasma heparin cofactor II activity is associated with reduced incidence of in-stent restenosis after percutaneous coronary intervention. Circulation. 2004;109:481–6. doi: 10.1161/01.CIR.0000109695.39671.37. [DOI] [PubMed] [Google Scholar]
- 94.Tollefsen DM. Insight into the mechanism of action of heparin cofactor II. Thromb Haemost. 1995;74:1209–14. [PubMed] [Google Scholar]
- 95.O’Keeffe D, Olson ST, Gasiunas N, Gallagher J, Baglin TP, Huntington JA. The heparin binding properties of heparin cofactor II suggest an antithrombin-like activation mechanism. J Biol Chem. 2004;279:50267–73. doi: 10.1074/jbc.M408774200. [DOI] [PubMed] [Google Scholar]
- 96.Buchanan MR, Brister SJ. Anticoagulant and antithrombin effects of intimatan, a heparin cofactor II agonist. Thromb Res. 2000;99:603–12. doi: 10.1016/s0049-3848(00)00276-0. [DOI] [PubMed] [Google Scholar]
- 97.Mungall D. Desmin 370 (Opocrin SpA/Alfa Wassermann) Drugs. 1999;2:579–83. [PubMed] [Google Scholar]
- 98.Pavao MS, Aiello KR, Werneck CC, Silva LC, Valente AP, Mulloy B, et al. Highly sulfated dermatan sulfates from Ascidians. Structure versus anticoagulant activity of these glycosaminoglycans. J Biol Chem. 1998;273:27848–57. doi: 10.1074/jbc.273.43.27848. [DOI] [PubMed] [Google Scholar]
- 99.Fonseca RJ, Mourao PA. Fucosylated chondroitin sulfate as a new oral antithrombotic agent. Thromb Haemost. 2006;96:822–9. [PubMed] [Google Scholar]
- 100.Church FC, Meade JB, Treanor RE, Whinna HC. Antithrombin activity of fucoidan: The interaction of fucoidan with heparin cofactor II, antithrombin III, and thrombin. J Biol Chem. 1989;264:3618–23. [PubMed] [Google Scholar]
- 101.Han X, Fiehler R, Broze GJ., Jr. Characterization of the protein Z-dependent protease inhibitor. Blood. 2000;96:3049–55. [PubMed] [Google Scholar]
- 102.Water N, Tan T, Ashton F, O’Grady A, Day T, Browett P, et al. Mutations within the protein Z-dependent protease inhibitor gene are associated with venous thromboembolic disease: a new form of thrombophilia. Br J Haematol. 2004;127:190–4. doi: 10.1111/j.1365-2141.2004.05189.x. [DOI] [PubMed] [Google Scholar]
- 103.Al-Shanqeeti A, van Hylckama Vlieg A, Berntorp E, Rosendaal FR, Broze GJ., Jr. Protein Z and protein Z-dependent protease inhibitor. Determinants of levels and risk of venous thrombosis. Thromb Haemost. 2005;93:411–3. doi: 10.1160/TH04-11-0715. [DOI] [PubMed] [Google Scholar]
- 104.Corral J, Gonzalez-Conejero R, Soria JM, Gonzalez-Porras JR, Perez-Ceballos E, Lecumberri R, et al. A nonsense polymorphism in the protein Z-dependent protease inhibitor increases the risk for venous thrombosis. Blood. 2006;108:177–83. doi: 10.1182/blood-2005-08-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kemkes-Matthes B, Nees M, Kuhnel G, Matzdorff A, Matthes KJ. Protein Z influences the prothrombotic phenotype in Factor V Leiden patients. Thromb Res. 2002;106:183–5. doi: 10.1016/s0049-3848(02)00181-0. [DOI] [PubMed] [Google Scholar]
- 106.Huntington JA, Kjellberg M, Stenflo J. Crystal structure of protein C inhibitor provides insights into hormone binding and heparin activation. Structure. 2003;11:205–15. doi: 10.1016/s0969-2126(02)00944-9. [DOI] [PubMed] [Google Scholar]
- 107.Laurell M, Christensson A, Abrahamsson PA, Stenflo J, Lilja H. Protein C inhibitor in human body fluids. Seminal plasma is rich in inhibitor antigen deriving from cells throughout the male reproductive system. J Clin Invest. 1992;89:1094–101. doi: 10.1172/JCI115689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aznar J, Espana F, Estelles A, Royo M. Heparin stimulation of the inhibition of activated protein C and other enzymes by human protein C inhibitor - influence of the molecular weightof heparin and ionic strength. Thromb Haemost. 1996;76:983–8. [PubMed] [Google Scholar]
