

Summary
Lipid rafts are cholesterol-enriched microdomains involved in cellular trafficking and implicated as portals for certain pathogens. We sought to determine whether the oral pathogen Porphyromonas gingivalis enters macrophages via lipid rafts, and if so, to examine the impact of raft entry on its intracellular fate. Using J774A.1 mouse macrophages, we found that P. gingivalis colocalizes with lipid rafts in a cholesterol-dependent way. Depletion of cellular cholesterol using methyl-β-cyclodextrin resulted in about 50% inhibition of P. gingivalis uptake, although this effect was reversed by cholesterol reconstitution. The intracellular survival of P. gingivalis was dramatically inhibited in cholesterol-depleted cells relative to untreated or cholesterol-reconstituted cells, even when infections were adjusted to allow equilibration of the initial intracellular bacterial load. P. gingivalis thus appeared to exploit raft-mediated uptake for promoting its survival. Consistent with this, lipid raft disruption enhanced the colocalization of internalized P. gingivalis with lysosomes. In contrast, raft disruption did not affect the expression of host receptors interacting with P. gingivalis, although it significantly inhibited signal transduction. In summary, P. gingivalis uses macrophage lipid rafts as signaling and entry platforms, which determine its intracellular fate to the pathogen’s own advantage.
Introduction
Host-pathogen cross-talk involves microbial interactions with the signaling apparatus of the infected cell, and in this regard lipid rafts serve as a major interface (Simons and Toomre, 2000). Lipid rafts are membrane microdomains rich in cholesterol, sphingolipids and glycosylphosphatidylinositol (GPI)-anchored proteins, which partition receptors for various cellular signaling and trafficking processes (Simons and Ikonen, 1997). The formation of lipid rafts is attributed to the property of sphingolipids and cholesterol to preferentially interact with each other resulting in their spontaneous separation from other phospholipids in the cell membrane. Moreover, cholesterol is believed to stabilize lipid rafts by filling the voids between the relatively bulky glycosphingolipids (Riethmuller et al., 2006).
Microbe-detecting molecules of the immune system such as Toll-like receptors (TLR) as well as T-cell and B-cell receptors are recruited to lipid rafts of activated cells, although both their recruitment and ability to signal are inhibited by cholesterol-depleting drugs (Pizzo and Viola, 2003; Pierce, 2002; Triantafilou et al., 2002b). This indicates that cholesterol-enriched membrane microdomains participate in the induction of both innate and adaptive immunity. Lipid rafts play additional roles in host-pathogen interactions since they are implicated as the sites of action of several bacterial toxins (Boesze-Battaglia et al., 2006; Fong et al., 2006; Badizadegan et al., 2004; Nagahama et al., 2003) and as portals of entry for certain intracellular pathogens (reviewed by Manes et al., 2003). For example, lipid raft integrity is essential for the uptake of mycobacteria by macrophages, whereas the cholesterol-depleting drug methyl-β- cyclodextrin (MCD) prevents this activity (Gatfield and Pieters, 2000).
Given the importance of lipid rafts in host defense signaling, it might seem paradoxical that intracellular pathogens, such as Salmonella typhimurium, Shigella flexneri, and Mycobacterium spp. have “selected” these microdomains for cell entry (Manes et al., 2003). Ironically, however, it could be the very significance of lipid rafts in host defense that may have rendered them targets for immune subversion by pathogens. Although there are only a few examples of lipid raft exploitation by microbes as a means to enhance their adaptive fitness, it has been suggested that the lipid-raft route may afford protection from the intracellular degradative lysosomal pathway (reviewed by Manes et al., 2003). In this respect, it has been proposed that internalized lipid rafts do not readily fuse with late endosomes and lysosomes (Simons and Gruenberg, 2000). Moreover, at least certain pathogens proactively attempt to prevent post-phagocytosis killing. For example, mycobacteria induce recruitment of a coat protein known as TACO (tryptophane aspartate-containing coat protein) which associates in a cholesterol-dependent way with the phagosomal membrane and prevents its fusion with lysosomes (Gatfield and Pieters, 2000). In stark contrast to lipid raft-mediated phagocytosis, opsonic phagocytosis through Fc receptors is not affected by cholesterol depletion and normally leads to pathogen degradation (Manes et al., 2003; Naroeni and Porte, 2002; Shin et al., 2000; Joiner et al., 1990).
The main objective of this study was to determine whether the oral pathogen Porphyromonas gingivalis enters macrophages through lipid rafts, and if so, to examine the impact of lipid raft entry on the intracellular fate of this bacterial organism. P. gingivalis is associated with chronic periodontitis and implicated as a contributory factor in systemic conditions such as atherosclerotic heart disease, on the basis of human epidemiological findings and experimental evidence in mouse models (reviewed by Gibson et al., 2006). Our focus on macrophages in this paper is based on the significance of this cell type in the innate host response in periodontitis and other chronic infections including atherosclerosis (Teng, 2006; Linton and Fazio, 2003). The rationale for investigating macrophage lipid rafts as a possible portal of entry for P. gingivalis was provided by recent findings from our group.
Specifically, we have previously compared the phagocytic activities of normal and pattern-recognition receptor (PRR)-deficient mouse macrophages and found that CD14, CD11b, and TLR2 are important for the uptake of P. gingivalis (Hajishengallis et al., 2006a). Although TLR2 is not known to be a bona fide phagocytic receptor, its involvement in P. gingivalis uptake is attributed to TLR2 inside-out signaling which activates the ligand-binding capacity of complement receptor-3 (CR3; CD11b/CD18) (Harokopakis et al., 2006; Harokopakis and Hajishengallis, 2005). TLR2 and CR3 are recruited to lipid rafts following activation with appropriate stimuli including P. gingivalis fimA-encoded fimbriae (Triantafilou et al., 2007; Hajishengallis et al., 2006b; Triantafilou et al., 2004), whereas CD14 constitutively resides in lipid rafts by virtue of being a GPI-anchored molecule (Triantafilou et al., 2002a). We have thus hypothesized that the macrophage uptake of P. gingivalis is mediated, at least partly, via lipid rafts. Previous reports provided suggestive evidence for P. gingivalis interactions with lipid rafts, but these studies did not involve professional phagocytic cells. Specifically, polystyrene beads coated with P. gingivalis outer membrane vesicles are taken up by HeLa epithelial cells via lipid rafts, whereas MCD inhibits uptake (Tsuda et al., 2005). Moreover, MCD inhibits P. gingivalis invasion of KB epithelial cells (Tamai et al., 2005). However, control experiments with cholesterol-reconstituted MCD-treated cells were not performed to rule out potential pleiotropic MCD effects and, furthermore, the biological significance of lipid raft-mediated entry was not addressed in these pioneering studies (Tamai et al., 2005; Tsuda et al., 2005). Our current findings from functional and imaging studies underscore the importance of macrophage lipid rafts as uptake and signaling platforms for P. gingivalis in a way that favors its survival.
Results
Disruption of lipid rafts suppresses the phagocytosis of P. gingivalis
To investigate a possible role of lipid rafts in P. gingivalis virulence, we used J774A.1 mouse macrophages which have been widely employed as a model for lipid raft-dependent host-microbial interactions, such as bacterial entry or toxin action (Bavdek et al., 2007; Maldonado-Garcia et al., 2004; Pucadyil et al., 2004; Naroeni and Porte, 2002; Norkin et al., 2001; Gatfield and Pieters, 2000). Although we have previously shown that J774A.1 macrophages readily take up P. gingivalis, the role of lipid rafts in this phagocytic activity was not addressed (Wang et al., 2007). We have thus examined the effect of the raft-disrupting agent MCD (Riethmuller et al., 2006) on the ability of J774A.1 macrophages to interact with and internalize P. gingivalis. This was determined using FITC-labeled P. gingivalis and a flow cytometric uptake assay (Wang et al., 2007), which can determine the levels of macrophage-associated bacteria (extracellularly attached or intracellularly located) or of phagocytosed bacteria (upon quenching extracellular fluorescence with trypan blue).
MCD (10 mM) significantly inhibited both the association of P. gingivalis with macrophages and its phagocytosis (p < 0.05; Fig. 1A). Interestingly, MCD displayed a stronger inhibitory effect on phagocytosis than on association (53% vs. 28%, respectively; Fig. 1A). This finding indirectly suggested that the attachment of P. gingivalis to macrophages may not have been affected to the same extent as its phagocytosis. Indeed when cytochalasin D was used to block phagocytosis (in order to specifically address bacterial adherence), P. gingivalis displayed almost comparable binding to untreated and MCD-treated macrophages (Fig. 1B). Specifically, attachment to MCD-treated cells was 87 ± 5 % of that to untreated cells, and the difference did not reach statistical significance (Fig. 1B).
