

Abstract
A key challenge in radiotherapy is to maximize radiation doses to cancer cells while minimizing damage to surrounding healthy tissue. As severe toxicity in a minority of patients limits the doses that can be safely given to the majority, there is interest in developing a test to measure an individual’s radiosensitivity before treatment. Variation in sensitivity to radiation is an inherited genetic trait and recent progress in genotyping raises the possibility of genome-wide studies to characterize genetic profiles that predict patient response to radiotherapy.
The success of radiotherapy in eradicating a tumour depends principally on the total radiation dose given, but the tolerance of the normal tissues surrounding the tumour limits this dose. There is significant variation between patients in the severity of toxicity following a given dose of radiotherapy. As a result, dose is sub-maximal in many individuals because current dose thresholds are set in order to limit toxicity in those who are the most sensitive.
Researchers have long recognized that genetic variation contributes to individual differences in radiotherapy toxicity. For instance, homozygous mutations in ataxia-telangiectasia mutated (ATM) are associated with extreme sensitivity to radiation1. Further research implicated the involvement of multiple genetic pathways and by the end of the 1990s an individual’s sensitivity to radiation was considered to be an inherited, polygenic trait.
Current interest lies in developing genetic profiles that predict a patient’s probability of suffering toxicity following radiotherapy2,3. Although limited in scale, early studies support this possibility. Before this can happen, the genetic determinants that explain a measurable proportion of radiation toxicity first have to be identified. Genome-wide association studies (GWAS) have already been successful in finding novel genetic variants that explain a useful proportion of the risk of developing some common diseases and traits4. Therefore it is likely that a GWAS approach could also be successful in the field of radiogenomics. Once identified, the way in which the different variants interact to give an overall risk must also be understood before a useful test could be developed.
This Perspective reviews progress made in the field of radiogenomics and updates the recent review by Bentzen5, who summarized the current understanding of radiation toxicity and its pathogenesis. We highlight some of the potential problems that must be addressed as we progress from a candidate gene approach to GWAS.
Importance of radiotherapy
Radiotherapy is the most important non-surgical modality for the curative treatment of cancer. In 2004 in the United States, nearly 1 million of the ∼1.4 million people who developed cancer were treated with radiation (ASTRO). Of the 10.9 million people diagnosed with cancer worldwide each year (International Agency for Research on Cancer), around 50% require radiotherapy, 60% of whom are treated with curative intent. Radiotherapy is also highly cost effective, accounting for only 5% of the total cost of cancer care6.
Substantial gains in the therapeutic ratio, that is, the balance between cure and toxicity of treatment (FIG. 1a), have been made with the development of new technologies such as image-guided radiotherapy7,8 and intensity-modulated radiotherapy9-12. Other approaches for improving therapeutic ratios include altering radiotherapy fractionation, for example the Conventional or Hypofractionated High Dose Intensity Modulated Radiotherapy in Prostate Cancer (CHHiP) trial and the European Organization for Research and Treatment of Cancer (EORTC) trial of hyperfractionation in head and neck cancer13,14. The use of concurrent chemotherapy has become standard practice for a number of cancers, and the addition of new, molecularly targeted agents in combination with radiotherapy is promising to improve cure rates further15.
Figure 1. Dose–response curves for radiotherapy.
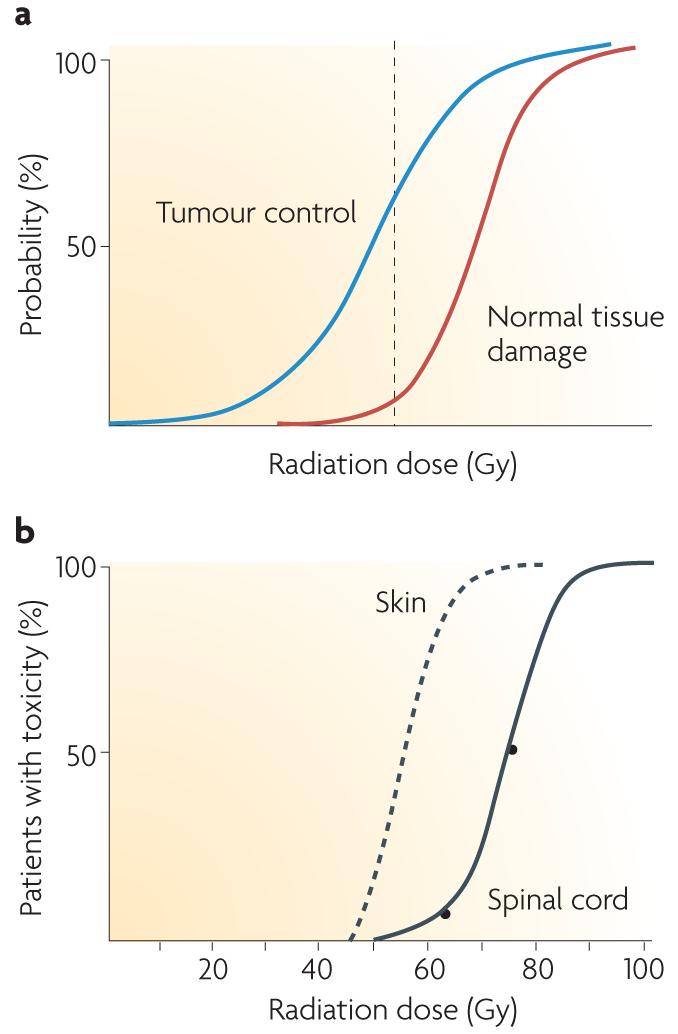
a | The probability of tumour cure increases with increasing radiation dose. As a small volume of normal tissue is unavoidably included in the radiation field, the probability of severe late normal tissue damage also increases. Radiotherapy schedules have developed to maximize cure while minimizing toxicity, and the dotted line shows a theoretical dose associated with ∼60% tumour control and ∼5% severe late toxicity. b | Cumulative frequency dose–response curves. The left-hand curve shows data for skin telangiectasia130; the right-hand curve is the putative dose–response curve for spinal cord necrosis131. The gradients of the two curves are similar, although the dose at which damage occurs is greater for the spinal cord than skin because of differences in target cells, tissue architecture and cell turnover. Interestingly, inbred animals have an even steeper gradient of the dose–response curve. The principal reason why human clinical data show shallower dose–response curves than inbred animals is inter-individual variability, that is, greater genetic variation, although animal studies are more carefully controlled for factors such as diet, age and co-morbidities than human studies.