- 109.Espana F, Estelles A, Fernandez P, Gilabert J, Sanchez-Cuenca J, Griffin J. Evidence for the regulation of urokinase and tissue type plasminogen activators by the serpin, protein C inhibitor, in semen and blood plasma. Thromb Hemost. 1993;70:989. [PubMed] [Google Scholar]
- 110.Pratt CW, Church FC. Heparin binding to protein C inhibitor. J Biol Chem. 1992;267:8789–94. [PubMed] [Google Scholar]
- 111.Ecke S, Geiger M, Resch I, Jerabek I, Sting L, Maier M, et al. Inhibition of tissue kallikrein by protein C inhibitor. Evidence for identity of protein C inhibitor with the kallikrein binding protein. J Biol Chem. 1992;267:7048–52. [PubMed] [Google Scholar]
- 112.Zechmeister-Machhart M, Hufnagl P, Uhrin P, Xu J, Geiger M, Binder BR. Molecular cloning and tissue distribution of mouse protein C inhibitor (PCI) Immunopharmacology. 1996;32:96–8. doi: 10.1016/0162-3109(95)00062-3. [DOI] [PubMed] [Google Scholar]
- 113.Nishii Y, Gabazza EC, Fujimoto H, Nakahara H, Takagi T, Bruno N, et al. Protective role of protein C inhibitor in monocrotaline-induced pulmonary hypertension. J Thromb Haemost. 2006 doi: 10.1111/j.1538-7836.2006.02174.x. in press. [DOI] [PubMed] [Google Scholar]
- 114.Uhrin P, Dewerchin M, Hilpert M, Chrenek P, Schofer C, Zechmeister-Machhart M, et al. Disruption of the protein C inhibitor gene results in impaired spermatogensis and male infertility. J Clin Invest. 2000;106:1531–9. doi: 10.1172/JCI10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kolbel T, Strandberg K, Mattiasson I, Stenflo J, Lindblad B. Activated protein C-protein C inhibitor complex: a new biological marker for aortic aneurysms. J Vasc Surg. 2006;43:935–9. doi: 10.1016/j.jvs.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 116.Bhiladvala P, Strandberg K, Stenflo J, Holm J. Early identification of acute myocardial infarction by activated protein C-protein C inhibitor complex. Thromb Res. 2006;118:213–9. doi: 10.1016/j.thromres.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 117.Nilsson G, Strandberg K, Astermark J, Vernersson E, Stenflo J, Berntorp E. The APC-PCI complex concentration predicts outcome of aortic surgery. Thromb Res. doi: 10.1016/j.thromres.2006.10.004. in press. [DOI] [PubMed] [Google Scholar]
- 118.Strandberg K, Stenflo J, Nilsson C, Svensson PJ. APC-PCI complex concentration is higher in patients with previous venous thromboembolism with Factor V Leiden. J Thromb Haemost. 2005;3:2578–80. doi: 10.1111/j.1538-7836.2005.01617.x. [DOI] [PubMed] [Google Scholar]
- 119.Carroll VA, Griffiths MR, Geiger M, Merlo C, Furlan M, Lammle B, et al. Plasma protein C inhibitor is elevated in survivors of myocardial infarction. Arterioscler Thromb Vasc Biol. 1997;17:114–8. doi: 10.1161/01.atv.17.1.114. [DOI] [PubMed] [Google Scholar]
- 120.Beatty K, Bieth J, Travis J. Kinetics of association of serine proteinases with native and oxidized alpha-1-proteinase inhibitor and alpha-1-antichymotrypsin. J Biol Chem. 1980;255:3931–4. [PubMed] [Google Scholar]
- 121.Burghaus B, Langer C, Thedieck S, Nowak-Gottl U. Elevated alpha1-antitrypsin is a risk factor for arterial ischemic stroke in childhood. Acta Haematol. 2006;115:186–91. doi: 10.1159/000090933. [DOI] [PubMed] [Google Scholar]
- 122.Owen MC, Brennan SO, Lewis JH, Carrell RW. Mutation of antitrypsin to antithrombin. Alpha1-antitrypsin Pittsburgh (358 Met to Arg), a fatal bleeding disorder. N Engl J Med. 1983;309:694–8. doi: 10.1056/NEJM198309223091203. [DOI] [PubMed] [Google Scholar]
