

Abstract
Melanoma antigen gene protein-A11 (MAGE-11) of the MAGE family of cancer-germline antigens increases androgen receptor (AR) transcriptional activity through its interaction with the AR NH2-terminal FXXLF motif. The present study investigated the regulatory mechanisms that control MAGE-11 expression during androgen deprivation therapy and prostate cancer progression. Studies include the CWR22 xenograft model of human prostate cancer, clinical specimens of benign and malignant prostate, and prostate cancer cell lines. MAGE-11 mRNA levels increased 100 to 1500 fold during androgen deprivation therapy and prostate cancer progression, with highest levels in the castration-recurrent CWR22 xenograft and clinical specimens of castration-recurrent prostate cancer. Pyrosequencing of genomic DNA from prostate cancer specimens and cell lines indicated the increase in MAGE-11 resulted from DNA hypomethylation of a CpG island in the 5´ promoter of the MAGE-11 gene. Sodium bisulfite sequencing of genomic DNA from benign and malignant prostate tumors and prostate cancer cell lines revealed DNA hypomethylation at individual CpG sites at the transcription start site were most critical for MAGE-11 expression. Cyclic AMP also increased MAGE-11 expression and AR transcriptional activity in prostate cancer cell lines. However, cyclic AMP did not alter DNA methylation of the promoter and its effects were inhibited by extensive DNA methylation in the MAGE-11 promoter region. Increased expression of the AR coregulator MAGE-11 through promoter DNA hypomethylation and cyclic AMP provides a novel mechanism for increased AR signaling in castration-recurrent prostate cancer.
Keywords: androgen receptor, prostate cancer, MAGE-11, DNA methylation, cancer-germline antigens
INTRODUCTION
The androgen receptor (AR) mediates androgen-stimulated growth of benign prostate and contributes to prostate cancer progression. Prostate cancers typically undergo remission during androgen deprivation therapy, but recur with castration-recurrent growth and a poor prognosis despite low circulating levels of testicular androgen. The CWR22 xenograft of human prostate cancer mimics clinical prostate cancer with regression after castration. Castration-recurrent growth of the CWR22 tumor begins ~120 days after castration to remove circulating testicular androgens (1–3). AR functions in prostate cancer as a transcriptional regulator that drives androgen-stimulated and castration-recurrent growth. Inhibition of prostate cancer growth by reducing AR levels provides evidence that AR is a central mediator of prostate cancer progression during androgen deprivation therapy, although the underlying mechanisms remain to be established (4–8).
Prostate cancer cells utilize a number of mechanisms mediated through the AR to promote tumor growth. One explanation for persistent AR activity in castration-recurrent cancer is the local tissue production of active androgens, which provides a hormonal stimulus for AR function (9, 10). AR transcriptional activity is mediated through interactions with coregulator proteins (11), and increased levels of critical coregulators, such as the SRC/p160 family of coactivators, appear to contribute to prostate cancer growth and progression (12). In a few instances, and more often in castration-recurrent prostate cancer, naturally occurring AR somatic mutations promote androgen-stimulated prostate cancer growth. Gain-of-function AR mutations can increase AR transcriptional activity by stabilizing interactions with SRC/p160 coactivators (13, 14).
One recently identified AR coactivator is melanoma antigen gene protein-A11 (MAGE-11) (15). MAGE-11 is a member of the MAGE-A subfamily of cancer-testis (or cancer-germline) antigens (16) that is expressed in normal tissues of the human male and female reproductive tracts, and in prostate cancer cells that express AR (15, 17). MAGE-11 is expressed only by human and nonhuman primates and is not present in the mouse or rat genomes, which suggests that the MAGE gene family is undergoing rapid evolution (18, 19). MAGE-11 increases AR transcriptional activity by binding the AR NH2-terminal FXXLF motif through mechanisms that include epidermal growth factor (EGF)-dependent phosphorylation and monoubiquitinylation of MAGE-11 (20). The AR NH2-terminal FXXLF motif also mediates the androgen-dependent AR N/C interaction with activation function 2 in the AR ligand binding domain (21). AR binding to MAGE-11 relieves competitive inhibition of AR transcriptional activity caused by the AR N/C interaction so that SRC/p160 coactivators are recruited more freely by activation function 2 (22, 23).
In the present study, MAGE-11 expression increased with time after castration as AR becomes reactivated during prostate cancer progression in the CWR22 xenograft model of human prostate cancer and in clinical prostate cancer specimens. In three different experimental settings, which include prostate cell lines, CWR22 xenografts and clinical specimens of benign and malignant prostate, increased expression of MAGE-11 resulted from hypomethylation of CpG sites directly proximal to the MAGE-11 transcriptional start site. In studies using prostate cancer cell lines, cyclic AMP (cAMP) signaling also increased MAGE-11 expression and AR transcriptional activity, but did not alter DNA methylation of the MAGE-11 promoter. The data suggest a mechanism whereby increased expression of MAGE-11 facilitates prostate cancer progression by enhancing AR-dependent tumor growth.
RESULTS
Increased MAGE-11 Expression during CWR22 Tumor Progression after Castration
Quantitative reverse transcription (RT)-PCR analysis of CWR22 tumor RNA obtained 2, 6 and 12 days after castration indicated increased levels of MAGE-11 mRNA in association with tumor progression (Fig. 1A). Highest levels of MAGE-11 mRNA occurred on day 120 or longer after castration when tumor growth recurred in the absence of circulating testicular androgens. MAGE-11 immunostaining was nuclear and most prominent on day 6 and 12 after castration (Fig. 2A). MAGE-11 protein levels also increased after castration when detected on immunoblots, with highest levels in the castration-recurrent CWR22 tumor (Fig. 2B). CWR22 tumors are heterogeneous during remission after castration with regions undergoing massive involution. Measurements of both MAGE-11 mRNA and protein indicated increased expression of MAGE-11 in the CWR22 xenograft during progression to castration-recurrent growth.
Figure 1. Increase in MAGE-11 mRNA during CWR22 prostate cancer progression relative to AR, TIF2 and p300.
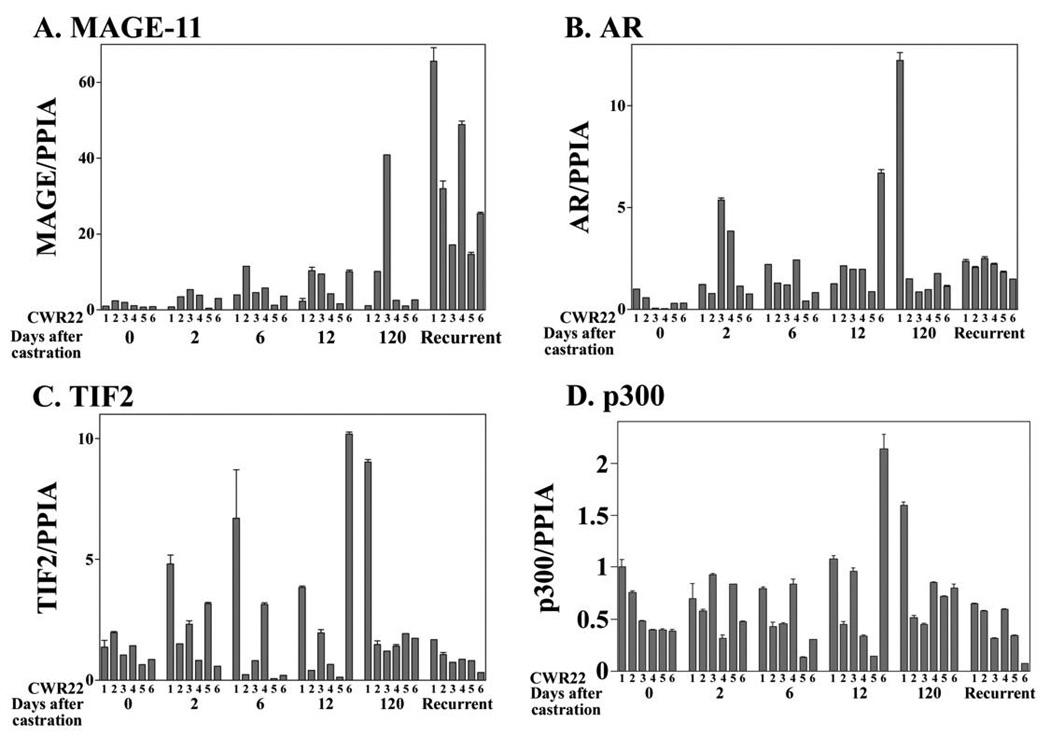
(A) MAGE-11, (B) AR, (C) TIF2 and (D) p300 mRNA levels were determined relative to peptidylprolyl isomerase A (PPIA) using quantitative RT-PCR of CWR22 xenografts excised from 6 animals per group of intact nude mice (0 day) and on days 2, 6, 12 and 120 or longer after castration. Castration-recurrent tumors obtained 120 days or longer after castration were characterized by growth in the absence of circulating testicular androgens.
Figure 2. Increase in MAGE-11 protein during CWR22 prostate cancer progression.