With improved cure rates, survivorship issues become increasingly important. The long-term toxicity that is associated with cancer treatment negatively affects quality of life, so strategies aimed at toxicity reduction are important. Although there are still gains that can be achieved through technical advances, altered fractionation and new drug combinations, it is ultimately the radiosensitivity of the few that will limit our ability to further maximize patients’ toxicity-free survival, so understanding the genetics of radiosensitivity is crucial.
Radiation toxicity
The clinical manifestations of either acute (occurring during or within weeks of treatment) or late (occurring 6 months to many years later) radiation toxicity are well documented (FIG. 2), although the mechanistic basis for the separation of early and late effects has changed considerably in recent years5. This distinction provides a useful framework with which to describe radiotherapy toxicity in terms that might be useful for wider genomic studies.
Figure 2. The toxicity of radiotherapy.

Acute effects occur during or shortly after completion of treatment and are usually reversible and not generally considered dose-limiting. They occur in rapidly proliferating tissues, such as skin, gastrointestinal tract and the haematopoietic system. Early reactions tend to be relatively insensitive to changes in the radiation dose per fraction but are sensitive to the time over which radiation is delivered. Protracted treatment reduces acute toxicity but can compromise tumour control. Late effects manifest 6 months to several years after radiotherapy. The long time frame prevents titration of radiation dose against toxicity in individual patients, and the relationship between acute and late effects remains unclear32,132,133. As late side effects can be permanent, they provide the basis for dose constraints to radiation toxicity. Late effects typically occur in more slowly proliferating tissues, such as kidney, heart and central nervous system. The pathogenesis includes fibrosis, atrophy and vascular damage. Other important late normal tissue side effects include hormone deficiencies, infertility and second malignancies. Late toxicity tends to be more sensitive to changes in the radiation dose per fraction than acute reacting tissues and less sensitive to the overall treatment time.
Late effects can be irreversible and limit the dose in radical radiotherapy regimens; monitoring late toxicity is therefore crucial in assessing therapeutic benefit and follow-up must be sufficiently long.
Pathogenesis of normal tissue damage
Although the functional and structural tolerance of normal tissue to radiotherapy is contextual (BOX 1), studies of cell and tissue response to ionizing radiation have led to an improved understanding of the pathogenesis of radiation toxicity5. Recent radiobiology research suggests that normal tissue injury is a dynamic and progressive process. The deposition of energy results in DNA damage and changes in the microenvironment through chemokines, inflammatory cytokines, fibrotic cytokines, altered cell–cell interactions, influx of inflammatory cells and the induction of reparative and restorative processes5. Therefore, genes involved in DnA damage recognition as well as signalling, apoptosis, proliferation and inflammatory processes may have a role in the development of normal tissue damage. TABLE 1 summarizes some of the many genes that are considered important in the pathogenesis of radiotherapy toxicity5,16-23.
Box 1 | normal tissue tolerance.
The extent of structural damage to a tissue generally depends on cell radiosensitivity. The relationship between anatomical and structural radiation damage and failure of organ function is different for different organs, and depends more on organ physiology than on cell survival. For example, ulceration of the small bowel is an example of structural damage from radiotherapy that can lead to small bowel obstruction (functional damage). In radiobiological studies, the volume effect is defined by the relationship between the radiation dose that causes a given probability of radiation toxicity and the irradiated volume of the investigated tissue or organ121. Tissues with a large reserve capacity (functional tolerance) show a large volume effect. This is the case for the lung, where a small volume receiving a high dose is tolerated. In other tissues, such as the spinal cord, volume effects are small and a modest amount of damage from a radiation hot spot results in unacceptable toxicity. In many tissues, such as the breast or soft tissue of the pelvis, an incidence of significant (severe or grade 3) normal tissue damage of up to 5% is often regarded as acceptable. Therefore, treatment doses can be relatively high. The desired therapeutic ratio (the ratio of the probability of tumour cell kill to the probability of normal tissue complications) also shifts according to whether the treatment is radical, adjuvant or palliative. For radical treatments, an increase in toxicity is acceptable to increase the chance of cure, whereas for palliative treatments toxicity must be minimal.