- 123.Travis J, Matheson NR, George PM, Carrell RW. Kinetic studies on the interaction of alpha1-proteinase inhibitor (Pittsburgh) with trypsin-like serine proteinases. Biol Chem Hoppe Seyler. 1986;367:853–9. doi: 10.1515/bchm3.1986.367.2.853. [DOI] [PubMed] [Google Scholar]
- 124.Vidaud D, Emmerich J, Alhenc-Gelas M, Yvart J, Fiessinger JN, Aiach M. Met 358 to Arg mutation of alpha 1-antitrypsin associated with protein C deficiency in a patient with mild bleeding tendency. J Clin Invest. 1992;89:1537–43. doi: 10.1172/JCI115746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Heeb MJ, Bischoff R, Courtney M, Griffin JH. Inhibition of activated protein C by recombinant alpha-1-antitrypsin variants with substitution of arginine or leucine for methionine(356) J Biol Chem. 1990;265:2365–9. [PubMed] [Google Scholar]
- 126.Aoki N. Genetic abnormalities of the fibrinolytic system. Semin Thromb Hemost. 1984;10:42–50. doi: 10.1055/s-2007-1004406. [DOI] [PubMed] [Google Scholar]
- 127.Collen D, Wiman B. Turnover of antiplasmin, the fast-acting plasmin inhibitor of plasma. Blood. 1979;53:313–24. [PubMed] [Google Scholar]
- 128.Miles LA, Plow EF, Donnelly KJ, Hougie C, Griffin JH. A bleeding disorder due to deficiency of alpha 2-antiplasmin. Blood. 1982;59:1246–51. [PubMed] [Google Scholar]
- 129.Holmes WE, Nelles L, Lijnen HR, Collen D. Primary structure of human alpha 2-antiplasmin, a serine protease inhibitor (serpin) J Biol Chem. 1987;262:1659–64. [PubMed] [Google Scholar]
- 130.van Mourik JA, Lawrence DA, Loskutoff DJ. Purification of an inhibitor of plasminogen activator (antiactivator) synthesized by endothelial cells. J Biol Chem. 1984;259:14914–21. [PubMed] [Google Scholar]
- 131.Sprengers ED, Kluft C. Plasminogen activator inhibitors. Blood. 1987;69:381–7. [PubMed] [Google Scholar]
- 132.Fay WP. Plasminogen activator inhibitor 1, fibrin, and the vascular response to injury. Trends Cardiovasc Med. 2004;14:196–202. doi: 10.1016/j.tcm.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 133.Lawrence DA, Palaniappan S, Stefansson S, Olson ST, Francis-Chmura AM, Shore JD, et al. Characterization of the binding of different conformational forms of plasminogen activator inhibitor-1 to vitronectin. Implications for the regulation of pericellular proteolysis. J Biol Chem. 1997;272:7676–80. doi: 10.1074/jbc.272.12.7676. [DOI] [PubMed] [Google Scholar]
- 134.Keijer J, Ehrlich HJ, Linders M, Preissner KT, Pannekoek H. Vitronectin governs the interaction between plasminogen activator inhibitor 1 and tissue-type plasminogen activator. JBC. 1991;266:10700–7. [PubMed] [Google Scholar]
- 135.Zhou A, Huntington JA, Pannu NS, Carrell RW, Read RJ. How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat Struct Biol. 2003;10:541–4. doi: 10.1038/nsb943. [DOI] [PubMed] [Google Scholar]
- 136.Heimark RL, Kurachi K, Fujikawa K, Davie EW. Surface activation of blood coagulation, fibrinolysis and kinin formation. Nature. 1980;286:456–60. doi: 10.1038/286456a0. [DOI] [PubMed] [Google Scholar]
- 137.Sharp AM, Stein PE, Pannu NS, Carrell RW, Berkenpas MB, Ginsburg D, et al. The active conformation of plasminogen activator inhibitor 1, a target for drugs to control fibrinolysis and cell adhesion. Structure. 1999;7:111–8. doi: 10.1016/S0969-2126(99)80018-5. [DOI] [PubMed] [Google Scholar]
- 138.Nar H, Bauer M, Stassen JM, Lang D, Gils A, Declerck PJ. Plasminogen activator inhibitor 1. Structure of the native serpin, comparison to its other conformers and implications for serpin inactivation. J Mol Biol. 2000;297:683–95. doi: 10.1006/jmbi.2000.3604. [DOI] [PubMed] [Google Scholar]
- 139.Stout TJ, Graham H, Buckley DI, Matthews DJ. Structures of active and latent PAI-1: a possible stabilizing role for chloride ions. Biochemistry. 2000;39:8460–9. doi: 10.1021/bi000290w. [DOI] [PubMed] [Google Scholar]