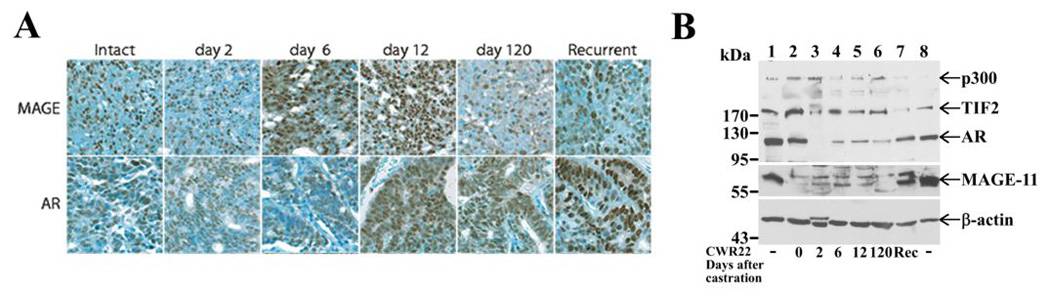
(A) MAGE-11 and AR immunostaining was analyzed in formalin-fixed, paraffin-embedded sections of the CWR22 xenograft excised from intact non-castrated mice, and on days 2, 6, 12 and 120 after castration and longer for the castration-recurrent tumor, using MAGE-11 antibody MagAb94-108 (8 µg/ml) and AR PG21 (Upstate, 1:150 dilution). Brown reaction product indicates immunoreactivity against a toluidine blue counterstain. Original magnification 400X. (B) Immunoblots of MAGE-11, AR, TIF2 and p300 were assayed using CWR22 xenograft extracts prepared from intact-7 (day 0, lane 2), day 2 castrate-3 (lane 3), day 6 castrate-2 (lane 4), day 12 castrate-2 (lane 5), day 120 castrate-3 (lane 6) and castration-recurrent-4 (Rec), according to the numbering of RNA analysis in Fig. 1. Protein extracts (100 µg/lane) were analyzed using antibodies as described in Methods. Combined extracts from COS cells transfected with pSG5-MAGE-11, pCMV-AR, pSG5-TIF and pSG5-HA-p300 served as protein controls (lanes 1 and 8) and endogenous β-actin served as a loading control.
In parallel studies, AR mRNA levels also increased in the CWR22 tumor on day 2 after castration and further at later times after castration in some samples (Fig. 1B). However, unlike the striking increase in MAGE-11 mRNA in the castration-recurrent CWR22 tumor, AR mRNA levels remained relatively constant after castration with few exceptions. On the other hand, AR immunostaining (Fig. 2A) and immunoblotting (Fig. 2B) showed increased AR protein in the castration-recurrent CWR22 tumor, as reported previously (24). AR coactivators TIF2 and p300 mRNA levels were variable and did not correlate with tumor progression (Fig. 1C and D). Immunostaining (data not shown) and immunoblots (Fig. 2B) indicated relatively constant levels of TIF2 and p300 during CWR22 tumor regression and regrowth, although p300 declined in the castration-recurrent CWR22 tumor.
The data indicated that both MAGE-11 and AR mRNA and protein levels increase during CWR22 tumor progression after castration. In the castration-recurrent CWR22 prostate cancer xenograft, MAGE-11 mRNA increased to a greater extent in association with prominent immunostaining of AR and MAGE-11.
MAGE-11 Expression in Clinical Specimens of Benign and Malignant Prostate
To investigate whether the increase in MAGE-11 during progression of the CWR22 xenograft is a characteristic feature of prostate cancer progression, tissue specimens of benign, androgen-stimulated and castration-recurrent prostate cancer were analyzed. MAGE-11 mRNA levels were 10–1500 fold greater in 4 of 11 clinical specimens of castration-recurrent prostate cancer than in androgen-stimulated benign prostate or prostate cancer (Fig. 3A). In one specimen, a Gleason score of 4+5=9 castration-recurrent prostate cancer from patient-5 (CR-CaP-5) obtained 49 months after the initiation of androgen deprivation therapy, MAGE-11 mRNA levels increased by 1500 fold (Fig. 3A). A prostate-specific antigen serum level of 199 ng/dl in CR-CaP-5 was indicative of AR signaling, even though AR mRNA was undetectable (Fig. 3B). AR mRNA levels also increased by 10–100 fold in 4 of 11 castration-recurrent prostate cancer specimens. However, castration-recurrent prostate cancer specimens with increased levels of AR mRNA had lower levels of MAGE-11 mRNA. In agreement with the results of CR-CaP-5, the data suggest an inverse relationship between MAGE-11 and AR mRNA in clinical specimens of castration-recurrent prostate cancer (Fig. 3C). MAGE-11 and AR protein levels both increased in specimens of castration-recurrent prostate cancer (Fig. 3D).
Figure 3. MAGE-11 and AR expression in clinical specimens of benign and malignant prostate.
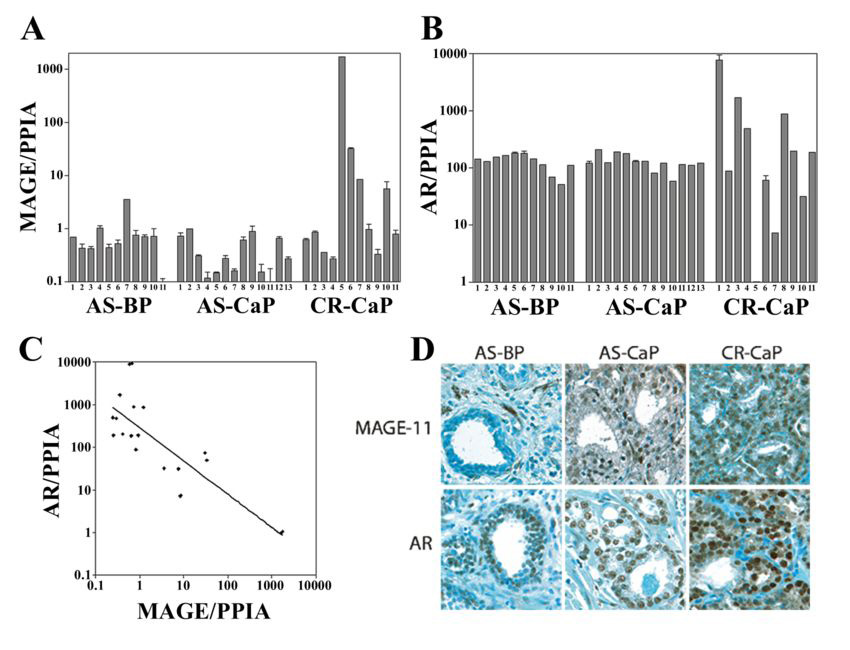
(A) MAGE- 11 and (B) AR mRNA levels relative to peptidylprolyl isomerase A (PPIA) are shown on a semi-log plot determined using quantitative RT-PCR from clinical specimens of androgen-stimulated benign prostate (AS-BP), androgen-stimulated prostate cancer (AS-CaP), and castration-recurrent prostate cancer (CR-CaP). (C) Inverse relationship between AR and MAGE-11 mRNA levels in patient specimens of castration-recurrent prostate cancer. AR and MAGE-11 mRNA levels were determined in duplicate using quantitative PCR of RNA extracted from castration-recurrent prostate cancer specimens. Each of the duplicate data points from the analysis of castration-recurrent specimens in Fig. 2A and B were used to construct the graph. Correlation coefficient R2 = 0.66. (D) MAGE-11 and AR immunostaining of benign and malignant human prostate tissue was performed using formalin-fixed, paraffin-embedded sections of androgen-stimulated benign prostate (AS-BP), androgen-stimulated prostate cancer (AS-CaP), and castration-recurrent prostate cancer (CR-CaP), using MAGE-11 antibody MagAb94-108 (8 µg/ml) and AR antibody PG21 (1:150 dilution). Original magnification 400X.
The results indicated that ~30% of clinical specimens of castration-recurrent prostate cancer have elevated levels of MAGE-11 or AR mRNA, which suggests the presence of a compensatory relationship controlling AR transcriptional activity.
MAGE-11 Promoter DNA Hypomethylation in the CWR22 Xenograft
The molecular basis for increased expression of MAGE-11 during prostate cancer progression was investigated further based on evidence that DNA hypomethylation occurs for other cancer-germline antigens (16, 25). The MAGE-11 gene is located at Xq28 on the human X chromosome (18) and contains a classical CpG island flanking the transcription start site (Fig. 4A). Pyrosequencing of 10 CpG sites ~200 bp before the transcription start site indicated 5% DNA methylation of the MAGE-11 promoter in CWR22 xenografts excised from intact mice, and on days 2, 6 and 12 after castration (Fig. 4B). In the CWR22 castration-recurrent tumor, DNA methylation decreased further to 2%.
Figure 4. DNA hypomethylation of the MAGE-11 promoter during CWR22 prostate cancer xenograft progression.