Fig. 1. Cholesterol depletion inhibits the ability of mouse macrophages to phagocytose P. gingivalis.
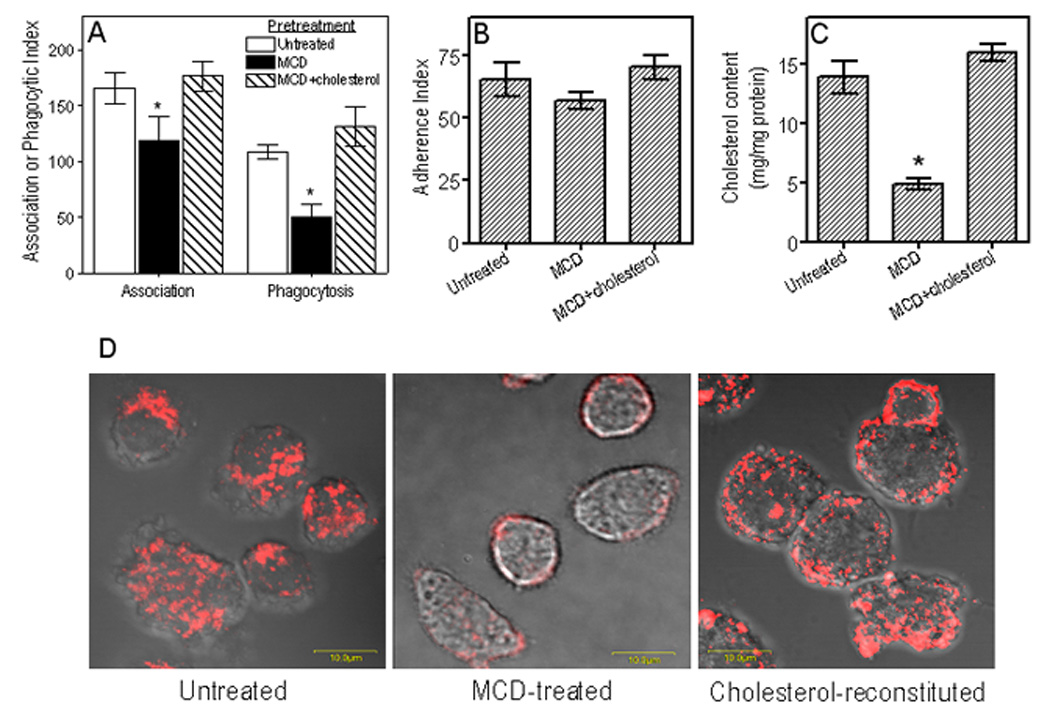
(A) J774A.1 macrophages were pretreated for 30 min with 10 mM MCD to deplete cholesterol, or were pretreated for 30 min with 10 mM MCD followed by addition of 1 mM cholesterol for an additional 30 min. The MCD- and MCD/cholesterol-pretreated macrophages, as well as cells pretreated with medium only, were subsequently incubated for 30 min with FITC-labeled P. gingivalis (MOI = 10:1). Association (i.e., representing both adherence and phagocytosis) or phagocytic indices were determined by flow cytometry, as outlined in Experimental Procedures, using the formula (% positive cells × MFI)/100. (B) The adherence index was calculated as above with the exception that the cells were additionally treated with cytochalasin D (5 µg/ml) to prevent phagocytosis and allow for monitoring of adherence only. (C) Levels of cellular cholesterol in untreated and MCD-treated cells were quantified using the Amplex Red cholesterol assay kit. (D) Visualization of lipid rafts in untreated, MCD-treated, and cholesterol reconstituted MCD-treated cells by confocal microscopy using staining with Alexa Fluor 594-CTB followed by anti-CTB antibody to crosslink lipid rafts into distinct patches on the cell membrane. Numerical data are presented as means ± SDs (n = 3), from one of three independent sets of experiments that yielded similar results. Asterisks indicate statistically significant (p < 0.05) inhibition of indicated activity (A,B) or significant (p < 0.05) reduction of cholesterol content (C) in MCD-treated compared to untreated cells.
The observed inhibitory effects of MCD on association and phagocytosis (Fig. 1A) were strictly dependent on cholesterol depletion. First, we confirmed the ability of MCD to extract cholesterol. Indeed, MCD extracted 65% of cellular cholesterol, although the cholesterol content was readily restored by adding exogenous cholesterol to MCD-treated cells (Fig. 1C). Moreover, cholesterol reconstitution of MCD-treated macrophages effectively reversed the inhibitory effects of MCD (Fig. 1A), thus indicating that the MCD effects are mediated through cholesterol extraction and cannot be attributed to non-specific toxic effects.
To determine whether the observed levels of cholesterol depletion by MCD were sufficient to disrupt lipid rafts (and thus to attribute the inhibited phagocytosis of P. gingivalis to disruption of lipid raft organization), we used a confocal microscopy-based approach. In untreated cells, lipid rafts were readily visualized after staining of the lipid raft marker GM1 ganglioside with Alexa Fluor 594-labeled cholera toxin subunit B (CTB), followed by anti-CTB antibody to crosslink them into distinct patches (Fig. 1D, left). However, lipid rafts were hardly detectable in MCD-treated cells indicating that they were disrupted (Fig. 1D, middle). On the other hand, cholesterol reconstitution restored the appearance of lipid rafts (Fig. 1D, right). In summary, these data show that cholesterol depletion diminishes the functionality of lipid rafts and inhibits macrophage phagocytosis of P. gingivalis, although its cell attachment is not significantly influenced.
Cholesterol-dependent colocalization of P. gingivalis with lipid rafts
In view of the above findings implicating lipid rafts in the phagocytosis of P. gingivalis, we sought to demonstrate that P. gingivalis interacts directly with lipid rafts. Specifically, we examined whether this pathogen colocalizes with GM1 ganglioside, an established lipid raft marker (Triantafilou et al., 2002a), stained with Alexa Fluor 594-CTB. Confocal microscopy confirmed that FITC-labeled P. gingivalis bacteria (green) attached to or entered macrophages, and revealed evident colocalization between GM1-containing membrane microdomains (red) and bacteria (appearing yellow) (Fig. 2 A1,A2). However, some bacteria (remaining green) did not show colocalization with GM1, suggesting that macrophage-P. gingivalis interactions may also involve non-raft membrane regions. In Fig. 2A2 for example, neighboring bacteria stained differently (yellow vs. green) indicating respectively the presence or lack of GM1 colocalization. In contrast to the above, P. gingivalis did not colocalize with GM1 in cholesterol-depleted cells (Fig. 2B); therefore, under these conditions, its observed uptake may involve a lipid raft-independent route. However, cholesterol reconstitution of MCD-treated cells restored the ability of P. gingivalis to colocalize with GM1 (Fig. 2C), confirming the importance of cholesterol for functional lipid rafts. Quantification of the degree of colocalization under the different experimental conditions examined revealed that P. gingivalis was about five times less likely to colocalize with lipid rafts in cholesterol-depleted cells than in untreated or cholesterol-reconstituted cells (Table 1). Although cytochalasin D is known to inhibit phagocytosis (Deshpande et al., 1998), it had no effect on the ability of P. gingivalis to colocalize with lipid rafts (Fig. 2D and Table 1). These findings, taken together with the results of Fig. 1, indicate that P. gingivalis interacts with lipid rafts on the cell surface and subsequently traffics intracellularly. However, P. gingivalis phagocytosis can additionally proceed via non-raft membrane regions. We next determined the impact of the lipid raft route of entry on the intracellular fate of P. gingivalis.
Fig. 2. P. gingivalis colocalizes with lipid rafts in a cholesterol-dependent way.