- 140.Hagglof P, Bergstrom F, Wilczynska M, Johansson LB, Ny T. The reactive-center loop of active PAI-1 is folded close to the protein core and can be partially inserted. J Mol Biol. 2004;335:823–32. doi: 10.1016/j.jmb.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 141.Rezaie AR. Role of exosites 1 and 2 in thrombin reaction with plasminogen activator inhibitor-1 in the absence and presence of cofactors. Biochemistry. 1999;38:14592–9. doi: 10.1021/bi9913303. [DOI] [PubMed] [Google Scholar]
- 142.de Fouw NJ, van Hinsbergh VWM, de Jong YF, Haverkate F, Bertina RM. The interaction of activated protein C and thrombin with the plasminogen activator inhibitor released from human endothelial cells. Thromb Haemost. 1987;57:176–82. [PubMed] [Google Scholar]
- 143.Sakata Y, Loskutoff DJ, Gladson CL, Hekman CM, Griffin JH. Mechanism of protein C-dependent clot lysis: role of plasminogen activator inhibitor. Blood. 1986;68:1218–23. [PubMed] [Google Scholar]
- 144.Mertens I, Verrijken A, Michiels JJ, van der Planken M, Ruige JB, van Gaal LF. Among inflammation and coagulation markers, PAI-1 is a true component of the metabolic syndrome. Int J Obes (Lond) 2006;30:1308–14. doi: 10.1038/sj.ijo.0803189. [DOI] [PubMed] [Google Scholar]
- 145.Bourcier T, Libby P. HMG CoA reductase inhibitors reduce plasminogen activator inhibitor-1 expression by human vascular smooth muscle and endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:556–62. doi: 10.1161/01.atv.20.2.556. [DOI] [PubMed] [Google Scholar]
- 146.Wolfrum S, Jensen KS, Liao JK. Endothelium-dependent effects of statins. Arterioscler Thromb Vasc Biol. 2003;23:729–36. doi: 10.1161/01.ATV.0000063385.12476.A7. [DOI] [PubMed] [Google Scholar]
- 147.Oishi K, Shirai H, Ishida N. Identification of the circadian clock-regulated E-box element in the mouse plasminogen activator inhibitor-1 gene. J Thromb Haemost. 2007;5:428–31. doi: 10.1111/j.1538-7836.2007.02348.x. [DOI] [PubMed] [Google Scholar]
- 148.Kaikita K, Fogo AB, Ma L, Schoenhard JA, Brown NJ, Vaughan DE. Plasminogen activator inhibitor-1 deficiency prevents hypertension and vascular fibrosis in response to long-term nitric oxide synthase inhibition. Circulation. 2001;104:839–44. doi: 10.1161/hc3301.092803. [DOI] [PubMed] [Google Scholar]
- 149.Moore JH, Smolkin ME, Lamb JM, Brown NJ, Vaughan DE. The relationship between plasma t-PA and PAI-1 levels is dependent on epistatic effects of the ACE I/D and PAI-1 4 G/5 G polymorphisms. Clin Genet. 2002;62:53–9. doi: 10.1034/j.1399-0004.2002.620107.x. [DOI] [PubMed] [Google Scholar]
- 150.Moreno R, Fernandez C, Sanchez-Recalde A, Galeote G, Calvo L, Alfonso F, et al. Clinical impact of in-stent late loss after drug-eluting coronary stent implantation. Eur Heart J. doi: 10.1093/eurheartj/ehl423. in press. [DOI] [PubMed] [Google Scholar]
- 151.Joner M, Finn AV, Farb A, Mont EK, Kolodgie FD, Ladich E, et al. Pathology of drug-eluting stents in humans: delayed healing and late thrombotic risk. J Am Coll Cardiol. 2006;48:193–202. doi: 10.1016/j.jacc.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 152.Muldowney JA, III, Stringham JR, Levy SE, Gleaves LA, Eren M, Piana RN, et al. Antiproliferative agents alter vascular plasminogen activator inhibitor-1 expression: a potential prothrombotic mechanism of drug-eluting stents. Arterioscler Thromb Vasc Biol. 2007;27:400–6. doi: 10.1161/01.ATV.0000254677.12861.b8. [DOI] [PubMed] [Google Scholar]