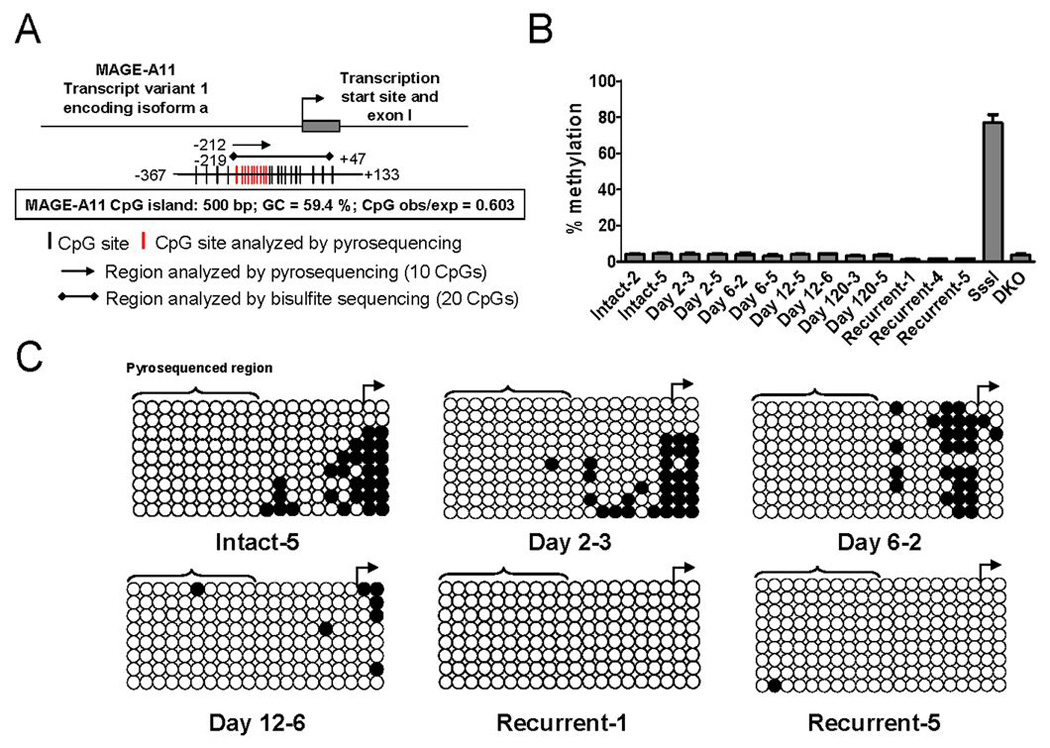
(A) The MAGE-A11 5´ promoter region and CpG island as predicted using http://www.uscnorris.com/cpgislands2/cpg.aspx in the range 148575477 to 148605507 on the reverse complement strand (Accession # NC-000023) are shown. CpG island characteristics are indicated. CpG sites in the 5´ CpG island of MAGE-11 (−367 to +133 relative to the transcription start site) are indicated by black vertical lines, and CpG sites analyzed using pyrosequencing by red vertical lines. Regions analyzed using sodium bisulfite pyrosequencing and standard sodium bisulfite sequencing, and the number of CpG sites analyzed using each method are indicated. (B) Sodium bisulfite pyrosequencing of MAGE-11 promoter DNA methylation in CWR22 xenografts before and after castration. Shown is the percent DNA methylation averaged over 10 analyzed CpG sites in the MAGE-11 promoter region indicated in red in (A). Human genomic DNA modified with bacterial CpG methylase SssI and DNMT-deficient HCT116 colorectal cancer cell DNA (DKO) served as positive and negative controls for MAGE-11 5´ region DNA methylation, respectively. Each bar represents the average ± SE of four independent pyrosequencing analyses. (C) Sodium bisulfite sequencing of the MAGE-11 promoter DNA methylation in CWR22 xenografts before and after castration. Tumors were analyzed from intact mice (intact-5) and on day 2 after castration (day 2 castrate-3), day 6 after castration (day 6 castrate-2), day 12 after castration (day 12 castrate-6) and during castration-recurrent tumor growth more than 120 days after castration (recurrent-1 and recurrent-5), with samples designations according to Fig. 1. Open circles indicate unmethylated CpG sites, filled circles are methylated CpG sites, and rows are individually sequenced alleles. The transcription start site is shown by the right arrow and the region analyzed using pyrosequencing in (B) is indicated by brackets.
Methylation of individual CpG sites was investigated further using sodium bisulfite sequencing of 20 CpG sites that included 10 additional downstream sites in closer proximity to the transcription start site. Unmethylated (open circles) and methylated (filled circles) CpG sites were determined for multiple alleles in CWR22 tumors on different days after castration (Fig. 4C). Hypomethylation at sites more distal to the transcription start site appeared to account for the overall low percentage of DNA methylation in the CWR22 tumors, whereas CpG sites near the transcription start site were methylated in intact CWR22 tumors and on day 2 after castration (Fig. 4B and C). At later times after castration and in the castration-recurrent CWR22 tumor, DNA methylation sites near the transcription start site were hypomethylated.
The results suggested that hypomethylation at CpG sites in direct proximity to the transcription start site is associated with increased expression of MAGE-11 during CWR22 tumor progression.
MAGE-11 Promoter DNA Hypomethylation in Clinical Specimens of Prostate Cancer
To extend this analysis to clinical specimens, pyrosequencing of the MAGE-11 promoter was performed using benign and malignant prostate tissue to investigate further the dependence of MAGE-11 expression on promoter DNA methylation during prostate cancer progression after castration. Pyrosequencing analysis revealed that MAGE-11 methylation levels decreased in castration-recurrent prostate cancer compared to androgen-stimulated benign prostate or androgen-stimulated prostate cancer (Fig. 5A). In particular, the MAGE-11 methylation level in CR-CaP-5, a castration-recurrent prostate cancer specimen that had 1500 fold higher MAGE-11 mRNA levels than benign prostate (Fig. 3A), was reduced to 5% compared to ~70% DNA methylation in androgen-stimulated benign prostate and androgen-stimulated prostate cancer specimens (Fig. 5A). DNA hypomethylation of CR-CaP-5 was evident at all CpG sites within the pyrosequenced region that was different than androgen-stimulated benign prostate (AS-BP-3) and androgen-stimulated prostate cancer (AS-CaP-3) (Fig. 5B). The trend of relative uniformity in methylation levels at individual CpG sites across the pyrosequenced region was consistent across all samples analyzed (data not shown). Sodium bisulfite sequencing of CR-CaP-5 indicated almost complete loss of DNA methylation, compared to dense DNA methylation of the MAGE-11 promoter in androgen-stimulated benign prostate (AS-BP-9) (Fig. 5C). CR-CaP-4 MAGE-11 mRNA levels were 1500 fold less than CR-CaP-5 (Fig. 3A) and dense methylation of CpG sites was retained in close proximity to the transcription start site, although more distal CpG sites were hypomethylated (Fig. 5C, middle panel).
Figure 5. DNA hypomethylation of the MAGE-11 promoter region in clinical specimens of benign and malignant prostate.
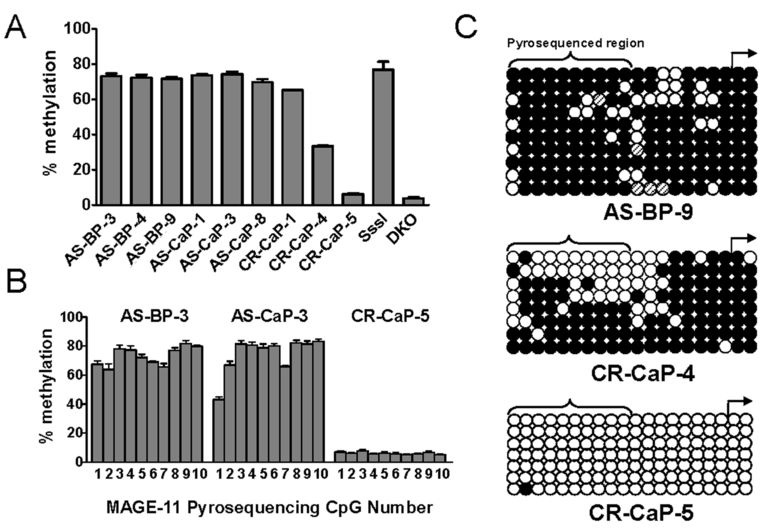
(A) Sodium bisulfite pyrosequencing analysis of MAGE-11 promoter DNA methylation in androgen-stimulated benign prostate (AS-BP), androgen-stimulated prostate cancer (AS-CaP), and castration-recurrent prostate cancer (CR-CaP) specimens. Shown is the percent DNA methylation averaged over 10 CpG sites in the MAGE-11 promoter region indicated in red in Fig. 4A. Human genomic DNA modified with bacterial CpG methylase SssI and DNMT-deficient HCT116 colorectal cancer cells DNA (DKO) served as positive and negative controls for MAGE-11 5´ region DNA methylation, respectively. Each bar represents the average ± SE of four independent pyrosequencing analyses. (B) DNA methylation at individual CpG sites in the MAGE-11 promoter region determined using pyrosequencing. Representative examples of AS-BP-3, AS-CaP-3 and CRCaP- 5 are shown. CpG sites are numbered 1–10 starting at the 5´ site (nucleotide position -192 relative to the transcription start site). Each bar represents the average ± SE of four independent pyrosequencing analyses. (C) Sodium bisulfite sequencing of the MAGE-11 promoter in human specimens of androgen-stimulated benign prostate and castration-recurrent prostate cancer. Samples include AS-BP-9, CR-CaP-4 and CR-CaP-5, and are designated according to Fig. 2. Open and filled circles indicate unmethylated and methylated CpG sites, respectively, and rows indicate individually sequenced alleles. Methylation status of CpG sites shown with hashed lines could not be determined accurately. The transcription start site is shown by the right arrow and the region analyzed using pyrosequencing in (A) and (B) is indicated by brackets.
The results from clinical specimens of benign prostate and prostate cancer provided further evidence that DNA hypomethylation of CpG sites proximal to the MAGE-11 transcription start site is correlated with increased expression of MAGE-11 in association with prostate cancer progression to castration-recurrent growth.
MAGE-11 Expression in Prostate Cancer Cell lines
The direct relationship between increased expression of MAGE-11 and promoter DNA hypomethylation near the transcription start site, prompted a study of cell lines that express AR and MAGE-11 at different levels. MAGE-11 mRNA levels were 10 to 100 fold higher in LAPC-4 prostate cancer cells than in LNCaP, LNCaP-C4-2 or CWR-R1 prostate cancer cells, each of which had high levels of AR mRNA (Fig. 6A). These results suggested a more direct relationship between AR and MAGE-11 mRNA than was seen in clinical specimens of prostate cancer. MAGE-11 mRNA levels were 10 to 1000 fold higher in LAPC-4, LNCaP, LNCaP-C4-2 and CWR-R1 cells than in benign PWR-1E and RWPE-2 prostate cells, DU145 or PC-3 prostate cancer cells, or CV1 or COS-1 cells. In the majority of prostate cancer cell lines, AR mRNA levels were 100 to 10,000 times higher than MAGE-11.
Figure 6. MAGE-11 expression in benign and malignant cell lines.