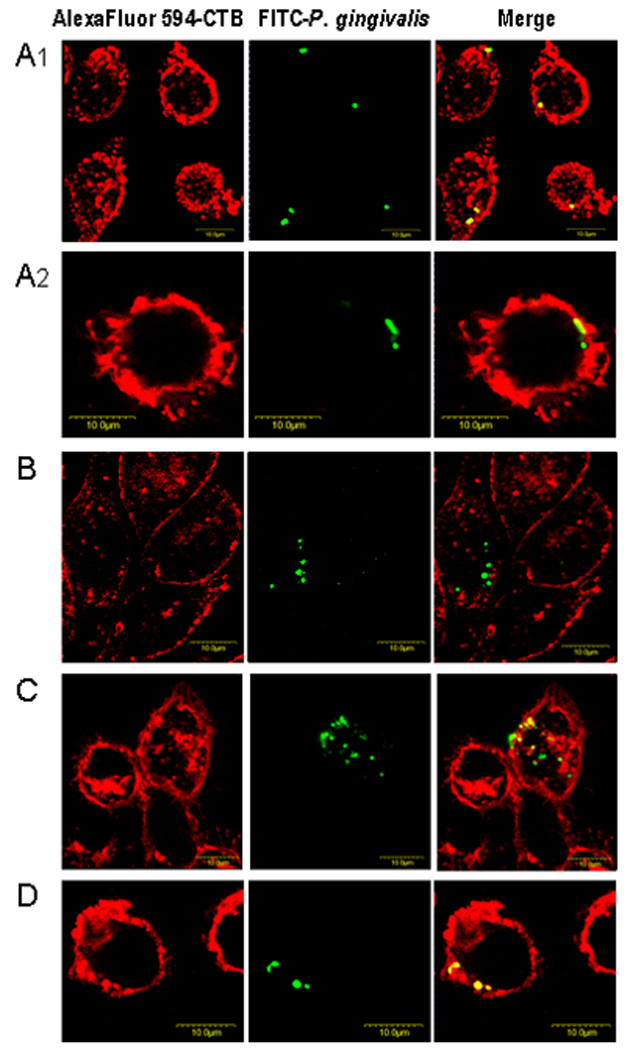
J774A.1 mouse macrophages, cultured on glass coverslips, were pretreated for 30 min at 37°C with medium only (A1 and A2) or with 10 mM MCD to deplete cholesterol (B), or were pretreated for 30 min with 10 mM MCD followed by addition of 1 mM cholesterol for an additional 30 min (C). Another group of J774A.1 cells was pretreated for 30 min at 37°C with 5 µg/ml cytochalasin D to prevent phagocytosis (D). All groups were subsequently exposed to FITC-labeled P. gingivalis 33277 (MOI = 10:1). Infection was allowed to proceed for 15 min and unattached bacteria were removed by washing, followed by cell fixation and staining for GM1 (lipid raft marker) with Alexa Fluor 594-labeled CTB. The cells were then examined by confocal microscopy.
Table 1.
Quantification of P. gingivalis colocalization with lipid rafts in J774A.1 macrophages a
| Cell condition | % Colocalization b |
|---|---|
| Untreated | 76.7 ± 13.3 |
| Cholesterol depleted c | 13.7 ± 6.2 * |
| Cholesterol reconstituted d | 67.3 ± 13.1 |
| Impaired phagocytosis e | 70.1 ± 19.9 |
The colocalization of FITC-P. gingivalis 33277 with Alexa Fluor 594-CTB—stained GM1(lipid raft marker) was quantified using ImageJ intensity correlation analysis. See Fig. 2 legend for experimental details.
Means ± SD of 15–20 examined cells from three independent experiments.
By pretreatment with 10 mM MCD.
By addition of 1 mM cholesterol to MCD-pretreated cells.
By pretreatment with 5 μg/ml cytochalasin D.
p < 0.05 compared to all other conditions.
Cholesterol depletion suppresses the intracellular survival of P. gingivalis
We have previously shown that P. gingivalis resists killing by human monocytes or mouse macrophages, wherein it can persist for at least 72 h (Wang et al., 2007). We have now determined whether lipid rafts play a role in the ability of P. gingivalis for intracellular persistence in J774A.1 macrophages. For this purpose, MCD was used to deplete the cells of cholesterol and disrupt lipid rafts. MCD-treated, MCD-untreated, as well as cholesterol-reconstituted MCD-treated cells were then infected with P. gingivalis (MOI = 10:1) and viable internalized bacteria (colony forming units; CFU) were enumerated using an antibiotic protection-based survival assay. MCD treatment resulted in about 50% reduction in recovered P. gingivalis CFU after 1.5-h incubation (compared to untreated cells; p < 0.05, Fig. 3A). The inhibitory effect of MCD was reversed in cholesterol-reconstituted cells, confirming that MCD acts in a cholesterol-specific way. Interestingly, following overnight incubation (15 h), viable P. gingivalis bacteria were hardly recoverable from MCD-treated macrophages which displayed 20 times lower CFU levels compared to untreated or cholesterol-reconstituted cells (p < 0.05; Fig. 3A). Although cholesterol depletion appeared to accelerate the intracellular clearance of P. gingivalis, we modified the assay in a way that would allow us to conclusively determine whether lipid rafts influence P. gingivalis intracellular survival. Specifically, we ensured that MCD-treated and untreated cells would contain comparable numbers of starting intracellular bacteria. In preliminary flow cytometric uptake experiments, using 2-fold serial increases in the numbers of bacteria added to MCD-treated cells, we found that at a MOI of 80:1, MCD-treated cells display comparable phagocytosis to that seen in untreated cells incubated with bacteria at a MOI of 10:1 (Fig. 3B, inset). We next performed an intracellular survival assay (Fig. 3B) using differential MOI values as specified above. Although there were no significant differences between MCD-treated and control groups at 1.5 h regarding the numbers of intracellular viable bacteria (Fig. 3B), there was a dramatic and statistically significant reduction of viable counts in MCD-treated cells at 15 hr relative to untreated cells or cholesterol-reconstituted MCD-treated controls (Fig. 3B). Moreover, MCD-treated cells did not harbor any viable bacteria 24 hr later (39-hr time point), in stark contrast to the control groups (Fig. 3B). These data clearly indicate that cholesterol depletion promotes the intracellular clearance of P. gingivalis. Taken together with the earlier findings of this study (Fig. 1 and Fig. 2), it may be concluded that the lipid raft route of entry influences the intracellular fate of P. gingivalis to the pathogen’s own advantage.
Fig. 3. Cholesterol depletion suppresses the ability of P. gingivalis for intracellular persistence in mouse macrophages.

(A) J774A.1 macrophages were pretreated for 30 min with 10 mM MCD to deplete cholesterol, or were pretreated for 30 min with 10 mM MCD followed by addition of 1 mM cholesterol for an additional 30 min. The MCD- and MCD/cholesterol-pretreated macrophages, as well as cells pretreated with medium only, were subsequently incubated with P. gingivalis (MOI=10:1) for the indicated time points. (B) Similar intracellular survival assay, which was modified to ensure comparable numbers of starting intracellular bacteria in MCD-treated and control cells, by using a MOI of 80:1 for MCD-treated cells (the inset confirms similar uptake at 30-min postinfection, determined by the flow cytometric uptake assay). The persistence of viable internalized bacteria was determined using an antibiotic protection-based survival assay. Data are means ± SD (n = 3) from typical experiments performed three (A) or two (B) times yielding similar findings. Asterisks indicate significantly (p < 0.05) reduced recovery of viable CFU from MCD-treated compared to MCD-untreated or cholesterol-reconstituted cells.
Cholesterol depletion promotes the colocalization of P. gingivalis with lysosomes
As a possible mechanism whereby certain pathogens may exploit lipid rafts, it has been proposed that lipid rafts mediate an uptake pathway which does not readily fuse with lysosomes (Nagahama et al., 2003). If this mechanism applies to P. gingivalis, we would expect to see increased trafficking of this pathogen to lysosomes upon cholesterol depletion. We have thus examined the colocalization of FITC-labeled P. gingivalis with LysoTraker Red-labeled lysosomes in MCD-treated and untreated J774A.1 macrophages. As would be expected from the hypothesis, confocal microscopy revealed increased colocalization of green fluorescent P. gingivalis and red fluorescent lysosomes (manifested as yellow spots) in MCD-treated compared to untreated control macrophages (Fig. 4 A–C). Whereas green fluorescent bacteria could readily be found intracellularly in untreated control cells (along with yellow spots indicating fusion with lysosomes) (Fig. 4A), “green” bacteria were either absent or rarely found within MCD-treated cells which contained mostly yellow spots (Fig. 4B,C). However, “green” bacteria could be found attached extracellularly in MCD-treated (Fig. 4C) as well as in untreated control cells (Fig. 4A). Cholesterol-reconstituted MCD-treated cells resembled untreated control cells in that both “green” intracellular bacteria (viable) and yellow spots (probably representing nonviable bacteria) could readily be found within the same cell (Fig. 4D). Quantification of the degree of colocalization of P. gingivalis with lysosomes revealed that an overwhelming majority (about 90%) of internalized bacteria traffic to lysosomes in cholesterol-depleted cells, in contrast to untreated or cholesterol-reconstituted cells which roughly contain comparable numbers of lysosome-associated and non-associated bacteria (Table 2). In contrast to P. gingivalis, another periodontal pathogen, Aggregatibacter actinomycetemcomitans (A.a), cannot be recovered in a viable state from macrophages as it is readily killed intracellularly (Wang et al., 2007). A.a. was thus used as a control and was found exclusively associated with lysosomes (yellow spots) (Fig. 4E). In conclusion, cholesterol depletion enhances the trafficking of P. gingivalis to the lysosomes, suggesting that intact lipid rafts allow a relatively safe entry of this pathogen into macrophages.