- 153.Chambers SK, Ivins CM, Carcangiu ML. Plasminogen activator inhibitor-1 is an independent poor prognostic factor for survival in advanced stage epithelial ovarian cancer patients. Int J Cancer. 1998;79:449–54. doi: 10.1002/(sici)1097-0215(19981023)79:5<449::aid-ijc1>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 154.Costantini V, Sidoni A, Deveglia R, Cazzato OA, Bellezza G, Ferri I, et al. Combined overexpression of urokinase, urokinase receptor, and plasminogen activator inhibitor-1 is associated with breast cancer progression: an immunohistochemical comparison of normal, benign, and malignant breast tissues. Cancer. 1996;77:1079–88. doi: 10.1002/(sici)1097-0142(19960315)77:6<1079::aid-cncr12>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 155.Vaughan DE. PAI-1 and atherothrombosis. J Thromb Haemost. 2005;3:1879–83. doi: 10.1111/j.1538-7836.2005.01420.x. [DOI] [PubMed] [Google Scholar]
- 156.Ma J, Weisberg A, Griffin JP, Vaughan DE, Fogo AB, Brown NJ. Plasminogen activator inhibitor-1 deficiency protects against aldosterone-induced glomerular injury. Kidney Int. 2006;69:1064–72. doi: 10.1038/sj.ki.5000201. [DOI] [PubMed] [Google Scholar]
- 157.Gils A, Declerck PJ. The structural basis for the pathophysiological relevance of PAI-I in cardiovascular diseases and the development of potential PAI-I inhibitors. Thromb Haemost. 2004;91:425–37. doi: 10.1160/TH03-12-0764. [DOI] [PubMed] [Google Scholar]
- 158.Xiang G, Schuster MD, Seki T, Witkowski P, Eshghi S, Itescu S. Downregulated expression of plasminogen activator inhibitor-1 augments myocardial neovascularization and reduces cardiomyocyte apoptosis after acute myocardial infarction. J Am Coll Cardiol. 2005;46:536–41. doi: 10.1016/j.jacc.2005.04.047. [DOI] [PubMed] [Google Scholar]
- 159.Bjorquist P, Ehnebom J, Inghardt T, Hansson L, Lindberg M, Linschoten M, et al. Identification of the binding site for a low-molecular-weight inhibitor of plasminogen activator inhibitor type 1 by site-directed mutagenesis. Biochemistry. 1998;37:1227–34. doi: 10.1021/bi971554q. [DOI] [PubMed] [Google Scholar]
- 160.Einholm AP, Pedersen KE, Wind T, Kulig P, Overgaard MT, Jensen JK, et al. Biochemical mechanism of action of a diketopiperazine inactivator of plasminogen activator inhibitor-1. Biochem J. 2003;373:723–32. doi: 10.1042/BJ20021880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Elokdah H, Abou-Gharbia M, Hennan JK, McFarlane G, Mugford CP, Krishnamurthy G, et al. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: design, synthesis, and preclinical characterization. J Med Chem. 2004;47:3491–4. doi: 10.1021/jm049766q. [DOI] [PubMed] [Google Scholar]
- 162.Folkes A, Roe MB, Sohal S, Golec J, Faint R, Brooks T, et al. Synthesis and in vitro evaluation of a series of diketopiperazine inhibitors of plasminogen activator inhibitor-1. Bioorg Med Chem Lett. 2001;11:2589–92. doi: 10.1016/s0960-894x(01)00508-x. [DOI] [PubMed] [Google Scholar]
- 163.Friederich PW, Levi M, Biemond BJ, Charlton P, Templeton D, van Zonneveld AJ, et al. Novel low-molecular-weight inhibitor of PAI-1 (XR5118) promotes endogenous fibrinolysis and reduces postthrombolysis thrombus growth in rabbits. Circulation. 1997;96:916–21. [PubMed] [Google Scholar]
- 164.Gils A, Stassen JM, Nar H, Kley JT, Wienen W, Ries UJ, et al. Characterization and comparative evaluation of a novel PAI-1 inhibitor. Thromb Haemost. 2002;88:137–43. [PubMed] [Google Scholar]
- 165.Leik CE, Su EJ, Nambi P, Crandall DL, Lawrence DA. Effect of pharmacologic plasminogen activator inhibitor-1 inhibition on cell motility and tumor angiogenesis. J Thromb Haemost. 2006;4:2710–5. doi: 10.1111/j.1538-7836.2006.02244.x. [DOI] [PubMed] [Google Scholar]