Table 1. Candidate genes involved in the pathogenesis of radiation toxicity.
| Mechanism | examples of genes involved | Refs |
|---|---|---|
| DNA damage repair | ||
| Damage sensing | MRE11A, RAD50, NBN, H2AFX, TP53BP1, BRCA1, MDC1 | 16,17 |
| Mediator proteins | RB1, cyclins (e.g. CCNE1, CCND1, CCNB1), CDKs (e.g. CDK7, CDK10) | 16,17 |
| Cell cycle checkpoint control | CDK inhibitors (e.g. CDKN2C, CCKND2), ATM, ATR, TP53, CHEK2 | 16,17 |
| NHEJ | XRCC6, XRCC5, PRKDC, XRCC4, LIG4 | 16,17 |
| HR | RAD51, BRCA1, BRCA2, XRCC3 | 16,17 |
| Base excision repair | XRCC1, APEX, OGG1 | 16,17 |
| Radiation fibrogenesis | ||
| Apoptosis | TP53, BCL2, caspases (e.g. CASP3), BIRC5, RHOB, TP53INP1 | 18,19 |
| Pro-inflammatory cytokines | TNF, IL1A, IL6 | 18,19 |
| Pro-fibrotic proteins | TGFB1, TGFB2, TGFB3, CTGF | 5 |
| Smad signalling pathway | Receptor-regulated SMADs (SMAD1, SMAD2, SMAD3, SMAD5, SMAD8) | 5 |
| DNA-binding TFs, co-regulators and co-receptors | Type I receptor (SMAD4), inhibitory sMADs (SMAD6,SMAD7), NFKB1 | 5 |
| Increased ECM and collagen deposition | TGF transmembrane receptors TGFBR1 and TGFBR2 | 5 |
| Oxidative stress | ||
| Antioxidant enzymes | Superoxide dismutases (e.g. SOD1) | 20 |
| Renin-angiotensin system | AGT | 21 |
| Endothelial cell damage | ||
| Chemokines recruiting monocytes | Rho proteins, Rho kinases, CTGF, HSP27, ZYX | 22,23 |
| Cytokines | FGF2 | 22,23 |
| Growth factors | VEGF | 22,23 |
AGT, angiotensinogen; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related protein; BIRC5, baculoviral IAP repeat-containing protein 5; CDK, cyclin-dependent kinase; CTGF, connective tissue growth factor; ECM, extracellular matrix; FGF2, fibroblast growth factor 2; HR, homologous recombination; HSP27, heat shock protein 27; IL, interleukin; LIG4, DNA ligase 4; MDC1, mediator of DNA damage checkpoint protein 1; MRE11A, mitotic recombination 11A; NBN, nibrin; NHEJ, non-homologous end joining; NFKB1, nuclear factor-κB; OGG1, 8-oxoguanine DNA glycosylase; PRKDC, DNA-dependent protein kinase catalytic subunit; RB1, retinoblastoma 1; SOD, superoxide dismutase; TF, transcription factor; TGFB, transforming growth factor-β; TGFBR, TGFβ receptor; TNF, tumour necrosis factor; TP53BP1, TP53 binding protein 1; TP53INP1, tumour protein p53-inducible nuclear protein 1; VEGF, vascular endothelial growth factor ZYX, zyxin.
Variation in patient response
variation in normal tissue reactions has been observed since the earliest days of radiotherapy24 and follows an approximately Gaussian distribution for both acute and late effects25. A number of factors influence a patient’s likelihood of developing toxicity, which may complicate the relationship between genotype and toxicity. These confounding factors are related to physics (total dose, dose per fraction and volume irradiated, irradiation site and dose inhomogeneity), additional treatment (use of concomitant chemotherapy or surgery), patient characteristics (age, use of cigarettes, haemoglobin level and co-morbid conditions such as diabetes, hypertension, vascular and connective tissue diseases) and chance (Poisson statistics)26,27. The investigation of underlying genetic determinants of toxicity, therefore, requires careful control of as many of these factors as possible, and particularly radiotherapy dose inhomogeneity28. Patients in formal, carefully controlled clinical trials are ideal subjects29.
As total radiation dose increases so does the likelihood of developing toxicity (FIG. 1a,b). Radiation dose–response relationships for normal tissues have a threshold at low doses — which produce no reaction — and saturate at high doses (FIG. 1b). There is evidence that normal tissue radiation dose–response relationships are steep; this means that small changes in dose result in relatively large differences in toxicity30. The steep dose–response relationship was illustrated in the results of the UK START trial A that randomized 2,236 breast cancer patients to receive 50 Gy in 25 fractions or 39 or 42.9 Gy in 13 fractions, with all schedules given over 5 weeks. The risk of developing late toxicity — photographic change in breast appearance — was 24% for patients who received 39 Gy in 13 fractions compared with 36% for those allocated 42.9 Gy in 13 fractions (p < 0.001): a 50% increase in toxicity associated with a 3.9 Gy escalation in dose31.
The dose–response relationships for late toxicities such as skin telangiectasia and spinal cord damage are illustrated in FIG. 1b. It has been shown that the dose–response curve for telangiectasia can be broken down into a series of steep curves, each of which would apply to a subpopulation of patients stratified according to intrinsic radiosensitivity32. This means that if these subpopulations were identified before treatment, a substantial therapeutic benefit could be achieved.
Despite the various factors influencing the development of radiation toxicity, it has been estimated that as much as 80% of the variation in normal tissue reactions between patients cannot be accounted for by known factors and is likely to be genetic33. This provides the basis for examining the genetic variation that underlies individual variation in normal tissue response. Although the sigmoid dose–response has been understood for over 70 years24, and the estimates of the genetic contribution for over 10 (REF. 33), only now in the era of high-throughput analyses is it possible to investigate the genetic basis of radiation toxicity.