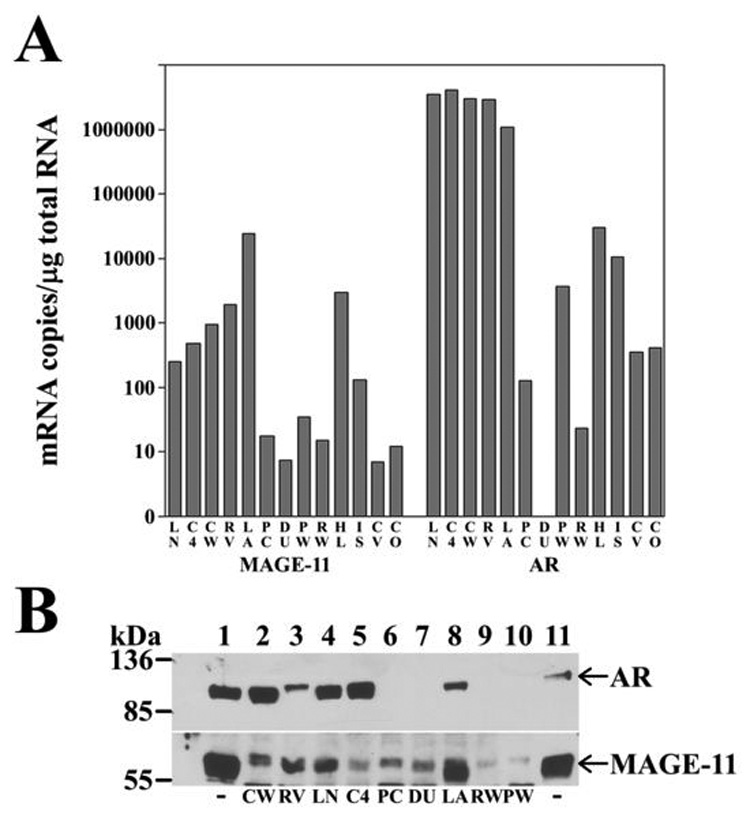
(A) MAGE-11 and AR mRNA levels were determined using quantitative PCR for human prostate cancer cell lines LNCaP (LN), LNCaP-C4-2 (C4), CWRR1 (CW), CWR22-RV1 (RV), LAPC-4 (LA), PC-3 (PC), DU145 (DU), benign human prostate cell lines PWR- 1E (PW) and RWPE-2 (RW), human cervical carcinoma HeLa (HL), human endometrial cancer Ishikawa (IS), monkey kidney CV1 (CV) and COS-1 cells (CO). mRNA amounts were normalized to 1 µg total RNA. (B) Immunoblot of MAGE-11 and AR is shown for extracts (50 µg protein/lane) of CWR-R1 (lane 2), CWR22-RV1 (lane 3), LNCaP (lane 4), LNCaP-C4-2 (lane 5), PC-3 (lane 6), DU145 (lane 7), LAPC-4 (lane 8), RWPE-2 (lane 9) and PWR-1E (lane 10), with abbreviations as in (A). Cells were cultured in serum containing media and analyzed on a 10% acrylamide gel containing SDS. The blot was probed using AR32 (2 µg/lane, top panel) and combined MAGE-11 antibodies 94–108, 59–79 and 13–26 (10 µg/ml each, bottom panel). Combined extracts of COS cells transfected with pSG5-MAGE-11 and pCMV-AR (lanes 1 and 11) were included as controls.
MAGE-11 protein expression determined using immunoblot analysis correlated with MAGE-11 mRNA levels. Highest levels of MAGE-11 protein were in LAPC-4 cells, with lowest levels in RWPE-2 and PWR-1E benign prostate cells (Fig. 6B). AR protein in CWR-R1, CWR-RV1, LNCaP, LNCaP-C4-2 and LAPC-4 prostate cancer cell extracts (Fig. 6B) correlated with AR mRNA levels (Fig. 6A). Under these assay conditions, AR protein was not detected in PC-3 or DU145 prostate cancer cells, nor in PWR-1E and RWPE-2 benign human prostate cells. Except for DU145 cells, in which AR mRNA was not detected, AR mRNA was expressed in benign prostate cells (Fig. 6A), but at levels too low to detect on immunoblots (Fig. 6B), and too low to activate androgen responsive genes in transient reporter gene assays (data not shown).
As found in the CWR22 xenograft and tissue specimens of benign and malignant prostate, MAGE-11 mRNA levels correlated directly with DNA methylation status of the MAGE-11 promoter in the majority of cell lines. Pyrosequencing of 10 CpG sites preceding the MAGE-11 transcription start site demonstrated 5% DNA methylation in LAPC-4 cells, which had the highest levels of MAGE-11 mRNA and protein (Fig. 6 and Fig. 7A). MAGE-11 CpG site methylation was 8 to 17% in LNCaP, LNCaP-C4-2 and CWR-R1 cells, 50 to 65% in DU145, CV1 and COS cells, 35% in PWR-1E and RWPE-2, and 12% in PC-3 cells (Fig. 7A). Representative examples of the extent of DNA methylation at the 10 individual CpG sites interrogated by pyrosequencing are shown for LAPC-4 and DU145 cells (Fig. 7B).
Figure 7. MAGE-11 promoter DNA methylation status in benign and malignant cell lines.
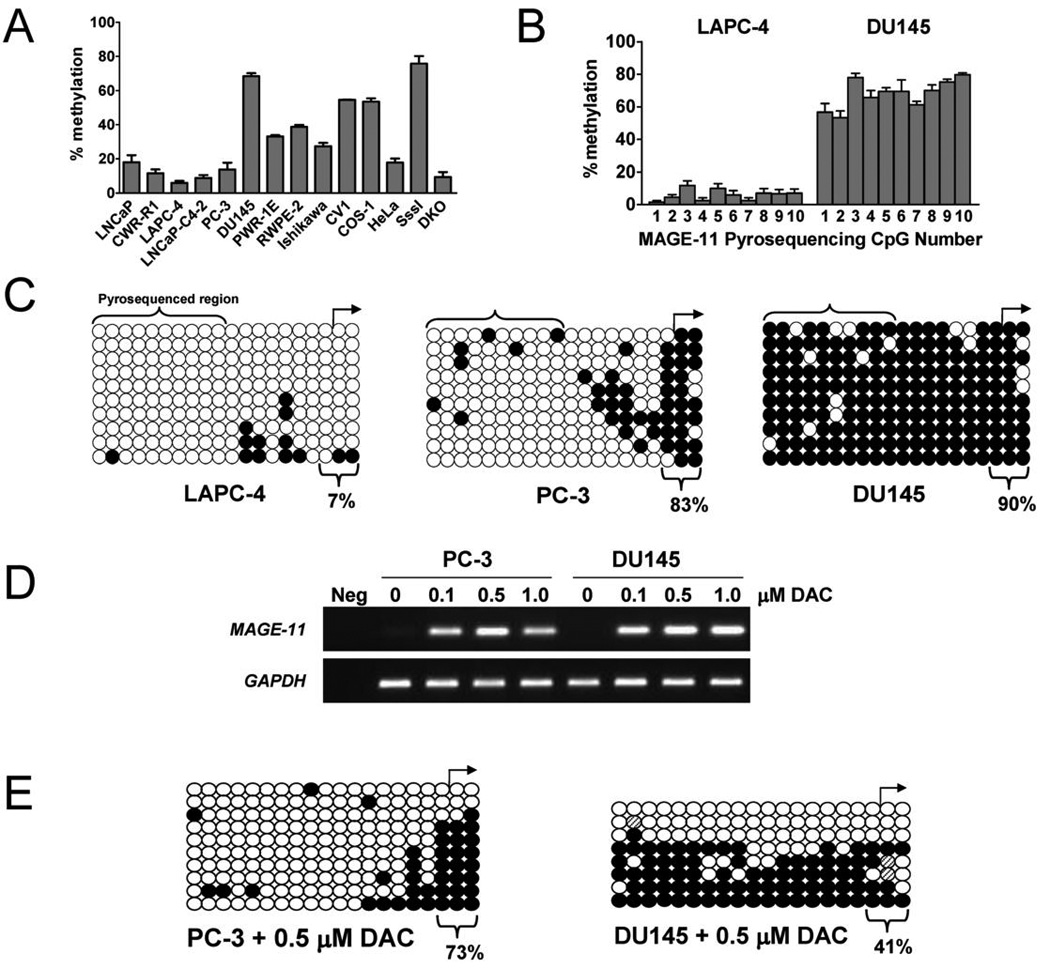
(A) Sodium bisulfite pyrosequencing of MAGE-11 promoter DNA methylation in human and monkey benign and malignant cell lines. Shown is percent DNA methylation averaged over 10 CpG sites in the MAGE-11 promoter region indicated in red in Fig. 4A for human prostate cancer cell lines LNCaP, CWR-R1, LAPC-4, LNCaP-C4-2, PC-3, DU145, benign human prostate cell lines PWR-1E and RWPE-2, human endometrial cancer Ishikawa, monkey kidney CV1 and COS-1, and human cervical cancer HeLa cells. Human genomic DNA modified with bacterial CpG methylase SssI and DNMT-deficient HCT116 colorectal cancer cells DNA (DKO) served as positive and negative controls for MAGE-11 5´ region DNA methylation, respectively. Each bar represents the average ± SE of four independent pyrosequencing analyses. (B) DNA methylation at individual CpG sites in the MAGE-11 promoter region in LAPC-4 and DU145 cells determined using pyrosequencing. CpG sites are numbered 1–10 starting at the 5´ site (nucleotide position −192 relative to the transcription start site). Each bar represents the average ± SE of four independent pyrosequencing analyses. (C) Sodium bisulfite sequencing of the MAGE-11 promoter in LAPC-4, PC-3 and DU145 cells. Open and filled circles indicate unmethylated and methylated CpG sites, respectively, and rows indicate sequenced alleles. The transcription start site is shown by the right arrow and the region analyzed using pyrosequencing in (A) and (B) is indicated by brackets. The percent methylation of three CpG sites proximal to the transcriptional start site is indicated and takes into account all sequenced alleles for each sample. (D) Activation of MAGE-11 expression by DNA demethylation using 5-aza-2´-deoxycytidine (DAC). PC-3 and DU145 cells were treated at 0 and 48 h with 0, 0.1, 0.5 and 1 µM DAC in phosphate buffered saline and RNA was harvested for analysis at 120 h (5 days post treatment). MAGE-11 expression levels were determined using RT-PCR of total RNA extracted with Trizol. RT-PCR amplification of glyceraldehyde 3- phosphate dehydrogenase (GAPDH) served as control for cDNA input. Neg indicates a no template control reaction. (E) Sodium bisulfite sequencing of the MAGE-11 promoter in PC-3 (left panel) and DU145 cells (right panel) following 0.5 µM DAC five days post treatment. Open and filled circles indicate unmethylated and methylated CpG sites, respectively, and rows indicate sequenced alleles. The transcription start site is shown by the right arrow. The percent methylation of three CpG sites proximal to the transcriptional start site is indicated and takes into account all sequenced alleles for each sample.