Fig. 4. Cholesterol depletion promotes the colocalization of P. gingivalis with lysosomes.
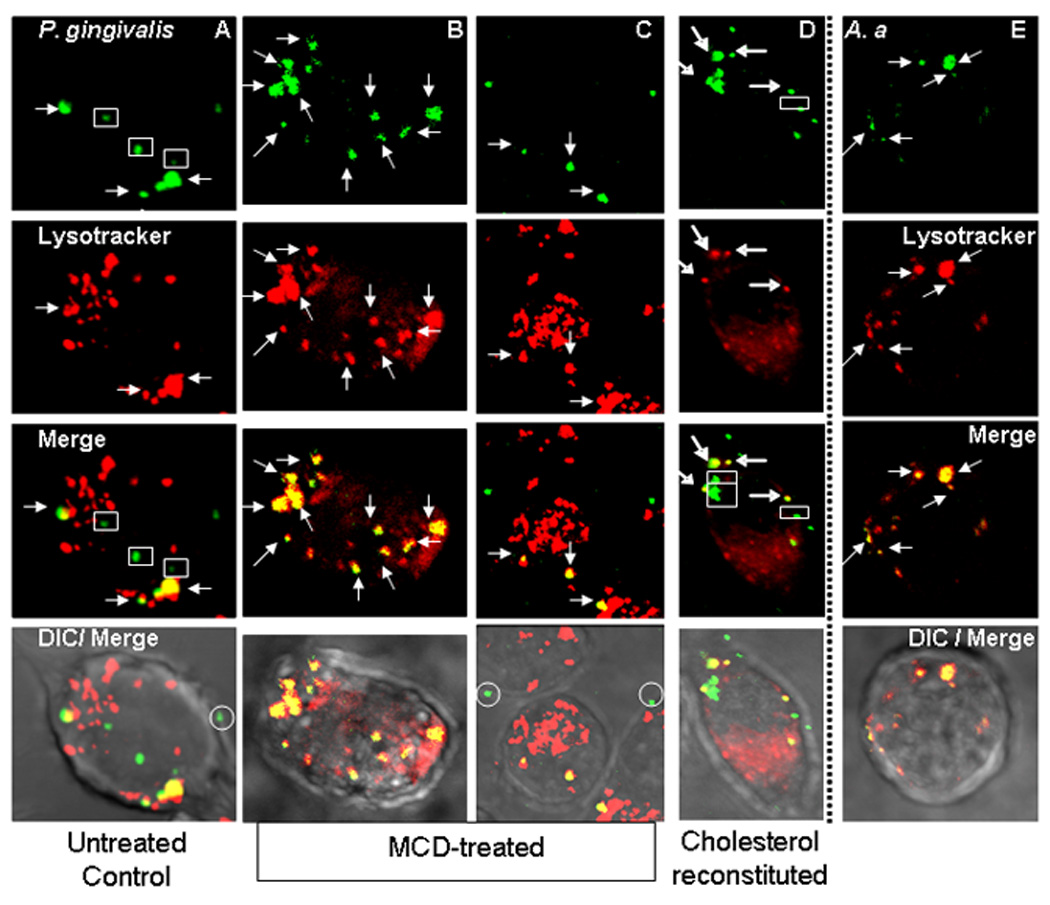
J774A.1 macrophages were pretreated with medium only (A,E), with 10 mM MCD to deplete cholesterol (B,C), or with MCD followed by cholesterol reconstitution (D). The cells were subsequently exposed to FITC-labeled P. gingivalis (A–D) or to FITC-labeled A. actinomycetemcomitans (A.a) (E) at a MOI of 10:1 for 30 min. After washing, incubation was allowed to proceed for an additional 90 min at 37°C. Subsequently, lysosomes were stained with LysoTraker Red for 15 min followed by cell fixation and imaging by confocal microscopy. Arrows indicate green fluorescent bacteria or red fluorescent lysosomes which have colocalized (manifested as yellow spots in merged images), whereas rectangles enclose green fluorescent bacteria that escaped lysosomal colocalization. In DIC/Merge, circles enclose green fluorescent bacteria that remained extracellualrly attached on the cell surface. Although typically MCD-treated cells had fewer internalized bacteria (as in C) than untreated controls, also shown is an image (B) with comparable bacterial numbers with untreated control cells (A) for better comparison of the bacterial intracellular fate.
Table 2.
Quantification of P. gingivalis colocalization with lysosomes in J774A.1 macrophages a
| Cell condition | % Colocalization b |
|---|---|
| Untreated | 47.7 ± 22.1 |
| Cholesterol depleted c | 89.6 ± 8.8 * |
| Cholesterol reconstituted d | 42.4 ± 19.7 |
The colocalization of FITC-P. gingivalis 33277 with LysoTraker Red-labeled lysosomes was quantified using ImageJ intensity correlation analysis. See Fig. 4 legend for experimental details.
Means ± SD of 15–20 examined cells from three independent experiments.
By pretreatment with 10 mM MCD.
By addition of 1 mM cholesterol to MCD-pretreated cells.
p < 0.05 compared to all other conditions.
Cholesterol depletion inhibits P. gingivalis-induced cytokine production without affecting cell surface interactions or expression of signaling receptors
We next determined whether intact lipid raft function and/or phagocytosis are required for P. gingivalis-induced signaling. MCD pretreatment of J774A.1 macrophages resulted in significant (p < 0.05) inhibition of P. gingivalis-induced TNF-α and IL-6 production (>70% inhibition compared to untreated cells; Fig. 5A,B). Moreover, MCD significantly (p < 0.05) inhibited TLR2/1-dependent NF-κB activation by P. gingivalis in transfected human embryonic kidney (HEK)-293 cells (83% inhibition; Fig. 5C). However, cholesterol reconstitution of MCD-treated cells reversed the MCD inhibitory effects (Fig. 5). On the other hand, the inhibitory effects of MCD could not be attributed to its ability to suppress P. gingivalis phagocytosis. Indeed, pretreatment with cytochalasin D, which prevents phagocytosis by blocking actin polymerization (Deshpande et al., 1998), had no significant influence on P. gingivalis-induced cell activation (Fig. 5). That cytochalasin D inhibits the internalization of P. gingivalis was verified by confocal microscopy (e.g., see Fig. 2D). These results show that cellular cholesterol is important for maximal NF-κB activation and cytokine induction by P. gingivalis, although its phagocytosis is not essential in this respect. It can thus be concluded that P. gingivalis-induced signaling for cell activation is initiated at the cell surface in cholesterol-enriched lipid rafts.
Fig. 5. Lipid raft-dependent but phagocytosis-independent cytokine induction by P. gingivalis.

J774A.1 macrophages (A,B) or HEK293 cells transfected as indicated (C) were pretreated or not with MCD (10 mM) or cytochalasin D (5 µg/ml). Half of the MCD-treated groups were subsequently reconstituted with cholesterol (1 mM). The cells were stimulated or not with P. gingivalis (MOI = 10:1) and assayed for TNF-α (A) or IL-6 (B) by ELISA, or for NF-κB activation (C) reported as relative luciferase activity (RLA). Data are means ± SD (n = 3) from one of two sets of experiments with similar results. Asterisks show statistically significant (p < 0.05) inhibition of cell activation.
The inhibitory effect of MCD on P. gingivalis-induced cytokine production may be attributed to disruption of lipid rafts, which partition TLRs and other PRRs required for initiation of signal transduction (Triantafilou et al., 2002b; Simons and Ikonen, 1997). However, it was essential to rule out the possibility that MCD may cause loss or shedding of receptors involved in P. gingivalis-induced signaling, such as TLR1, TLR2, TLR6, CD11b, and CD14 (Hajishengallis et al., 2006b). We have thus examined the expression of these PRRs in MCD-treated or untreated cells. Flow cytometric analysis showed that MCD does not alter the expression of these receptors (Fig. 6), thus suggesting the notion that cholesterol depletion affects the receptor capacity for signaling rather than the receptors per se.
Fig. 6. Effect of cholesterol depletion on PRR expression.