Recording data
The investigation of normal tissue responses is complicated not only by the confounding factors summarized above but also because of the multiple end points involved (such as skin telangiectasia, bowel stricture or lung pneumonitis), which are determined by the site irradiated. For some tumours, several normal tissues can be irradiated, such as bowel, bladder and reproductive organs following radiotherapy to the pelvis. Therefore, tissue-specific end points, and often severities of effect, must be defined. FIGURE 2 illustrates that late radiation toxicity occurs through a variety of mechanisms, and these mechanisms lead to a variety of clinical end points.
Despite a recognized need for a standardized approach for reporting toxicity, a variety of scoring systems are used and toxicity remains generally under reported34. The RTOG (Radiation Therapy Oncology Group)–EORTC late effects scale was in common use before the publication of the LEnT SOMA (Late Effects normal Tissues: Subjective, Objective, Management and Analytic) system35,36. The LEnT SOMA scales were designed to provide a common platform for recording the late effects of radiotherapy but are now largely superseded by the national Cancer Institute (nCI) Common Terminology Criteria for Adverse Effects version 3.0 (CTCAE v3.0). CTCAE v3.0 incorporated LEnT SOMA items to become the first multimodality grading system for recording acute and late effects in oncology. There are also other published toxicity scales used for particular cancers, for example the Franco-Italian glossary for cervical cancer patients37 and the UCLA (University of California, Los Angeles) index for scoring late radiation toxicity in prostate cancer patients38. Analytical techniques have the potential to provide objective measures of response as a continuous rather than a dichotomized variable, especially if subclinical effects could be measured28,39. Unfortunately, this is an area of clinical research that has not received much attention.
Analysis of toxicity data
Toxicity is graded according to severity. Graded reactions may be dichotomized into a simple binary scale, such as none versus any late effects. Although associated with loss of information, dichotomization simplifies the analysis and interpretation of results31,40. Alternatively, graded response data can be analysed by logistic regression, which incorporates the timescale of the occurrence of radiation toxicity. The timescale is important for radiotherapy because late effects can manifest many years after irradiation and can be progressive41. Continuous monitoring of toxicity is preferable and, with the more widespread use of survival analysis (or actuarial) statistics, adjustments can be made for the actual number of patients still at risk42. Crude proportions such as the number of responders divided by the number of patients in a group are a poor description of treatment toxicity. Death from cancer is a censoring event that, if proper statistical methods are not applied, will reduce the level of toxicity detected. A standard approach is to estimate the toxicity-free survival using the Kaplan–Meier method. Cumulative incidence estimates provide an alternative to the Kaplan–Meier method for quantifying morbidity as a function of time43. Prevalence estimates as a function of time are also relevant44. Time-adjusted toxicity can be reported as the prevalence of an end point in surviving patients rather than the cumulative incidence or probability of experiencing a complication, which can overestimate the incidence of complications45. Prevalence estimates, however, are influenced by the management of toxicity and must address the problem of censored observations. Therefore, as a minimum, actuarial estimates of treatment toxicity should be calculated and, as a supplement, it may be useful to estimate cumulative incidence or prevalence of morbidity.
Measuring individual radiosensitivity
The first report of an individual with extreme sensitivity to radiation was published in 1975 (REF. 46). This landmark paper described a patient with ataxia-telangiectasia with severe reactions to radiotherapy. Fibroblasts cultured from the individual were approximately three times more sensitive to radiation than those from normal donors. Several more studies were published in the 1980s showing in vitro radiosensitivity of cells from patients with extreme reactions to radiotherapy, supporting the notion that clinically normal tissue toxicity is associated with cellular radiation sensitivity47-53.
Most of the initial studies that examined normal tissue response to radiotherapy took skin biopsies and cultured fibroblasts. The first study showed a correlation between in vitro radiosensitivity and normal tissue toxicity in patients with breast cancer54. Later studies included patients with a range of normal tissue reactions deemed to be within the expected (Gaussian) range of reactions55-58. However, two large studies in breast cancer patents failed to demonstrate any relationship between fibroblast sensitivity and normal tissue response59,60.
The lack of correlation between in vitro tests and clinical response may partially be explained by the laboratory techniques used, intra- and inter-assay variability, inadequate patient follow up, cell types used, small study sizes and confounding factors61. For example, in vitro cell, chromosome and DnA damage assays of radiosensitivity cannot account for variability in cytokine response, tissue remodelling and collagen deposition in whole tissues62. nevertheless, the largest and one of the few prospective studies measured lymphocyte radiosensitivity in patients with carcinoma of the cervix, and found this to be an independent prognostic factor for the probability of toxicity-free survival58. Importantly, the work showed that an individual’s sensitivity to radiation could be measured in a nontarget cell type, which is consistent with it being genetically determined.
During the 1990s there was interest in exploring potential rapid assays measuring, for example, DnA damage, chromosome damage or apoptosis. This work showed that there can be a relationship between these variables and toxicity. However, in general, cell-based and molecular assays lack the sensitivity, specificity and reproducibility that are required for a routine clinical test. In addition, such assays are generally not rapid enough to be used in a clinical setting. However, different approaches continue to be investigated, for example, an assay of T-lymphocyte apoptosis was recently shown to be predictive of radiation-induced late toxicity in patients with a variety of cancer types63. There are also studies aimed at generating gene expression profiles that predict a patient’s likelihood of developing radiation toxicity64,65, and work is being carried out using lymphoblast and fibroblastic cells to generate RnA signatures that predict the development of acute66-68 or late69-71 effects.