Sodium bisulfite sequencing of the MAGE-11 promoter in LAPC-4, PC-3 and DU145 provided further evidence that CpG site methylation at the transcription start site regulates MAGE-11 expression. Sodium bisulfite sequencing of CpG sites distal and proximal to the MAGE-11 transcription start site revealed extensive DNA hypomethylation in LAPC-4 cells that express high levels of MAGE-11 (Fig. 7C, left panel). In contrast, CpG sites were densely methylated in DU145 cells that express low levels of MAGE-11 (Fig. 7C, right panel). In PC-3 cells, which show low expression of MAGE-11 (Fig. 6), the MAGE-11 promoter was hypomethylated at CpG sites distal to the transcription start site, but hypermethylated at CpG sites proximal to the transcription start site (Fig. 7C, middle panel). The results provided further evidence that DNA methylation adjacent to the transcription start site may be sufficient to inhibit the expression of MAGE-11.
To test directly whether DNA methylation of CpG sites proximal to the transcription start site actively represses MAGE-11 expression, PC-3 and DU145 cells were treated with 5-aza-2´-deoxycytidine, a DNA methyltransferase inhibitor, and MAGE-11 expression was assessed. Treatment with 5-aza-2´-deoxycytidine increased MAGE-11 expression in both PC-3 and DU145 cells (Fig. 7D), in association with hypomethylation of CpG sites proximal to the transcription start site (Fig. 7E).
Regulation of MAGE-11 Expression in Prostate Cancer Cell Lines by cyclic AMP
Regulation of MAGE-11 expression was investigated further based on evidence that cyclic AMP (cAMP) increases, and 17β-estradiol decreases, MAGE-11 expression in human endometrial cells (17). MAGE-11 mRNA levels increased up to 35 fold in a dose-dependent manner in response to dibutyryl-cAMP in LNCaP-C4-2, CWR22-RV1, LAPC-4, LNCaP, RWPE-2 and PWR-1E cells, as shown for LNCaP-C4-2 and PWR-1E cells (Fig. 8A and B). The cAMP-dependent increase in MAGE-11 mRNA was evident within 12 h in LNCaP cells, indicative of a transcriptional effect, was maximal by 24 h, and was not influenced by 17β-estradiol (Fig. 8C).
Figure 8. Cyclic-AMP-dependent increase in MAGE-11 expression.
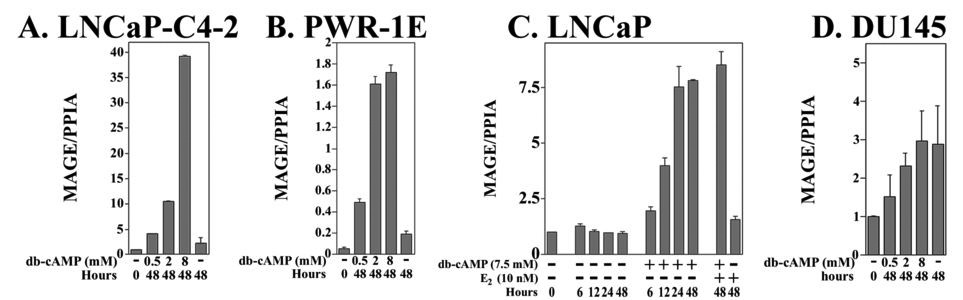
Dose-dependent increases in MAGE-11 mRNA levels in response to cAMP were measured in (A) LNCaP-C4-2, (B) PWR-1E cells, (C) LNCaP, and (D) DU145 cells, at the indicated times after treatment in 5% charcoal-stripped serum in the absence and presence of 0.5, 2 and 8 mM dibutyryl-cyclic AMP (db-cAMP). In (C), LNCaP cells were treated in the absence and presence of 7.5 mM db-cAMP for 0, 6, 12, 24 and 48 h, and for 48 h with 10 nM 17β-estradiol in the absence and presence of 7.5 mM db-cAMP. Total RNA was extracted and MAGE-11 mRNA levels were determined using RT-PCR. The data are representative of two independent experiments.
In DU145 cells where the MAGE-11 promoter was densely methylated (Fig. 7C), MAGE-11 mRNA levels did not increase in response to cAMP relative to a 48 h control (Fig. 8D). The inability of cAMP to increase MAGE-11 expression in DU145 cells suggests that extensive promoter DNA methylation blocks cAMP signaling, and cAMP may not alter DNA methylation of the MAGE-11 promoter. DNA methylation-dependent inhibition of cAMP regulation was supported further by studies in CWR-RV1 and LNCaP-C4-2 cells, where the extent of DNA methylation of the MAGE-11 promoter CpG sites determined by sodium bisulfite sequencing, was unchanged after 48 h treatment with 8 mM dibutyryl-cAMP (data not shown). Furthermore, cAMP did not increase MAGE-11 mRNA levels in DU145 cells after treatment with the DNA methyltransferase inhibitor, 5-aza-2´-deoxycytidine, although a small potentiation effect was apparent in PC-3 cells (data not shown).
The results suggested that hypomethylation within the MAGE-11 promoter near the transcription start site is necessary but not sufficient for cAMP-dependent up-regulation of MAGE-11.
cAMP Regulation of AR Transcriptional Activity
To determine whether the cAMP-dependent increase in MAGE-11 is associated with an increase in AR transcriptional activity, AR and a prostate-specific antigen-luciferase reporter gene were coexpressed in LNCaPC4-2 and DU145 prostate cancer cells, two cell lines that differ in the extent of MAGE-11 promoter DNA methylation, expression and responsiveness to cAMP. AR transactivation increased up to 25 fold in response to dibutyryl-cAMP in a dose-dependent manner in LNCaP-C4-2 cells in the absence and presence of 1 nM DHT (Fig. 9A). In contrast, increased AR transactivation in DU145 cells required higher concentrations of cAMP and was evident only in the presence of DHT (Fig. 9B).
Figure 9. Effects of cyclic-AMP on AR transcriptional activity.
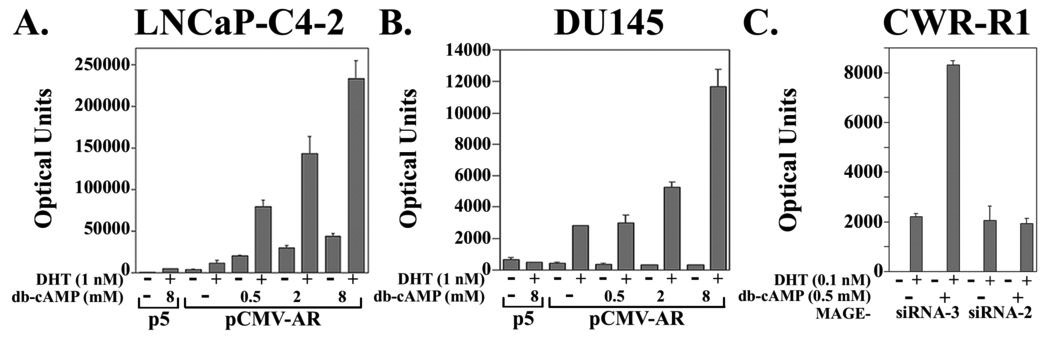
Dose-dependence of androgen-dependent and androgen-independent AR transcriptional activity in response to cAMP was measured in (A) LNCaP-C4-2, and (B) DU145 prostate cancer cells. Cells were transfected in 12 well plates using Effectene (Qiagen) with 10 ng/well pSG5 (p5) empty vector or 10 ng/well pCMV-AR with 0.25 µg/well PSA-Enh-Luc in 5% charcoal-stripped serum medium. Cells were treated for 24 h in 5% charcoal-stripped serum containing media in the absence and presence of 1 nM DHT and 0, 0.5, 2 and 8 mM db-cAMP as indicated. Luciferase activity is representative of three independent experiments. (C) Inhibition of cAMP-stimulated endogenous AR transcriptional activity in CWR-R1 cells using MAGE-11 siRNA. CWR-R1 cells (1.6 × 105/well in 12 well plates) were plated in serum containing growth medium and transferred the next day into antibiotic-free medium containing 5% charcoal-stripped serum. Cells were transfected using Lipofectamine-2000 Reagent (Invitrogen) with 0.1 µg MMTVΔ-421–-364-Luc/well (an MMTV-Luc reporter with deletion of a negative response element) in the presence of 2 nM MAGE-11 siRNA-3, which does not inhibit MAGE-11 expression, and 2 nM MAGE-11 siRNA-2, which reduces MAGE-11 expression (20). Cells were placed the next day in serum-free, phenol red-free medium, and 24 h later the medium was replaced with serum-free medium with and without 0.1 nM DHT and 0.5 mM dibutyryl-cAMP (db-cAMP). Cells were cultured for 24 h and luciferase activity was measured. The results are representative of four independent experiments.