- 166.Liang A, Wu F, Tran K, Jones SW, Deng G, Ye B, et al. Characterization of a small molecule PAI-1 inhibitor, ZK4044. Thromb Res. 2005;115:341–50. doi: 10.1016/j.thromres.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 167.Pedersen KE, Einholm AP, Christensen A, Schack L, Wind T, Kenney JM, et al. Plasminogen activator inhibitor-1 polymers, induced by inactivating amphipathic organochemical ligands. Biochem J. 2003;372:747–55. doi: 10.1042/BJ20021868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rogers SJ, Pratt CW, Whinna HC, Church FC. Role of thrombin exosites in inhibition by heparin cofactor II. J Biol Chem. 1992;267:3613–7. [PubMed] [Google Scholar]
- 169.Izaguirre G, Zhang W, Swanson R, Bedsted T, Olson ST. Localization of an antithrombin exosite that promotes rapid inhibition of factors Xa and IXa dependent on heparin activation of the serpin. J Biol Chem. 2003;278:51433–40. doi: 10.1074/jbc.M309266200. [DOI] [PubMed] [Google Scholar]
- 170.Olson ST, Swanson R, Raub-Segall E, Bedsted T, Sadri M, Petitou M, et al. Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Comparison with heparin and low-molecular-weight heparin. Thromb Haemost. 2004;92:929–39. doi: 10.1160/TH04-06-0384. [DOI] [PubMed] [Google Scholar]
- 171.Björk I, Olson ST. Antithrombin: a bloody important serpin. Adv Exp Med Biol. 1997;425:17–33. [PubMed] [Google Scholar]
- 172.Mauray S, de Raucourt E, Talbot JC, Dachary-Prigent J, Jozefowicz M, Fischer AM. Mechanism of factor IXa inhibition by antithrombin in the presence of unfractionated and low molecular weight heparins and fucoidan. Biochem Biophys Acta. 1998;1387:184–94. doi: 10.1016/s0167-4838(98)00120-4. [DOI] [PubMed] [Google Scholar]
- 173.VanDeerlin VMD, Tollefsen DM. The N-terminal acidic domain of heparin cofactor II mediates the inhibition of α-thrombin in the presence of glycosaminoglycans. J Biol Chem. 1991;266:20223–31. [PubMed] [Google Scholar]
- 174.Rezaie AR, Manithody C, Yang L. Identification of factor Xa residues critical for interaction with protein Z-dependent protease inhibitor: both active site and exosite interactions are required for inhibition. J Biol Chem. 2005;280:32722–8. doi: 10.1074/jbc.M505517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pratt CW, Macik BG, Church FC. Protein C inhibitor: purification and proteinase reactivity. Thromb Res. 1989;53:595–602. doi: 10.1016/0049-3848(89)90149-7. [DOI] [PubMed] [Google Scholar]
- 176.Shen L, Villoutreix BO, Dahlback B. Involvement of Lys 62(217) and Lys 63(218) of human anticoagulant protein C in heparin stimulation of inhibition by the protein C inhibitor. Thromb Haemost. 1999;82:72–9. [PubMed] [Google Scholar]
- 177.Travis J, Johnson D. Human α1-proteinase inhibitor. Meth Enzymol. 1981;80:754–64. [Google Scholar]
- 178.Glasscock LN, Gerlitz B, Cooper ST, Grinnell BW, Church FC. Basic residues in the 37-loop of activated protein C modulate inhibition by protein C inhibitor but not by alpha(1)-antitrypsin. Biochim Biophys Acta. 2003;1649:106–17. doi: 10.1016/s1570-9639(03)00164-x. [DOI] [PubMed] [Google Scholar]
- 179.Hopkins PCR, Crowther DC, Carrell RW, Stone SR. Development of a novel recombinant serpin with potential antithrombotic properties. J Biol Chem. 1995;270:11866–71. doi: 10.1074/jbc.270.20.11866. [DOI] [PubMed] [Google Scholar]
- 180.Longstaff C, Gaffney PJ. Serpin-serine protease binding kinetics: alpha 2-antiplasmin as a model inhibitor. Biochemistry. 1991;30:979–86. doi: 10.1021/bi00218a014. [DOI] [PubMed] [Google Scholar]
- 181.Hekman CM, Loskutoff DJ. Kinetic analysis of the interactions between plasminogen activator inhibitor 1 and both urokinase and tissue plasminogen activator. Arch Biochem Biophys. 1988;262:199–210. doi: 10.1016/0003-9861(88)90182-8. [DOI] [PubMed] [Google Scholar]