Finding relevant genetic variation
Rare radiosensitivity syndromes that are characterized by Mendelian inheritance of germline mutations in genes involved in the detection of DnA damage or DnA repair, such as ataxia-telangiectasia, nijmegen breakage syndrome, Fanconi’s anaemia and Bloom’s syndrome, illustrate that specific genes can influence the radiosensitivity of tissues. However, these syndromes are confined to individual families and are probably of little relevance when assessing radiosensitivity in the vast majority of cancer patients.
It is clear that inherited genetic variation is important in determining an individual’s sensitivity to radiation. However, the complexity of the genetic model that underlies this is unknown and ranges from a single rare mutation with a large effect (such as a mutation in ATM) to a combination of multiple common variants that together confer increased sensitivity — the common-variant–common-disease model. In reality, it is likely that a range of alleles of differing frequencies and differing effect sizes are important.
Genetic association case–control studies, in which allele frequencies in individuals with the phenotype of interest are compared with the allele frequencies in those without the phenotype, are an efficient way of finding common variants that have modest effects. In these studies, single nucleotide polymorphisms (SnPs) are used as genetic markers to screen the known (and most of the unknown) common genetic variants in a given region.
Although the use of SnPs is well established in such association studies, other forms of genetic variation also exist. Copy number variation (Cnv) is a structural genomic variant that results in confined copy number changes in a specific chromosomal region. It is estimated that at least 10% of the genome is subject to copy number variation and it has been postulated that Cnvs may account for phenotypical variability in genetic disease by altering levels of gene expression, disrupting coding sequences or by interrupting long-range gene regulation72. Cnvs are less numerous than SnPs but can affect up to several megabases of DnA per variant, which adds up to a significant proportion of the genome. There is evidence to suggest that there is limited overlap between the two forms of genetic variation and therefore they can be used in conjunction with each other to screen different parts of the human genome73. Improved detection of Cnvs is anticipated within the next few years74 and the SnP arrays that are used for genome-wide scans are being developed so that SnPs can be used to tag Cnvs.
In addition, recent interest has focused on epigenetic modification of the genome, for example, DnA methylation of cytosine residues that lie within the CpG dinucleotide75. It is possible that epigenetic mechanisms may be important in the response of both tumour cells and normal tissues to radiotherapy.
Until recently, SnP association studies were limited to the candidate gene approach, in which biological pathways thought to be involved in the phenotype of interest were identified and only variants in genes encoding proteins in those pathways were studied. With recent developments in high-throughput genotyping, it is now possible to genotype up to 1 million SnPs that together represent all the common variation across the genome in a single, albeit large, experiment: a GWAS (BOX 2).
Box 2 | Genome-wide association studies.
Recombination occurs during meiosis as crossover between paired chromosomes. This crossover tends to occur at recombination hot spots. Chromosomes may be divided into haplotype blocks, separated by recombination hot spots. Within each block, alleles of multiple single nucleotide polymorphisms (SNPs) are inherited together as a single unit and the presence of one allele may predict the presence of several others. They are therefore said to be in linkage disequilibrium with each other. The entire genome can be screened using a small set of carefully selected marker SNPs, which can serve as proxies for many others. The aim is to find SNP markers that tag the known (and most of the unknown) common genetic variants in a given region (gene or haplotype block or genome). The functional variant may be a SNP or could be a copy number variant, deletion or insertion.
GWAS provide an empirical approach for identifying all moderate risk alleles without the need for prior knowledge of position or function122,123. To keep the false positives within acceptable bounds, significance levels of p < 10−7 are needed to provide strong evidence of association123.
GWAS have successfully identified several common, modest-risk variants for a variety of widespread complex diseases4,83,100,124-127. However, GWAS have been unsuccessful for other phenotypes. For example, few loci have been found for hypertension123,128,129 despite the fact that heritability appears to be in the order of 15-35% (REF. 128).
The main limitation of GWAS is their relatively high cost. One approach to limit costs is to use a staged study design in which a subset of the samples are genotyped for the set of genome-wide tagging SNPs and only the most strongly associated SNPs are genotyped in the remainder of the samples. Staged approaches have been used by most of the GWAS reported to date.
A major disadvantage of the candidate gene approach is that a gene would only be examined if its biology and that of the trait being considered are sufficiently well understood. The precise functions of the majority of genes identified by the Human Genome Project are still unknown and the molecular pathogenesis of normal tissue response to radiation is complex and not fully understood. GWAS avoid the limitation of a lack of prior knowledge and can even identify novel disease loci in regions of the genome that seem to contain no protein-coding genes.
Radiogenomics studies published to date have adopted the candidate gene approach to look for variations in genes involved in DnA repair (such as ATM, BRCA1, BRCA2 and TP53), antioxidant enzymes such as superoxide dismutase 2 (SOD2) and cytokines such as TGFB1. These studies have been reviewed recently3 and are summarized in Supplementary information S1 (table).
So far it has not been possible to demonstrate unequivocal links between genotype and radiation toxicity. TGFB1 variants and late toxicity in the breast provides a good example: initial studies suggested a relationship40,76-79, but subsequent studies demonstrated no consistent relationship2,78,79. This does not mean TGFB1 is unimportant, but rather that variation in TGFB1 itself is not a significant determinant of late radiotherapy change in the breast. If TGFB1 is involved in normal tissue response and toxicity, which appears likely from its biology, then it is possible that genes regulating or modulating its expression or activity may also be linked with normal tissue response.