The results suggested that increased AR transcriptional activity in response to cAMP results at least in part from increased expression of MAGE-11. To test this further, MAGE-11 siRNA was used to reduce expression in CWR-R1 prostate cancer cells, where endogenous AR transcriptional activity can be measured using an MMTV-luciferase reporter vector. The cAMP-dependent increase in AR transcriptional activity was retained in CWR-R1 cells treated with MAGE-11 siRNA-3, which does not alter MAGE-11 expression, but was lost after treatment with MAGE-11 siRNA-2, which reduces MAGE-11 expression (20) (Fig. 9C).
The results raised the possibility that the cAMP-dependent increase in AR transcriptional activity occurs through mechanisms that include increased expression of MAGE-11. On the other hand, the ability of higher concentrations cAMP to increase AR transcriptional activity in DU145 cells provideed evidence that additional cAMP-dependent mechanisms increase AR transcriptional activity possibly independent of MAGE-11.
DISCUSSION
MAGE-11 Expression during Prostate Cancer Progression
This report provides evidence that increased expression of the AR selective coregulator MAGE-11 represents a novel mechanism to promote AR-dependent growth of prostate cancer. In three experimental settings, MAGE-11 expression increased in prostate cancer as a result of MAGE-11 promoter DNA hypomethylation at the transcription start site or in response to cAMP. In clinical specimens of castration-recurrent prostate cancer, the MAGE-11 promoter region was hypomethylated, and correlated with increased expression of MAGE-11. MAGE-11 mRNA levels were highest in clinical specimens of castration-recurrent prostate cancer, where CpG sites were hypomethylated directly proximal to the transcription start site. In the CWR22 xenograft model of human prostate cancer, the MAGE-11 gene was hypomethylated in the androgen-stimulated tumor prior to castration, which suggests that the androgen-stimulated CWR22 tumor model represents an advanced stage of the disease. However, with progression to castration-recurrent growth, MAGE-11 expression increased ~100 fold in the CWR22 xenograft, and correlated with hypomethylation at CpG sites proximal to the transcription start site of the MAGE-11 promoter. MAGE-11 expression levels in cell lines similarly correlated with promoter DNA methylation proximal to the transcription start site, and hypomethylation was associated with increased expression. The ability of DNA methylation to inhibit MAGE-11 expression was especially evident among the prostate cancer cell lines. Most striking was the DU145 cell line, where MAGE-11 expression was low and the promoter was densely methylated.
While the predominant mechanism for increased expression of MAGE-11 during prostate cancer progression to castration-recurrent growth appears to be hypomethylation at the transcription start site, MAGE-11 gene expression was also up-regulated by cAMP. cAMP increased MAGE-11 expression in prostate cancer cells that were hypomethylated more extensively in the MAGE-11 promoter region that appeared to be independent of a change in DNA methylation. In contrast, the lack of cAMP stimulation of MAGE-11 expression in DU145 cells suggested that cAMP does not override the inhibitory effects of an extensively methylated MAGE-11 promoter. The results suggest that basal expression of MAGE-11 is blocked by DNA methylation near the transcription start site, some extent of hypomethylation in the MAGE-11 distal promoter is required for up-regulation by cAMP, and additional cell specific factors likely contribute MAGE-11 gene regulation by cAMP. MAGE-11 promoter DNA in clinical specimens of benign prostate tissue was methylated extensively, but only methylated partially in the PWR-1E and RWPE-2 cell lines that derive from benign prostate. These findings make it difficult to predict whether MAGE-11 expression is sensitive to cAMP stimulation in benign prostate tissue under normal physiological conditions.
The cAMP-dependent increase in AR transcriptional activity appears to result in part to an increase in the expression of MAGE-11. Depletion of MAGE-11 levels in CWR-R1 cells using siRNA that inhibits MAGE-11 expression, inhibited the cAMP-dependent increase in AR transcriptional activity. However, cAMP also increased AR transcriptional activity when AR was expressed in DU145 cells, a cell line that lacks endogenous AR and MAGE-11, and where MAGE-11 mRNA levels were not increased by cAMP. AR transcriptional activity was increased by cAMP in LNCaP-C4-2 cells in the absence and presence of androgen, which may reflect the increased expression of MAGE-11. The data suggest that additional mechanisms, possibly involving other coregulatory proteins (26), contribute to the cAMP-dependent increase in AR transcriptional activity.
Relationship between AR and MAGE-11
AR transcriptional activity is increased through several mechanisms involving MAGE-11. Through its interaction with the AR NH2-terminal FXXLF motif, MAGE-11 increases AR recruitment of SRC/p160 coactivators by activation function 2 in the AR ligand binding domain and inhibition of the AR N/C interaction (15). The effects of MAGE-11 on AR transcriptional activity are enhanced by EGF signaling, and MAGE-11 stabilizes AR in the absence or at low levels of androgen (15, 17, 20). The stabilizing effect of MAGE-11 on AR under conditions of low androgen raises the possibility that increases in MAGE-11 in the CWR22 tumor on days 6 and 12 after castration, and in the castration-recurrent tumor, provides a mechanism to increase AR levels in the low androgen environment in the absence of the stabilizing effects of the androgen-dependent AR N/C interaction. AR stabilization by MAGE-11 in castration-recurrent prostate cancer may contribute to the apparent increase in AR protein when AR mRNA levels remained relatively constant after castration. Greater levels of AR could increase the sensitivity to low circulating androgen levels associated with androgen deprivation therapy in the treatment of prostate cancer. Increased levels of MAGE-11 may also increase ligand-independent AR transactivation, as suggested by studies using the LNCaP-C4-2 prostate cancer cell line.
The apparent inverse relationship between AR and MAGE-11 mRNA levels identified in clinical specimens of castration-recurrent prostate cancer suggests a compensatory relationship between AR and MAGE-11 at the level of gene regulation. This relationship also was seen in some samples of the CWR22 xenograft, but not in the prostate cancer cell lines. Present evidence indicates that MAGE-11 expression is not regulated by androgen (data not shown), and increased expression of MAGE-11 is associated with prostate cancer progression to recurrent growth. Hypomethylation and cAMP signaling may account for the increased expression of MAGE-11 during prostate cancer progression, and contribute to AR-mediated castration-recurrent growth of prostate cancer. The molecular basis for the difference in relative levels of AR and MAGE-11 mRNA between castration-recurrent tumor specimens and prostate cancer cell lines remains to be established.
Epigenetic Regulation of MAGE-11
Increased DNA hypomethylation at CpG sites in the MAGE-11 promoter and the associated increase in MAGE-11 expression was greatest in castration-recurrent prostate cancer in the CWR xenograft model and clinical specimens of prostate cancer. Epigenetic abnormalities in gene silencing and activation observed for MAGE-11 are now well established markers for cancer development (27). These results demonstrate that hypomethylation of specific CpG sites proximal to the transcription start site is most critical in regulating MAGE- 11 expression, whereas more distal hypomethylation may influence regulation by cAMP. The lack of a cAMP-dependent increase in MAGE-11 associated with the densely methylated 5´ CpG island in DU145 cells suggests that MAGE-11 promoter DNA hypomethylation distal to the transcription start site is required for cAMP stimulation of MAGE-11 expression. On the other hand, recent genome-wide studies on DNA methylation, histone modification patterns and gene expression, support the importance of epigenetic marks proximal to the transcription start site in gene regulation (28, 29). Localized DNA hypomethylation may alter chromatin structure at the MAGE-11 transcription start site even when flanking DNA is hypermethylated.
Cancer cells exhibit global decreases in 5-methylcytosine and DNA hypomethylation to various extents depending on tumor stage and type, which may contribute to chromosomal instability and acquisition of mutations (30, 31). Growth promoting genes can be activated by DNA hypomethylation in tumors (32). Although relatively little is known about the mechanisms of genome-wide or gene-specific DNA hypomethylation, recent evidence suggests that the expression of DNA methyltransferase 3b (DNMT-3b) isoforms or aberrant expression of the BORIS/CTCFL imprinting-related protein plays a role (33, 34). Germline-specific genes, such as MAGE-11 and other members of the MAGE family, become methylated during normal development (35) and some members of the family of cancer-germline genes undergo DNA hypomethylation in cancer (16, 30). Germline antigen gene activation in cancer has led to the development of cancer vaccines targeting these antigens (16). DNA hypomethylation at CpG sites in the MAGE-11 promoter associated with the onset of castration-recurrent prostate cancer may have critical consequences to tumor growth based on the ability of MAGE-11 to increase AR transcriptional activity in the presence and especially in the relative absence of androgen. The AR coregulator MAGE-11 may be a viable therapeutic and/or vaccine target in some cases of castration-recurrent prostate cancer.
MATERIALS AND METHODS
Clinical Prostate and CWR22 Xenograft Sample Preparation
Serially transplanted androgen-dependent and nonmetastatic CWR22 human prostate cancer xenografts derived from a primary human prostate cancer were propagated in athymic nu/nu mice (24, 36). Some animals were castrated and testosterone pellets removed after tumors reached ~0.75 g. CWR22 tumors from intact mice and on days 2, 6, 12 and 120 or longer after castration when castration-recurrent tumors develop, were formalin-fixed and paraffin-embedded for immunocytochemistry or frozen for DNA and RNA extraction. Patient specimens of androgen-stimulated benign prostate were obtained by radical prostatectomy from African and Caucasian Americans aged 47 to 72 years. Clinically localized androgen-stimulated prostate cancer was obtained from radical prostatectomy specimens from African and Caucasian Americans 44 to 62 years of age, and castration-recurrent prostate cancer (Gleason score 3+5=8 to 5+5=10) was obtained from transurethral resection specimens from men aged 61 to 86 years who suffered urinary retention from local recurrence. Prostatectomy specimens were macrodissected (37) to enrich for benign or malignant epithelial cells when their content was < 80%. This method resulted in an average of 92% malignant epithelial cells in androgen-stimulated prostate cancer specimens (9). Tissues were stored in liquid nitrogen or formalin-fixed and paraffin-embedded for immunocytochemistry. Animal procedures and patient sample collections were performed in accordance with institutional guidelines.