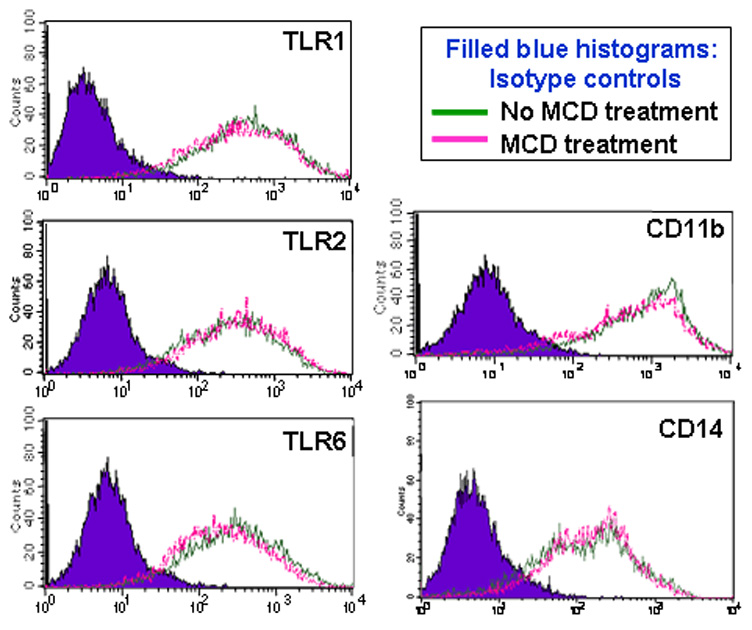
J774A.1 macrophages were treated or not with MCD (10 mM) for 30 min. Flow cytometry was then used to analyze expression of surface PRRs (TLR1, TLR2, TLR6, CD11b, and CD14) using appropriate FITC-labeled mAbs. The histograms shown are from one of three independent experiments that yielded similar findings.
That MCD predominantly affects signaling rather than cell surface interactions was also supported by additional, independent experiments. Since MCD appeared to affect the adherence of P. gingivalis to J774A.1 macrophages only slightly, we decided to determine the effect of MCD on the binding of purified fimbriae, a major adhesin of P. gingivalis encoded by the fimA gene (Hajishengallis, 2007; Lamont and Jenkinson, 1998). By comparing MCD-treated to untreated J774A.1 macrophages, we found that MCD had no significant influence on the binding of fimbriae (Fig. 7A) although, by great contrast, dramatically inhibited cytokine induction by fimbriae in a cholesterol-specific way (results shown for TNF-α in Fig. 7B; similar findings obtained for IL-6 [not shown]). The findings from Fig. 5–Fig. 7, taken together with the rest of the study, underscore the importance of cholesterol-enriched lipid rafts as signaling and entry platforms for P. gingivalis.
Fig. 7. Cholesterol depletion has minimal effect on macrophage binding but dramatically inhibits cytokine induction by P. gingivalis fimbriae.

J774A.1 macrophages were pretreated for 30 min with 10 mM MCD to deplete cholesterol, or were pretreated for 30 min with 10 mM MCD followed by addition of 1 mM cholesterol for an additional 30 min. The MCD- and MCD/cholesterol-pretreated macrophages, as well as cells pretreated with medium only, were subsequently used as follows: (A) The various groups were incubated for 30 min with the indicated concentrations of biotinylated P. gingivalis fimbriae and binding was measured as cell-associated fluorescence (in relative fluorescence units [RFU]) upon staining with streptavidin-FITC. (B) Similarly treated groups were incubated overnight with the indicated concentrations of P. gingivalis fimbriae and induction of TNF-α production in culture supernatants was measured by ELISA. Results are shown as means ± SD (n = 3) from one of two sets of experiments that yielded similar findings. Asterisks indicate statistically significant (p < 0.05) differences in cytokine induction compared to medium-only pretreated cells.
Discussion
To persist in a hostile host environment, pathogens have developed mechanisms to evade or subvert immune defenses aiming to control or eliminate them. Our findings implicate lipid rafts in the macrophage uptake of P. gingivalis, and show for the first time that this entry route impacts on the intracellular fate of the pathogen in a way that may promote its adaptive fitness. Our results are also consistent with the notion that cholesterol is crucial for the integrity and signaling capacity of lipid rafts (Goluszko and Nowicki, 2005), since in cholesterol-depleted cells P. gingivalis does not colocalize with lipid rafts and loses significant cytokine-inducing ability. The latter effect (inhibition of intracellular signaling) is not attributed to suppressed uptake of P. gingivalis. Indeed, cytochalasin D, which inhibits phagocytosis without affecting P. gingivalis-lipid rafts interactions, does not influence TLR2-dependent NF-κB activation and cytokine induction by this organism. Although inhibition of signaling by lipid raft disruption is not unique to P. gingivalis challenge and has been seen with other bacterial agonists (Liang et al., 2007b; Triantafilou et al., 2002a), it is of interest to note that rafts have also been implicated in negative regulation of signaling, as seen in response to Pseudomonas aeruginosa (Grassme et al., 2003).
Interestingly, cholesterol depletion had a much greater inhibitory effect on the phagocytosis of P. gingivalis than on its binding to macrophages. This preferential effect on phagocytosis may be related to observations that cholesterol depletion inhibits cytoskeletal rearrangements (Riff et al., 2005), which are necessary for phagocytic function (Niedergang and Chavrier, 2004). The inhibition of cytoskeletal rearrangements may in turn be related to diminished capacity for signal transduction in cholesterol depleted-cells. For example, cholesterol depletion results in reduced TLR activation (Triantafilou et al., 2002a) as seen in this study with reduced P. gingivalis-induced TLR2/1 signaling. Strikingly, TLR signaling plays an important role in the induction of cytoskeletal rearrangements promoting endocytosis (West et al., 2004). In cholesterol-depleted cells, therefore, inhibition of lipid raft-dependent TLR signaling may not simply suppress cytokine induction but may account, at least partly, for impaired phagocytosis.
At least for epithelial cells, it could be argued that microbes tend to invade through lipid rafts simply because these microdomains are concentrated on the apical side of polarized epithelial cells (Simons and Toomre, 2000), i.e., on the side which directly faces the microbial challenge. However, even in macrophages where rafts and non-raft regions are similarly available for interactions, certain pathogens still prefer to enter via lipid rafts as shown in this and previous studies (reviewed by Manes et al., 2003). This strongly suggests that the lipid-raft route of entry may provide an advantage to at least some pathogenic organisms. Indeed, we found that cholesterol depletion promotes the intracellular killing of P. gingivalis, even when MOI values are adjusted to allow equilibration of the initial intracellular bacterial load in cholesterol-depleted and control macrophages. These findings support similar claims about lipid raft exploitation by certain other pathogens (Manes et al., 2003; Duncan et al., 2002; Watarai et al., 2002; Gatfield and Pieters, 2000). It could thus be speculated that P. gingivalis-containing phagosomes originating from lipid rafts may follow a different intracellular fate than phagosomes emanating from non-raft regions of the cell membrane. In this regard, it is thought that internalized lipid rafts may not readily fuse with lysosomes (Simons and Gruenberg, 2000). This concept is readily supported by our findings that cholesterol depletion results in increased colocalization of P. gingivalis with lysosomes.
The specific molecular events underlying the capacity of P. gingivalis to avoid targeting to lysosomes following lipid raft-mediated uptake by macrophages are currently uncertain. At least in human coronary artery endothelial cells, P. gingivalis was shown to traffic from early phagosomes to autophagosomes which, however, do not appear to acquire cathepsins that would indicate formation of autolysosomes (Dorn et al., 2002; Dorn et al., 2001). Consistent with this observation, P. gingivalis appeared to replicate within autophagosomes which also contained undegraded cytoplasmic material (Dorn et al., 2002; Dorn et al., 2001). Whether P. gingivalis localizes within autophagosomes in macrophages and, if so, what signals are used to manipulate autophagy remain intriguing questions. However, subversion of autophagy in macrophages has actually been demonstrated for another organism. Indeed, Legionella pneumophila modifies the autophagosomal compartment to establish a niche that is permissive for intracellular replication (Dubuisson and Swanson, 2006; Swanson and Isberg, 1996). Interestingly, recent evidence suggests that pathogens which enter macrophages through lipid rafts, tend to localize in autophagosomes (Amer et al., 2005).
The capacity of P. gingivalis for lipid raft-mediated uptake and increased intracellular persistence is intriguing in the context of the epidemiological and mechanistic link between periodontitis and systemic diseases such as atherosclerosis (Pussinen et al., 2006; Desvarieux et al., 2005; Gibson et al., 2004). Remarkably, viable P. gingivalis has been demonstrated in atherosclerotic plaques (Kozarov et al., 2005). Conceivably, the persistence of P. gingivalis within macrophages may be sufficient to allow this organism to co-opt the migration potential of macrophages, facilitating relocation to systemic tissues and infection of more permissive cells (e.g., endothelial cells). The notion that macrophages might be exploited as “Trojan horses” for P. gingivalis systemic dissemination is an intriguing hypothesis that warrants further investigation. However, the capacity of P. gingivalis to exit initially infected host cells and then to enter and multiply within new host epithelial or vascular cells has been documented (Li et al., 2008; Yilmaz et al., 2006).