It would also be possible to identify rare variants using an association study design. However, although many rare genetic variants are known, the catalogue of such variants is not comprehensive and is unlikely to be complete for several years. In addition, approaches using tagging SnPs are inefficient for rare variants, which tend to have arisen recently in human history and so are poorly tagged by the older, common SnPs that are presently used in association studies. Finally, huge sample sizes are needed to identify rare variants associated with phenotypes unless risks are large. Consequently, it is likely that the search for genetic determinants of radiation sensitivity will focus on common variation for the foreseeable future.
The way ahead
One of the key lessons to emerge from the early association studies of radiation sensitivity and other complex phenotypes is the need to apply stringent criteria in the interpretation of statistical significance. This is because the probability that any one SnP is associated with the phenotype is small given the large number of SnPs in the genome, only a small proportion of which can be associated with the phenotype. Consequently large sample sizes are essential and can often only be achieved by multicentre collaboration. An example of such a study is the Genetic Pathways for the Prediction of the Effects of Irradiation (GEnEPI) project launched by the European Society for Therapeutic Radiation and Oncology. This project will catalogue the available patient data with a record of material obtained, including blood, tumour material and relevant normal tissue samples, from patients undergoing curative radiotherapy for a variety of different cancers at multiple centres across Europe. The data and material from this project will be available for genetic research80.
RAPPER (Radiogenomics: Assessment of Polymorphisms for Predicting the Effects of Radiotherapy), a component project within GEnEPI, is a large multicentre UK collaboration, studying the relationship between late radiotherapy morbidity and SnP genotype81 (ESTRO). The project recruits patients from large national and single-centre radiotherapy trials and includes patients with different cancer types to obtain representation of the full range of normal tissue responses to radiotherapy. Any future application of genotyping in the clinic should be applicable to all patients irrespectively of tumour type or of the patient-related factors that are used as eligibility criteria in clinical trials to reduce patient heterogeneity. The use of different cancer types should also enable the discovery of genetic variants that affect radiation response irrespectively of tumour type or position. FIGURE 3 illustrates a proposed design for a GWAS of radiation toxicity.
Figure 3. Proposed design for a radiation toxicity genome-wide association study (GWAS).

In the first phase of a staged approach a full set of tagged single nucleotide polymorphisms (tag-SNPs; 600,000) is chosen that comprehensively captures common variations across the genome. This set is genotyped using a whole genome chip in a relatively small population and at a liberal p-value threshold, to identify a subset of SNPs with putative associations. As the phenotype of interest exhibits a range of responses, including intermediate levels, this will give greater power than a case–control study of the same size. Phase II takes the top 5% of SNPs identified in phase I or those passing an initial threshold (or filter) and re-tests these SNPs using a custom-designed oligonucleotide array in a larger independent population sample, thus significantly increasing efficiency and reducing genotyping costs.
The NCI-National Human Genome Research Institute (NCI-NHGRI) Working Group on Replication in Association Studies has published a comprehensive set of guidelines on reporting and evaluating initial genotype–phenotype associations and for establishing replication82. Any study of radiogenomics should aim to comply with these guidelines. The initial study would need to be large and powerful, and careful replication studies would have to be performed to validate any positive findings. This could be achieved by collaboration between groups interested in the field of radiogenomics. Collaborations can overcome many of the disadvantages of a disconnected set of underpowered studies, and can establish whether the findings can be generalized by providing a greater diversity of populations. In addition, differences in linkage disequilibrium relationships across populations can sometimes be used to narrow the region of interest for later genetic and possibly functional analysis82,83.
Given the success of GWAS for other phenotypes, it seems likely that similar studies using available large sample sets will provide the highest chance of identifying common genetic variants that determine radiation sensitivity. However, there are several differences between the phenotypes for which GWAS have been successful and the phenotype of radiation sensitivity. For example, genome-wide studies of disease susceptibility have a binary outcome measure: patients are diagnosed with the disease or not. Methods for analysing quantitative traits (those with a range of values) are being developed84,85, and GWAS have already been successful for some quantitative traits such as obesity86-89, height90-92 and serum lipid levels93-96.
Genotype–phenotype associations that have been replicated widely have often used clearly defined phenotypes classified by standard and widely accepted criteria, such as diabetes and age-related macular degeneration. However, in many circumstances, our current approaches for defining and assaying phenotypes may not make optimal use of genotype data97. Some association studies have reported on intermediate phenotypes that predict disease (also known as endophenotypes). Often, intermediate phenotypes are more objective than the disease diagnosis. For example, a deficit in working memory can be used as an endophenotype of schizophrenia98, and heritable electrocardiographic and heart rate variability measures and serum lipid levels are associated with and can predict adverse cardiovascular outcomes, including sudden cardiac death and myocardial infarction99. Many genetic mapping studies of diseases are already using a wide variety of intermediate, quantitative phenotypes related to those diseases97,100,101.
One potential problem, however, is that there may be only limited evidence for the heritability of such traits, a prerequisite in most genetic studies. Unfortunately, it is unlikely that prospective toxicity data can be collected for members of the same family, making the heritability of a radiosensitive trait difficult to establish. Scott et al. used a chromosome damage assay to investigate the radiosensitivity of first-degree relatives of 16 radiation-sensitive and eight radiation-tolerant breast cancer survivors102. Of first-degree relatives of sensitive patients, 62% were also radiosensitive, compared with 7% first-degree relatives of radiation-tolerant patients103. Unfortunately, this assay did not transfer well between laboratories, but several recent studies have demonstrated that chromosomal radiosensitivity of lymphocytes is largely determined by genetic factors104-108.