Cell Culture
Cells were maintained in media containing phenol red, penicillin (100 U/ml), streptomycin (100 µg/ml) and 2 mM L-glutamine. LNCaP prostate cancer cells were maintained in RPMI-1640 (Cellgro) containing 1 mM sodium pyruvate and 10% fetal bovine serum (FBS, Sigma). LNCaP-C4-2 prostate cancer cells were cultured in Dulbecco’s modified Eagle medium (DMEM)-F12 containing 5% FBS, 5 µg/ml insulin, 13.65 pg/ml triiodothyronine, 5 µg/ml apotransferrin, 0.244 µg/ml d-biotin and 25 µg/ml adenine hemisulfate (Sigma). CWRR1 cells derived from the castration-recurrent CWR22 prostate cancer xenograft (12) were maintained in DMEM with high glucose, 5 µl 90 mg/ml linoleic acid (Sigma), 0.6 g nicotinamide (Sigma), 0.5 ml insulin-transferin-selenium mix (Roche), 10 ml FBS and 10 ng/ml EGF in 500 ml medium. Serum-free CWR-R1 cell medium was Improved Zinc Optimal with additives without serum, phenol red and EGF. CWR22-RV1 cells derived from the CWR22 human prostate xenograft (American Type Culture Collection) were maintained in RPMI-1640 medium (Cellgro) containing 10% FBS, 1 mM sodium pyruvate, 4.5 g/L glucose, 1.5 g/L sodium bicarbonate and 10 mM Hepes, pH 7.2. LAPC-4 prostate cancer cells were grown in RPMI-1640 with 10% FBS and 1 nM methyltrienolone. PC-3 cells were cultured in DMEM-F12 with 10% FBS. DU145 cells were propagated in Eagle’s Minimum Essential Medium with 10% FBS. Benign human prostate PWR-1E and RWPE-2 cells were maintained in keratinocyte serum-free and phenol red-free medium containing 2.5 µg human recombinant EGF and 25 mg bovine pituitary extract per 500 ml. HeLa human cervical cancer cells were maintained in Eagle’s Minimum Essential Medium with 10% FBS. Ishikawa human endometrial cancer cells were propagated in Minimum Essential Medium with 10% FBS. Monkey kidney CV1 and COS-1 cells were cultured in DMEM with high glucose, 20 mM Hepes, pH 7.2 and 10% bovine calf serum (Hyclone).
Cells cultured in serum-free or 5% charcoal-stripped serum (Atlanta Biologicals) medium were treated with and without dibutyryl-cAMP (Sigma). Reporter gene assays were performed using Effectene (Qiagen) or FuGENE-6 Transfection Reagent (Roche Applied Science) with pCMV-AR and prostate-specific antigen-enhancer luciferase vector, PSA-Enh-Luc (14). Cells were transferred to phenol red-free, serum-free or 5% charcoal-stripped serum medium, and incubated 24 h with and without DHT or dibutyryl-cAMP and assayed for luciferase activity (14).
Real-time Reverse Transcription PCR
CWR22 tumors (50 to 150 mg) and cultured cells were extracted using TRIzol Reagent (Invitrogen). Tumors were disrupted using a Brinkmann polytron (Switzerland). RNA from patient samples was obtained by extracting 0.1 ml AllPrep DNA/RNA extraction kit lysate (Qiagen) with 3 ml TRIzol. First strand cDNA was prepared using SuperScript II reverse transcriptase (Invitrogen). PCR primers and fluorogenic probes for peptidylprolyl isomerase A (PPIA, cyclophilin A) and human AR and MAGE-11 were described (17), however sequence homology for African green monkey COS and CV1 cells could not be confirmed. Primers and probes for transcriptional intermediary factor 2 (TIF2) (Hs00197990-m1) from Assays-On-Demand (Applied Biosytems, 99 bp amplicon) span the exon 14–15 junction at 3193 (NM006540.2). p300 primers and probes (Hs00914232-m1, Applied Biosytems, 138 base pair amplicon) span the exon 9-10 junction at assay location 2279 (NM001429.2). PCR reactions (20 µl) contained complementary DNA from 0.4 µg total RNA for AR, TIF2, MAGE-11 and PPIA, or 0.04 µg total RNA for p300 and PPIA, 4 µl Light Cycler TaqMan Master mix (Roche) and 0.5 µl 20X TaqMan Mix (Applied Biosystems). Thermal cycler reactions were performed at 95°C for 10 min, 55 cycles at 95°C for 15 s, 60°C for 25 s and 72°C for 1 s. Standard curves were prepared as described (17).
Immunochemistry
Immunocytochemistry was performed on formalin-fixed, paraffin-embedded sections of CWR22 prostate cancer xenograft and clinical specimens of androgen-stimulated benign prostate, androgen-stimulated prostate cancer, and castration-recurrent prostate cancer (17). MAGE-11 antibody MagAb94-108 (8 µg/ml) recognizes amino acid residues 94–108 (17). Human p300 C-20 antibody was obtained from Santa Cruz (sc-585, 1:100 dilution). Tissue sections were treated with 0.01 M sodium citrate, pH 6.0, for 15 min in a microwave at high setting (38) to retrieve antigens for affinity purified rabbit polyclonal PG21 AR antibody (Upstate, 1:150 dilution) and mouse monoclonal human TIF2 IgG1 antibody (BD Transduction Laboratories, 1:300 dilution), Sections were blocked with 2% goat serum, incubated overnight at 4°C with primary antibody, blocked and incubated for 30 min with biotinylated secondary antibody (Vector Labs). For MAGE-11 immunostaining, CWR-22 tumor sections were incubated for 30 min with biotinylated secondary antibody and 30 min with avidin DH-biotinylated horseradish peroxidase H complex Vectastain Elite ABC kit (Vector Labs). For AR, TIF2 and p300, samples were incubated for 1 h with biotinylated secondary antibody, and 1 h with avidin DH-biotinylated horseradish peroxidase H complex Vectastain Standard ABC kit (Vector Labs). Benign prostate and prostate cancer slides were incubated using the Vectastain Standard ABC kit for MAGE-11 and AR. Slides were immersed in 3,3’-diaminobenzidine tetrahydrochloride (17), exposed to osmium vapors and counterstained with 0.05% toluidine blue in 30% ethanol, dehydrated, cleared in xylene and mounted with Permount (Fisher).
For immunoblots, cells were solubilized in 0.3–0.4 ml 1% Triton X-100, 1% deoxycholate, 0.1% SDS, 0.15 M NaCl, 0.5 mM EDTA, 50 mM NaF, 0.5 mM Na3VO4 and 50 mM Tris-HCl, pH 7.4, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 5 µg/ml leupeptin, 5 µg/ml pepstatin A and 5 µg/ml aprotinin, and sedimented at 4°C for 30 min at 20,000×g. Extracts from 0.1–0.2 g CWR22 tumor were lysed in 2 ml buffer with detergents added after homogenization using a Brinkmann polytron. Tissue extracts were sedimented at 4°C for 1 h at 27,000 rpm in a Ti60 Beckman ultracentrifuge rotor. Protein controls were from COS cells transfected with pSG5- MAGE-11 (15, 17), pCMVhAR (39), pSG5-TIF2 and pSG5-HA-p300. Protein concentration was determined using the BioRad assay and bovine serum albumin as standard.
Protein extracts from cells (50 µg) and tissues (100 µg) were separated on 10% acrylamide gels containing SDS. Nitrocellulose transfer blots (PerkinElmer) were probed with MAGE-11 antibodies MagAb94-108, MagAb59-79 and MagAb13–26 (10 µg/ml each) (17), AR32 rabbit polyclonal antipeptide antibody (2 µg/ml), mouse monoclonal TIF2 IgG1 antibody (BD Transduction Laboratories, 1:250 dilution), rabbit polyclonal p300 C-20 antibody (Santa Cruz sc-585, 1:800 dilution) and mouse monoclonal β-actin antibody (Abcam, 1:5000). Blots were incubated with primary antibody overnight at 4°C and with anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary IgG antibody (1:10,000 dilution, Amersham Biosciences) for 1 h at room temperature, and except MAGE-11 antibody, the secondary antibody was incubated at 4°C for 1 h. Signals were detected using chemiluminescence (SuperSignal West Dura Extended Duration Substrate, Pierce).
DNA Methylation Analyses
The 5´ end of MAGE-A11 transcript variant 1 (NCBI accession #NM-005366) contains a CpG island (500 bp, GC = 59.4%, CpG obs/exp = 0.6) as predicted by uscnorris.com/cpgislands2/cpg.aspx. Sodium bisulfite sequencing was performed as described (25) using genomic DNA from tissue samples (50 to 150 mg) extracted using the AllPrep DNA/RNA extraction kit (Qiagen). Frozen tissue was homogenized in RLT lysis buffer using a polytron. Extracts were centrifuged at 12,000×g for 3 min and stored at −80°C. Genomic DNA was sodium bisulfite converted using the EZ DNA methylation kit (Zymo Research, Orange, CA). Primers for amplification of the MAGE-11 CpG island were -219 forward 5´-GGAGGATTGAGGTATTTTTATG AT-3´ and +47 reverse 5´-CACTCTCAAAACA CCCCTCAAAA-3´. Sodium bisulfite pyrosequencing was performed as described (40) using MAGE-A11 forward PCR primer -219 5´-GGAGGATTGAGGTATTTTTA TGAT-3´, MAGE-A11 reverse PCR primer −70 5´-biotin-AAACTT CCCTAAATTTACAACAAA-3´ and MAGE-A11 sequencing primer -212 5´-TTGAGGTATT TTTATGATTT-3´. PCR cycling conditions were 95°C for 30 sec, 59°C for 30 sec and 72°C for 1 min for 45 cycles. Pyrosequencing was performed on duplicate samples repeated at least twice. Human genomic DNA modified with bacterial CpG methylase SssI (New England Biolabs, Beverly, MA) served as positive control for DNA methylation. Genomic DNA from DNMT1−/−, DNMT3b−/− HCT116 colorectal cancer cells was used as a negative DNA methylation control (41).