On the basis of published literature and our recent work investigating molecular interactions between P. gingivalis and PRRs, it is possible that CR3 may be at least one of potential receptors exploited by P. gingivalis in lipid rafts. First, CR3 is recruitable to lipid rafts upon activation with appropriate microbial stimuli, including P. gingivalis fimbriae (Nakayama et al., 2007; Hajishengallis et al., 2006b; Pfeiffer et al., 2001; Peyron et al., 2000). Second, we have shown that CR3 blockade or deficiency impairs macrophage phagocytosis of P. gingivalis and exerts an even more dramatic inhibitory effect on its intracellular survival (Wang et al., 2007). Mechanistically, this could be explained by findings that CR3 is not linked to vigorous microbicidal mechanisms (Lowell, 2006; Rosenberger and Finlay, 2003; Caron and Hall, 1998; Wright and Silverstein, 1983). It is tempting to speculate that this may in turn be attributed, at least partly, to the functioning of CR3 in lipid rafts, wherefrom derived phagosomes are thought to not readily lead to lysosomal degradation (Manes et al., 2003; Watarai et al., 2001; Simons and Gruenberg, 2000).
In summary, lipid rafts serve as signaling platforms and portals of entry for P. gingivalis into macrophages. Importantly, this route of entry appears to promote the pathogen’s capacity for survival and virulence. Our results and those of others (Manes et al., 2003; Naroeni and Porte, 2002; Watarai et al., 2001; Gatfield and Pieters, 2000) implicating lipid rafts in microbial virulence raise the possibility for lipid raft-oriented strategies as a means to counteract immune evasion and control bacterial infection.
Experimental Procedures
Reagents, cells, and bacteria
Cytochalasin D, MCD, and cholesterol were purchased from Sigma-Aldrich. Murine-specific mAbs to TLR1 (clone TR23), TLR2 (6C2), CD14 (Sa2-8), and CD11b (M1/70) and their isotype controls were obtained from e-Bioscience. Anti-TLR6 mAb (clone 418601) was from the R&D Systems. Alexa Fluor 594-labeled CTB and LysoTraker Red DND-99 were from Molecular Probes/Invitrogen. The reagents were used at effective concentrations determined in preliminary experiments or in previous publications (Wang et al., 2007; Hajishengallis et al., 2006a). The mouse macrophage cell line J774A.1 (ATCC TIB-67) was cultured at 37°C and 5% CO2 atmosphere, in RPMI 1640 (InVitrogen/Gibco) supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 10 mM HEPES, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 0.05 mM 2-mercaptoethanol. HEK-293 cells (ATCC CRL-1573) were cultured in Dulbecco's modified Eagle's medium containing 10% heat-inactivated FBS, 100 units/ml penicillin, and 100 µg/ml streptomycin (InVitrogen/Gibco). Cell viability was monitored using the CellTiter-Blue™ assay kit (Promega). None of the experimental treatments affected cell viability compared to medium-only control treatments. P. gingivalis ATCC 33277 was grown anaerobically at 37°C in modified GAM medium (contains 5 µg/ml hemin and 1 µg/ml menadione) (Nissui Pharmaceutical) and fimA-encoded fimbriae were purified as previously described (Harokopakis and Hajishengallis, 2005). The purified fimbriae preparations were free of any contaminating substances on silver-stained SDS-PAGE, and tested negative for endotoxin (< 6 EU/mg protein) as indicated by the quantitative Limulus amebocyte lysate assay (BioWhittaker, Walkersville, MD).
Fimbria binding assay
The binding of purified fimbriae to macrophages was performed using a fluorescent cell-based assay in 96-well plates as we previously described (Harokopakis and Hajishengallis, 2005). Briefly, biotinylated fimbriae (1 µg/ml) were allowed to bind for 30 min at 37°C, and bound protein was probed with FITC-labeled streptavidin. Upon washing, binding was determined by measuring cell-associated fluorescence on a microplate fluorescence reader (FL600, Bio-Tek Instruments, Winooski, VT) with excitation/emission wavelength settings of 485/530 nm.
Flow cytometry
Flow cytometric analysis was performed to assess the uptake of fluorescently labeled P. gingivalis (Wang et al., 2007). Briefly, J774A.1 macrophages were incubated at 37°C with FITC-labeled P. gingivalis at a MOI of 10:1 for 30 min. Phagocytosis was stopped by cooling the incubation tubes on ice. After cell washing to remove nonadherent bacteria, in some groups extracellular fluorescence (representing attached but not internalized bacteria) was quenched with 0.2% trypan blue. The cells were washed again, fixed with 1% paraforlmadehyde, and analyzed by flow cytometry (% positive cells for FITC-P. gingivalis and mean fluorescence intensity [MFI]) using the FACSCalibur and the CellQuest software (Becton-Dickinson). Association (i.e., representing both adherence and phagocytosis) or phagocytic indices were calculated using the formula (% positive cells × MFI)/100. Control experiments indicated that cytochalasin D-pretreated macrophages incubated with FITC-P. gingivalis and subsequently exposed to trypan blue did not show significant fluorescence, thus confirming that cytochalasin D blocks phagocytosis and that trypan blue effectively quenches extracellular fluorescence. Flow cytometry was also used to analyze the effect of MCD pretreatment on J774A.1 macrophage expression of surface receptors (TLR1, TLR2, TLR6, CD11b, and CD14) using appropriate FITC-labeled mAbs (see above).
Cell activation assays
NF-κB-dependent transcription of a luciferase reporter gene was determined as previously described in detail (Liang et al., 2007a; Hajishengallis et al., 2006b). Briefly, HEK-293 cells were transiently cotransfected with CD14 (pUNO-hCD14; InVivogen) and TLR1/TLR2 (pDUO-hTLR1/ TLR2; InVivogen) and with a NF-κB reporter system, comprising a NF-κB-dependent firefly luciferase plasmid (pNF-κB-Luc; Stratagene) and a Renilla luciferase transfection control (pRLnull; Promega). Two days post-transfection, the cells were stimulated for 6h with P. gingivalis (MOI = 10:1) and relative luciferase activity was calculated as the ratio of firefly luciferase activity to Renilla luciferase activity, to correct for transfection efficiency. The results were then normalized to those of unstimulated control cells transfected with reporter and empty vectors, the value of which was taken as 1. Induction of cytokine (TNF-α and IL-6) production in stimulated cell culture supernatants was measured as previously described (Hajishengallis et al., 2006b) using specific ELISA kits (eBioscience).
Antibiotic protection-based intracellular survival assay
The capacity of phagocytosed P. gingivalis for intracellular persistence in macrophages was determined by an antibiotic protection-based survival assay, as we previously described (Wang et al., 2007). Briefly, viable counts (CFU) of internalized P. gingivalis were determined by plating serial dilutions of macrophage lysates on blood agar plates subjected to anaerobic culture. Prior to macrophage lysis, extracellular nonadherent bacteria were removed by washing, while extracellular adherent bacteria were killed by addition of gentamicin (300 µg/ml) and metronidazole (200 µg/ml).
Confocal microscopy
Confocal laser scanning microscopy was used to determine colocalization of P. gingivalis with lysosomes. For this purpose, J774A.1 macrophages, cultured on glass coverslips, were infected with FITC-labeled P. gingivalis (MOI=10:1) for 30 min, washed, and incubation was allowed to proceed for an additional 90 min at 37°C. Subsequently, the cells were stained for 15 min with LysoTraker Red DND-99, a freely permeant and vital dye that stains acidified late endosomes and lysosomes (Basyuk et al., 2003). The cells were then fixed and imaged on an Olympus FV500 confocal microscope. Confocal microscopy was also used to demonstrate colocalization of P. gingivalis with the lipid raft marker, GM1 ganglioside. In this case, J774A.1 macrophages were exposed to FITC-labeled P. gingivalis (MOI = 10:1) and infection was allowed to proceed for 15 min at 37°C. Unattached bacteria were removed by washing. After fixation and staining for GM1 with Alexa Fluor 594-labeled CTB (1 µg/ml; 15 min), the cells were imaged as above. Shown in figure 2 and figure 4 are representative single optical sections as well as 2-color overlay (merge) confocal images. Where appropriate (Fig. 4), additional overlay with differential interference contrast (DIC) images is shown. The degree of colocalization of P. gingivalis with lipid rafts or lysosomes was quantified using the public domain Image J software with the Intensity Correlation Analysis plugin (NIH; http://rsb.info.nih.gov/ij).