The assessment of radiation toxicity is complicated. As already mentioned, there are a variety of patient- and treatment-related factors that influence the development of radiation toxicity, and there is considerable variability in the scoring of normal tissue toxicity. Analytical techniques for the assessment of radiation toxicity are limited to just a few examples28,39, so radiogenomic studies must rely on mainly clinical data as a measure of phenotype. However, it is difficult to ascertain how much a clinical score, such as pain, reflects a biological problem, especially because of the complexity of the pathogenesis of radiation toxicity (FIG. 2). Attention must focus, therefore, on obtaining detailed, accurate and complete information on study participants over an adequate follow-up period in order to maximize the chance of finding an association between radiation toxicity and genotype. The actuarial statistical analysis of radiation toxicity end points needs to be carefully considered. All research groups must provide sufficient detail for the definition of the phenotypes investigated to allow assessment of their validity and comparability across studies.
Tailoring treatment dose from genotype
The ultimate goal of radiogenomics is to develop a genetic risk profile for individualizing radiation dose prescriptions in order to optimize tumour control while reducing damage to normal tissues. Such a profile could divide patients into subgroups with different probabilities of developing toxicity, to permit irradiation up to the normal tissue tolerance for each subgroup. There would still be a range of toxic effects on normal tissue in each subgroup, but this would be of much smaller magnitude.
The loci identified to date from GWAS that are linked to cancer risk in breast, prostate, colorectal, lung and melanoma individually confer only a modest risk of malignancy, increasing the relative risk of cancer by less than 50% of the baseline population risk4. In most cases the associated SnP is a tagging SnP rather than a causal variant and so the effect size may be underestimated. However, in many diseases the effects of different loci may be multiplicative109. If enough loci are found, it is plausible that a set of SnPs will together explain a meaningful proportion of the genetic variance in radiosensitivity and will therefore have a predictive value for an individual’s risk of radiation toxicity.
As noted above, the mechanisms of radiation toxicity are poorly understood. If new loci predicting radiosensitivity can be identified, insight may be gained into the pathogenesis of radiation toxicity. It has also been postulated that the use of genetic profiling in complex diseases is most likely to be of benefit for phenotypes in which the pathogenic pathways are poorly understood110. This is true for radiosensitivity, for which there are insufficient defined risk factors that predict a patient’s likelihood of developing toxicity. This contrasts, for example, with cardiovascular disease, for which classical risk factors such as diabetes, smoking, hypercholesterolaemia and hypertension are already predictive of disease onset and genetic testing may not greatly improve on the predictive value of these classical risk factors.
The integration of genetic profiling into routine radiotherapy practice would allow an increase in tumour dose for radiation-tolerant patients, increasing their probability of local recurrence-free survival. For example, in many sites, a 1% increase in dose should increase the probability of tumour control by 1-2% (REFs 111,112). Thus, a dose increase in radiation-resistant patients of 20% could achieve a 20-40% increase in tumour control. Such radiation dose escalation is likely to have a positive effect on overall survival, as illustrated by the observation that one breast cancer death is prevented for every four local recurrences prevented with radiotherapy113. By contrast, a dose reduction of 20% in radiation-sensitive patients would virtually abolish serious side effects. Alternatively, if dose reduction is not acceptable, patients could be given hyperfractionated radiotherapy. This could be combined with radiation techniques such as intensity-modulated radiotherapy and brachytherapy, which produce a steep dose gradient allowing a tumoricidal dose while sparing normal tissue. If a relationship exists between tumour and normal tissue radiosensitivity, this will further enhance the potential of genetic profiling in the management of radiotherapy patients114,115. However, although there is some evidence for a link116-118, the area is not well researched.
There is increasing use of concurrent chemotherapy given with radiotherapy, which typically augments some toxicities. An understanding of the sensitivity of normal tissues to radiotherapy might allow better tailoring of chemotherapy dose in such combined schedules. Genetic variations that influence a patient’s metabolism of a chemotherapeutic drug (pharmacogenomics) are being studied119. It may be necessary to consider genetic variation involved in both chemotherapy and radiotherapy toxicity, as well as in tumour response, to provide a comprehensive personalization of cancer treatment.
Conclusion
Candidate gene studies have been largely unsuccessful in identifying the genetic variants underlying most phenotypes. By contrast, empirical GWAS, until recently prohibitively expensive, have proved fruitful in finding numerous genetic loci, which together explain a useful proportion of the genetic variance of a phenotype. It is likely that a similar approach is needed to identify the majority of common variants underlying an individual’s sensitivity to radiation and put them together to create a clinically useful pretreatment (profile) test.
Patients at both ends of the spectrum of normal tissue radiosensitivity are likely to benefit from genetic profiling. It has been estimated that 50% of patients might benefit through an increased tumour control in the 40% most radiation-tolerant patients and a decrease in morbidity in the 10% that are most sensitive32. It is possible that such an approach could improve survival rates by more than 10% (REF. 120).
Supplementary Material
Acknowledgements
The work of the authors is supported by Cancer Research UK, The Royal College of Radiologists, Breast Cancer Campaign, the National Institute for Health Research Cambridge Biomedical Research Centre, and Experimental Cancer Research Centre funding. G.C.B. is funded by a fellowship from Cancer Research UK and The Royal College of Radiologists. P.D.P.P. is a Cancer Research UK Senior Clinical Research Fellow.