Cell Treatments with 5-aza-2´-deoxycytidine
PC-3 and DU145 cells were treated with 0, 0.1, 0.5 and 1.0 µM 5-aza-2´-deoxycytidine (Sigma) in phosphate buffered saline at 0 and 48 h. RNA was harvested 120 h (5 days) post-treatment and analyzed for MAGE-11 using reverse transcription PCR (15).
ACKNOWLEDGMENTS
We thank Gail Grossman, Brian J. Kennerley, John T. Minges, Andrew T. Hnat and K. Michelle Cobb for excellent technical assistance, and O. Harris Ford, III, and the Immunoanalysis and Tumor Management Core Laboratory of the University of North Carolina at Chapel Hill Lineberger Comprehensive Cancer Center. The project was supported by R01-HD16910 from the National Institute of Child Health and Human Development, and P01-CA77739, R01-CA11674 and R21-CA128062 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Child Health and Human Development or the National Cancer Institute.
REFERENCES
- 1.Nagabhushan M, Miller CM, Pretlow TP, Giaconia JM, Edgehouse NL, Schwartz S, Kung HJ, de Vere White RW, Gumerlock PH, Resnick MI, Amini SB, Pretlow TG. CWR22: the first human prostate cancer xenograft with strongly androgen-dependent and relapsed strains both in vivo and in soft agar. Cancer Res. 1996;56:3042–3046. [PubMed] [Google Scholar]
- 2.Wainstein MA, He F, Robinson D, Kung HJ, Schwartz S, Giaconia JM, Edgehouse NL, Pretlow TP, Bodner DR, Kursh ED, Resnick MI, Amino SB, Pretlow TG. CWR22: androgen-dependent xenograft model derived from a primary human prostatic carcinoma. Cancer Res. 1994;54:6049–6052. [PubMed] [Google Scholar]
- 3.Kim D, Gregory CW, French FS, Smith GJ, Mohler JL. Androgen receptor expression and cellular proliferation during transition from androgen-dependent to recurrent growth after castration in the CWR22 prostate cancer xenograft. Am J Pathol. 2002;160:219–226. doi: 10.1016/S0002-9440(10)64365-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008–1013. [PubMed] [Google Scholar]
- 5.Furutani T, Takeyama K, Koutoku H, Ito S, Taniguchi N, Suzuki E, Kudoh M, Shibasaki M, Shikama H, Kato S. A role of androgen receptor protein in cell growth of an androgen-independent prostate cancer cell line. Biosci Biotechnol Biochem. 2005;69:2236–2239. doi: 10.1271/bbb.69.2236. [DOI] [PubMed] [Google Scholar]
- 6.Yuan X, Li T, Wang H, Zhang T, Barua M, Borgesi RA, Bubley GJ, Lu ML, Balk SP. Androgen receptor remains critical for cell-cycle progression in androgen-independent CWR22 prostate cancer cells. Am J Pathol. 2006;169:682–696. doi: 10.2353/ajpath.2006.051047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li TH, Zhao H, Peng Y, Beliakoff J, Brooks JD, Sun ZA. A promoting role of androgen receptor in androgen-sensitive and -insensitive prostate cancer cells. Nucleic Acids Res. 2007;35:2767–2776. doi: 10.1093/nar/gkm198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponguta LA, Gregory CW, French FS, Wilson EM. Site specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J Biol Chem. 2008;283:20989–21001. doi: 10.1074/jbc.M802392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohler JL, Gregory CW, Ford OH, Kim D, Weaver CM, Petrusz P, Wilson EM, French FS. The androgen axis in recurrent prostate cancer. Clin Can Res. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 10.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–4657. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 11.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 12.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–4319. [PubMed] [Google Scholar]
- 13.He B, Gampe RT, Hnat AT, Faggart JL, Minges JT, French FS, Wilson EM. Probing the functional link between androgen receptor coactivator and ligand binding sites in prostate cancer and androgen insensitivity. J Biol Chem. 2006;281:6648–6663. doi: 10.1074/jbc.M511738200. [DOI] [PubMed] [Google Scholar]
- 14.Askew EB, Gampe RT, Stanley TB, Faggart JL, Wilson EM. Modulation of androgen receptor activation function 2 by testosterone and dihydrotestosterone. J Biol Chem. 2007;282:25801–25816. doi: 10.1074/jbc.M703268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai S, He B, Wilson EM. Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol Cell Biol. 2005;25:1238–1257. doi: 10.1128/MCB.25.4.1238-1257.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–625. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 17.Bai S, Grossman G, Yuan L, Lessey BA, French FS, Young SL, Wilson EM. Hormone control and expression of androgen receptor coregulator MAGE-11 in human endometrium during the window of receptivity to embryo implantation. Mol Human Reprod. 2008;14:107–116. doi: 10.1093/molehr/gam080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogner UC, Wilke K, Steck E, Korn B, Poustka A. The melanoma antigen gene (MAGE) family is clustered in the chromosomal band Xq28. Genomics. 1995;29:725–731. doi: 10.1006/geno.1995.9945. [DOI] [PubMed] [Google Scholar]
- 19.Chomez P, De Backer O, Bertrand M, De Plaen E, Boon T, Lucas S. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res. 2001;61:5544–5551. [PubMed] [Google Scholar]
- 20.Bai S, Wilson EM. Epidermal growth factor-dependent phosphorylation and ubiquitinylation of MAGE-11 regulates its interaction with the androgen receptor. Mol Cell Biol. 2008;28:1947–1963. doi: 10.1128/MCB.01672-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem. 2000;275:22986–22994. doi: 10.1074/jbc.M002807200. [DOI] [PubMed] [Google Scholar]
- 22.He B, Bowen NT, Minges JT, Wilson EM. Androgen-induced NH2- and carboxyl-terminal interaction inhibits p160 coactivator recruitment by activation function 2. J Biol Chem. 2001;276:42293–42301. doi: 10.1074/jbc.M107492200. [DOI] [PubMed] [Google Scholar]
- 23.He B, Lee LW, Minges JT, Wilson EM. Dependence of selective gene activation on the androgen receptor NH2- and carboxyl-terminal interaction. J Biol Chem. 2002;277:25631–25639. doi: 10.1074/jbc.M202809200. [DOI] [PubMed] [Google Scholar]
- 24.Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, French FS. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58:5718–5724. [PubMed] [Google Scholar]
- 25.James SR, Link PA, Karpf AR. Epigenetic regulation of X-linked cancer/germline antigen genes by DNMT1 and DNMT3b. Oncogene. 2006;25:6975–6985. doi: 10.1038/sj.onc.1209678. [DOI] [PubMed] [Google Scholar]
- 26.Nazareth LV, Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. J Biol Chem. 1996;271:19900–19907. doi: 10.1074/jbc.271.33.19900. [DOI] [PubMed] [Google Scholar]
- 27.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 29.Liang G, Lin JC, Wei V, Yoo C, Cheng JC, Nguyen CT, Weisenberger DJ, Egger G, Takai D, Gonzales FA, Jones PA. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci USA. 2004;101:7357–7362. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 31.Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775:138–162. doi: 10.1016/j.bbcan.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 33.Vatolin S, Abdullaev Z, Pack SD, Flanagan PT, Custer M, Loukinov DI, Pugacheva E, Hong JA, Morse H, Schrump DS, Risinger JI, Barrett JC, Lobanenkov VV. Conditional expression of the CTCF-paralogous transcriptional factor BORIS in normal cells results in demethylation and derepression of MAGE-A1 and reactivation of other cancer-testis genes. Cancer Res. 2005;65:7751–7762. doi: 10.1158/0008-5472.CAN-05-0858. [DOI] [PubMed] [Google Scholar]
- 34.Ostler KR, Davis EM, Payne SL, Gosalia BB, Expósito-Céspedes J, Le Beau MM, Godley LA. Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene. 2007;26:5553–5563. doi: 10.1038/sj.onc.1210351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 36.Gregory CW, Johnson RT, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–2898. [PubMed] [Google Scholar]
- 37.Epstein JI. Evaluation of radical prostatectomy capsular margins of resection The significance of margins designated as negative, closely approaching, and positive. Am J Surg Pathol. 1990;14:626–632. doi: 10.1097/00000478-199007000-00003. [DOI] [PubMed] [Google Scholar]
- 38.Balaton AJ, Ochando F, Painchaud MH. Use of microwaves for enhancing or restoring antigens before immunohistochemical staining. Ann Pathol. 1993;13:188–189. [PubMed] [Google Scholar]
- 39.Lubahn DB, Joseph DR, Sar M, Tan JA, Higgs HN, Larson RE, French FS, Wilson EM. Mol Endocrinol. 1988;2:1265–1275. doi: 10.1210/mend-2-12-1265. [DOI] [PubMed] [Google Scholar]
- 40.Woloszynska-Read A, James SR, Link PA, Yu J, Odunsi K, Karpf AR. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun. 2007;7:21. [PMC free article] [PubMed] [Google Scholar]
- 41.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, Baylin SB, Vogelstein B. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]