MCD treatment and cholesterol reconstitution
To deplete macrophages of cholesterol using MCD and reconstituting cellular cholesterol in MCD-treated cells, we used a modification of previously published methodology (Lawrence et al., 2003; Christian et al., 1997). Briefly, J774A.1 macrophages were incubated in the presence of 10 mM methyl-β-cyclodextrin (MCD) for 30 min at 37°C to deplete the cells of cholesterol. The cells were washed and incubated for an additional 30 min with medium only or with 1 mM cholesterol. Subsequently, the cells (MCD-treated, MCD-treated plus cholesterol-reconstituted, and cells treated with medium only) were used in functional assays. To confirm depletion of cholesterol by MCD, cellular cholesterol was quantified using the fluorometric Amplex Red cholesterol assay kit (Molecular Probes/Invitrogen) and normalized to total protein concentration determined by the BCA Protein Assay (Pierce Biotechnology). The MCD concentration used in the study (10 mM) was decided based on preliminary experiments. These showed that the use of MCD at increasing concentrations (1–25 mM) reduced cellular cholesterol content in a dose-dependent manner, although concentrations >10 mM did not extract significantly more cholesterol than the 10 mM dose.
Statistical analysis
Data were evaluated by analysis of variance and the Dunnett multiple-comparison test using the InStat program (GraphPad Software). Where appropriate (comparison of two groups only), two-tailed t tests were also performed. Statistical differences were considered significant at the level of p < 0.05.
Acknowledgements
This work was supported by U.S. Public Health Service Grants DE015254 and DE018292 (to G.H.) from the NIDCR/NIH.
References
- Amer AO, Byrne BG, Swanson MS. Macrophages rapidly transfer pathogens from lipid raft vacuoles to autophagosomes. Autophagy. 2005;1:53–58. doi: 10.4161/auto.1.1.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badizadegan K, Wheeler HE, Fujinaga Y, Lencer WI. Trafficking of cholera toxin-ganglioside GM1 complex into Golgi and induction of toxicity depend on actin cytoskeleton. Am. J. Physiol. Cell Physiol. 2004;287:C1453–C1462. doi: 10.1152/ajpcell.00189.2004. [DOI] [PubMed] [Google Scholar]
- Basyuk E, Galli T, Mougel M, Blanchard JM, Sitbon M, Bertrand E. Retroviral genomic RNAs are transported to the plasma membrane by endosomal vesicles. Dev. Cell. 2003;5:161–174. doi: 10.1016/s1534-5807(03)00188-6. [DOI] [PubMed] [Google Scholar]
- Bavdek A, Gekara NO, Priselac D, Gutierrez Aguirre I, Darji A, Chakraborty T, et al. Sterol and pH interdependence in the binding, oligomerization, and pore formation of Listeriolysin O. Biochemistry. 2007;46:4425–4437. doi: 10.1021/bi602497g. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Besack D, McKay T, Zekavat A, Otis L, Jordan-Sciutto K, Shenker BJ. Cholesterol-rich membrane microdomains mediate cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal-distending toxin. Cell Microbiol. 2006;8:823–836. doi: 10.1111/j.1462-5822.2005.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid. Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- Deshpande RG, Khan MB, Genco CA. Invasion of aortic and heart endothelial cells by Porphyromonas gingivalis. Infect. Immun. 1998;66:5337–5343. doi: 10.1128/iai.66.11.5337-5343.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DR, Jr, Sacco RL, Papapanou PN. Periodontal microbiota and carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology Study (INVEST) Circulation. 2005;111:576–582. doi: 10.1161/01.CIR.0000154582.37101.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn BR, Dunn WA, Jr, Progulske-Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect. Immun. 2001;69:5698–5708. doi: 10.1128/IAI.69.9.5698-5708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn BR, Dunn WA, Jr, Progulske-Fox A. Bacterial interactions with the autophagic pathway. Cell Microbiol. 2002;4:1–10. doi: 10.1046/j.1462-5822.2002.00164.x. [DOI] [PubMed] [Google Scholar]
- Dubuisson JF, Swanson MS. Mouse infection by Legionella, a model to analyze autophagy. Autophagy. 2006;2:179–182. doi: 10.4161/auto.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan MJ, Shin JS, Abraham SN. Microbial entry through caveolae: variations on a theme. Cell. Microbiol. 2002;4:783–791. doi: 10.1046/j.1462-5822.2002.00230.x. [DOI] [PubMed] [Google Scholar]
- Fong KP, Pacheco CM, Otis LL, Baranwal S, Kieba IR, Harrison G, et al. Actinobacillus actinomycetemcomitans leukotoxin requires lipid microdomains for target cell cytotoxicity. Cell Microbiol. 2006;8:1753–1767. doi: 10.1111/j.1462-5822.2006.00746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 2000;288:1647–1650. doi: 10.1126/science.288.5471.1647. [DOI] [PubMed] [Google Scholar]
- Gibson FC, 3rd, Yumoto H, Takahashi Y, Chou HH, Genco CA. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J. Dent. Res. 2006;85:106–121. doi: 10.1177/154405910608500202. [DOI] [PubMed] [Google Scholar]
- Gibson FC, 3rd, Hong C, Chou HH, Yumoto H, Chen J, Lien E, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:2801–2806. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- Goluszko P, Nowicki B. Membrane cholesterol: a crucial molecule affecting interactions of microbial pathogens with mammalian cells. Infect Immun. 2005;73:7791–7796. doi: 10.1128/IAI.73.12.7791-7796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassme H, Jendrossek V, Riehle A, von Kurthy G, Berger J, Schwarz H, et al. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat.Med. 2003;9:322–330. doi: 10.1038/nm823. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G. Peptide mapping of a functionally versatile fimbrial adhesin from Porphyromonas gingivalis. Int. J. Pept. Res. Ther. 2007;13:533–546. [Google Scholar]
- Hajishengallis G, Wang M, Harokopakis E, Triantafilou M, Triantafilou K. Porphyromonas gingivalis fimbriae proactively modulate β2 integrin adhesive activity and promote binding to and internalization by macrophages. Infect. Immun. 2006a;74:5658–5666. doi: 10.1128/IAI.00784-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Tapping RI, Harokopakis E, Nishiyama S-I, Ratti P, Schifferle RE, et al. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell.Microbiol. 2006b;8:1557–1570. doi: 10.1111/j.1462-5822.2006.00730.x. [DOI] [PubMed] [Google Scholar]
- Harokopakis E, Hajishengallis G. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. Eur. J.Immunol. 2005;35:1201–1210. doi: 10.1002/eji.200425883. [DOI] [PubMed] [Google Scholar]
- Harokopakis E, Albzreh MH, Martin MH, Hajishengallis G. TLR2 transmodulates monocyte adhesion and transmigration via Rac1- and PI3K-mediated inside-out signaling in response to Porphyromonas gingivalis fimbriae. J. Immunol. 2006;176:7645–7656. doi: 10.4049/jimmunol.176.12.7645. [DOI] [PubMed] [Google Scholar]
- Joiner KA, Fuhrman SA, Miettinen HM, Kasper LH, Mellman I. Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor-transfected fibroblasts. Science. 1990;249:641–646. doi: 10.1126/science.2200126. [DOI] [PubMed] [Google Scholar]
- Kozarov EV, Dorn BR, Shelburne CE, Dunn WA, Jr, Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler. Thromb. Vasc. Biol. 2005;25:e17–e18. doi: 10.1161/01.ATV.0000155018.67835.1a. [DOI] [PubMed] [Google Scholar]
- Lamont RJ, Jenkinson HF. Life below the gum line: Pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JC, Saslowsky DE, Edwardson JM, Henderson RM. Real-time analysis of the effects of cholesterol on lipid raft behavior using atomic force microscopy. Biophys. J. 2003;84:1827–1832. doi: 10.1016/s0006-3495(03)74990-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Michel R, Cohen J, Decarlo A, Kozarov E. Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol. 2008;8:26. doi: 10.1186/1471-2180-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Wang M, Triantafilou K, Triantafilou M, Nawar HF, Russell MW, et al. The A subunit of Type IIb enterotoxin (LT-IIb) suppresses the proinflammatory potential of the B subunit and its ability to recruit and interact with TLR2. J. Immunol. 2007a;178:4811–4819. doi: 10.4049/jimmunol.178.8.4811. [DOI] [PubMed] [Google Scholar]
- Liang S, Wang M, Tapping RI, Stepensky V, Nawar HF, Triantafilou M, et al. Ganglioside GD1a is an essential coreceptor for toll-like receptor 2 signaling in response to the B subunit of Type IIb enterotoxin. J. Biol. Chem. 2007b;282:7532–7542. doi: 10.1074/jbc.M611722200. [DOI] [PubMed] [Google Scholar]
- Linton MF, Fazio S. Macrophages, inflammation, and atherosclerosis. Int. J. Obes. Relat. Metab. Disord. 2003;27 Suppl 3:S35–S40. doi: 10.1038/sj.ijo.0802498. [DOI] [PubMed] [Google Scholar]
- Lowell CA. Rewiring phagocytic signal transduction. Immunity. 2006;24:243–245. doi: 10.1016/j.immuni.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Maldonado-Garcia G, Chico-Ortiz M, Lopez-Marin LM, Sanchez-Garcia FJ. High-polarity Mycobacterium avium-derived lipids interact with murine macrophage lipid rafts. Scand. J. Immunol. 2004;60:463–470. doi: 10.1111/j.0300-9475.2004.01511.x. [DOI] [PubMed] [Google Scholar]
- Manes S, del Real G, Martinez AC. Pathogens: raft hijackers. Nat. Rev. Immunol. 2003;3:557–568. doi: 10.1038/nri1129. [DOI] [PubMed] [Google Scholar]
- Nagahama M, Hayashi S, Morimitsu S, Sakurai J. Biological activities and pore formation of Clostridium perfringens beta toxin in HL 60 cells. J. Biol. Chem. 2003;278:36934–36941. doi: 10.1074/jbc.M306562200. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Yoshizaki F, Prinetti A, Sonnino S, Mauri L, Takamori K, et al. Lyn-coupled LacCer-enriched lipid rafts are required for CD11b/CD18-mediated neutrophil phagocytosis of nonopsonized microorganisms. J. Leukoc. Biol. 2007;83:728–741. doi: 10.1189/jlb.0707478. [DOI] [PubMed] [Google Scholar]
- Naroeni A, Porte F. Role of cholesterol and the ganglioside GM(1) in entry and short-term survival of Brucella suis in murine macrophages. Infect. Immun. 2002;70:1640–1644. doi: 10.1128/IAI.70.3.1640-1644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedergang F, Chavrier P. Signaling and membrane dynamics during phagocytosis: many roads lead to the phagos(R)ome. Curr. Opin. Cell Biol. 2004;16:422–428. doi: 10.1016/j.ceb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Norkin LC, Wolfrom SA, Stuart ES. Association of caveolin with Chlamydia trachomatis inclusions at early and late stages of infection. Exp. Cell Res. 2001;266:229–238. doi: 10.1006/excr.2001.5202. [DOI] [PubMed] [Google Scholar]
- Peyron P, Bordier C, N'Diaye E-N, Maridonneau-Parini I. Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with gllycosylphosphatidylinositol-anchored proteins. J.Immunol. 2000;165:5186–5191. doi: 10.4049/jimmunol.165.9.5186. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Bottcher A, Orso E, Kapinsky M, Nagy P, Bodnar A, et al. Lipopolysaccharide and ceramide docking to CD14 provokes ligand-specific receptor clustering in rafts. Eur. J. Immunol. 2001;31:3153–3164. doi: 10.1002/1521-4141(200111)31:11<3153::aid-immu3153>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Pierce SK. Lipid rafts and B-cell activation. Nat. Rev. Immunol. 2002;2:96–105. doi: 10.1038/nri726. [DOI] [PubMed] [Google Scholar]
- Pizzo P, Viola A. Lymphocyte lipid rafts: structure and function. Curr. Opin.Immunol. 2003;15:255–260. doi: 10.1016/s0952-7915(03)00038-4. [DOI] [PubMed] [Google Scholar]
- Pucadyil TJ, Tewary P, Madhubala R, Chattopadhyay A. Cholesterol is required for Leishmania donovani infection: implications in leishmaniasis. Mol. Biochem. Parasitol. 2004;133:145–152. doi: 10.1016/j.molbiopara.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Pussinen PJ, Alfthan G, Jousilahti P, Paju S, Tuomilehto J. Systemic exposure to Porphyromonas gingivalis predicts incident stroke. Atherosclerosis. 2007;193:222–228. doi: 10.1016/j.atherosclerosis.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Riethmuller J, Riehle A, Grassme H, Gulbins E. Membrane rafts in host-pathogen interactions. Biochim. Biophys. Acta. 2006;1758:2139–2147. doi: 10.1016/j.bbamem.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Riff JD, Callahan JW, Sherman PM. Cholesterol-enriched membrane microdomains are required for inducing host cell cytoskeleton rearrangements in response to attaching-effacing Escherichia coli. Infect. Immun. 2005;73:7113–7125. doi: 10.1128/IAI.73.11.7113-7125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger CM, Finlay BB. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 2003;4:385–396. doi: 10.1038/nrm1104. [DOI] [PubMed] [Google Scholar]
- Shin JS, Gao Z, Abraham SN. Involvement of cellular caveolae in bacterial entry into mast cells. Science. 2000;289:785–788. doi: 10.1126/science.289.5480.785. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simons K, Gruenberg J. Jamming the endosomal system: lipid rafts and lysosomal storage diseases. Trends Cell Biol. 2000;10:459–462. doi: 10.1016/s0962-8924(00)01847-x. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Swanson MS, Isberg RR. Analysis of the intracellular fate of Legionella pneumophila mutants. Ann. N .Y. Acad. Sci. 1996;797:8–18. doi: 10.1111/j.1749-6632.1996.tb52944.x. [DOI] [PubMed] [Google Scholar]
- Tamai R, Asai Y, Ogawa T. Requirement for intercellular adhesion molecule 1 and caveolae in invasion of human oral epithelial cells by Porphyromonas gingivalis. Infect.Immun. 2005;73:6290–6298. doi: 10.1128/IAI.73.10.6290-6298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng YT. Protective and destructive immunity in the periodontium: Part 1-Innate and humoral immunity and the periodontium. J. Dent. Res. 2006;85:198–208. doi: 10.1177/154405910608500301. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell Sci. 2002a;115:2603–2611. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Brandenburg K, Gutsmann T, Seydel U, Triantafilou K. Innate recognition of bacteria: engagement of multiple receptors. Crit. Rev. Immunol. 2002b;22:251–268. [PubMed] [Google Scholar]
- Triantafilou M, Manukyan M, Mackie A, Morath S, Hartung T, Heine H, Triantafilou K. Lipoteichoic acid and toll-like receptor 2 internalization and targeting to the Golgi are lipid raft-dependent. J. Biol. Chem. 2004;279:40882–40889. doi: 10.1074/jbc.M400466200. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Gamper FGJ, Lepper PM, Mouratis MA, Schumann C, Harokopakis E, et al. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell. Microbiol. 2007;9:2030–2039. doi: 10.1111/j.1462-5822.2007.00935.x. [DOI] [PubMed] [Google Scholar]
- Tsuda K, Amano A, Umebayashi K, Inaba H, Nakagawa I, Nakanishi Y, Yoshimori T. Molecular dissection of internalization of Porphyromonas gingivalis by cells using fluorescent beads coated with bacterial membrane vesicle. Cell Struct. Funct. 2005;30:81–91. doi: 10.1247/csf.30.81. [DOI] [PubMed] [Google Scholar]
- Wang M, Shakhatreh M-AK, James D, Liang S, Nishiyama S-i, Yoshimura F, et al. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J. Immunol. 2007;179:2349–2358. doi: 10.4049/jimmunol.179.4.2349. [DOI] [PubMed] [Google Scholar]
- Watarai M, Makino S, Fujii Y, Okamoto K, Shirahata T. Modulation of Brucella-induced macropinocytosis by lipid rafts mediates intracellular replication. Cell.Microbiol. 2002;4:341–355. doi: 10.1046/j.1462-5822.2002.00195.x. [DOI] [PubMed] [Google Scholar]
- Watarai M, Derre I, Kirby J, Growney JD, Dietrich WF, Isberg RR. Legionella pneumophila is internalized by a macropinocytotic uptake pathway controlled by the Dot/Icm system and the mouse Lgn1 locus. J. Exp. Med. 2001;194:1081–1096. doi: 10.1084/jem.194.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, et al. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305:1153–1157. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- Wright SD, Silverstein SC. Receptors for C3b and C3bi promote phagocytosis but not the release of toxic oxygen from human phagocytes. J. Exp. Med. 1983;158:2016–2023. doi: 10.1084/jem.158.6.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Verbeke P, Lamont RJ, Ojcius DM. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect. Immun. 2006;74:703–710. doi: 10.1128/IAI.74.1.703-710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]