Footnotes
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
ATM | BRCA1 | BRCA2 | SOD2 | TGFB1 | TP53
OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
ataxia telangiectasia | Bloom’s syndrome | Fanconi’s anaemia | Nijmegen breakage syndrome
FURTHER INFORMATION
ASTRO: www.rtanswers.org
Cancer Research UK Department of Oncology at Strangeways: http://www.srl.cam.ac.uk/groups/dept_oncology_strangeways.htm
CTCAE v3.0: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf
ESTRO: http://www.estro.be/estro/index.cfm
International Agency for Research on Cancer: http://www-dep.iarc.fr/
International HapMap Project: http://www.hapmap.org/
The NIEHS SNPs Program: http://egp.gs.washington.edu/
SeattleSNPs: http://pga.gs.washington.edu
Contributor Information
Gillian C. Barnett, University of Cambridge Department of Oncology, Oncology Centre, Addenbrooke’s Hospital, Hills Road, Cambridge CB2 0QQ, UK & Department of Oncology, Strangeways Research Laboratories, Worts Causeway, Cambridge CB1 8RN, UK
Catherine M. L. West, Academic Radiation Oncology, University of Manchester, Christie Hospital, Manchester M20 4BX, UK
Alison M. Dunning, Department of Oncology, Strangeways Research Laboratories, Worts Causeway, Cambridge CB1 8RN, UK
Rebecca M. Elliott, Academic Radiation Oncology, University of Manchester, Christie Hospital, Manchester M20 4BX, UK
Charlotte E. Coles, University of Cambridge Department of Oncology, Oncology Centre, Addenbrooke’s Hospital, Hills Road, Cambridge CB2 0QQ, UK
Paul D. P. Pharoah, Department of Oncology, Strangeways Research Laboratories, Worts Causeway, Cambridge CB1 8RN, UK
Neil G. Burnet, University of Cambridge Department of Oncology, Oncology Centre, Addenbrooke’s Hospital, Hills Road, Cambridge CB2 0QQ, UK
References
- 1.Savitsky K, et al. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Mol. Genet. 1995;4:2025–2032. doi: 10.1093/hmg/4.11.2025. [DOI] [PubMed] [Google Scholar]
- 2.Andreassen CN. Can. risk of radiotherapy-induced normal tissue complications be predicted from genetic profiles? Acta Oncol. 2005;44:801–815. doi: 10.1080/02841860500374513. [DOI] [PubMed] [Google Scholar]
- 3.Alsner J, Andreassen CN, Overgaard J. Genetic markers for prediction of normal tissue toxicity after radiotherapy. Semin. Radiat. Oncol. 2008;18:126–135. doi: 10.1016/j.semradonc.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Easton DF, Eeles RA. Genome-wide association studies in cancer. Hum. Mol. Genet. 2008;17:R109–R115. doi: 10.1093/hmg/ddn287. [DOI] [PubMed] [Google Scholar]
- 5.Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nature Rev. Cancer. 2006;6:702–713. doi: 10.1038/nrc1950. [DOI] [PubMed] [Google Scholar]
- 6.Ringborg U, et al. The Swedish Council on Technology Assessment in Health Care (SBU) systematic overview of radiotherapy for cancer including a prospective survey of radiotherapy practice in Sweden 2001 — summary and conclusions. Acta Oncol. 2003;42:357–365. doi: 10.1080/02841860310010826. [DOI] [PubMed] [Google Scholar]
- 7.Dawson LA, Sharpe MB. Image-guided radiotherapy: rationale, benefits, and limitations. Lancet Oncol. 2006;7:848–858. doi: 10.1016/S1470-2045(06)70904-4. [DOI] [PubMed] [Google Scholar]
- 8.van Herk M. Different styles of image-guided radiotherapy. Semin. Radiat. Oncol. 2007;17:258–267. doi: 10.1016/j.semradonc.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Glatstein E. Intensity-modulated radiation therapy: the inverse, the converse, and the perverse. Semin. Radiat. Oncol. 2002;12:272–281. doi: 10.1053/srao.2002.32433. [DOI] [PubMed] [Google Scholar]
- 10.Hong TS, Ritter MA, Tome WA, Harari PM. Intensity-modulated radiation therapy: emerging cancer treatment technology. Br. J. Cancer. 2005;92:1819–1824. doi: 10.1038/sj.bjc.6602577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moran JM, Elshaikh MA, Lawrence TS. Radiotherapy: what can be achieved by technical improvements in dose delivery? Lancet Oncol. 2005;6:51–58. doi: 10.1016/S1470-2045(04)01713-9. [DOI] [PubMed] [Google Scholar]
- 12.Ten Haken RK, Lawrence TS. The clinical application of intensity-modulated radiation therapy. Semin. Radiat. Oncol. 2006;16:224–231. doi: 10.1016/j.semradonc.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Dearnaley D, et al. Conventional or hypofractionated high dose intensity modulated radiotherapy in prostate cancer: Preliminary report on acute and late toxicity. A phase III multicentre trial (CHHIP); ASCO Proceedings 2007; 2007; Abstract 303. [Google Scholar]
- 14.Bourhis J, et al. Hyperfractionated or accelerated radiotherapy in head and neck cancer: a meta-analysis. Lancet. 2006;368:843–854. doi: 10.1016/S0140-6736(06)69121-6. [DOI] [PubMed] [Google Scholar]
- 15.Bonner JA, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 16.O’Driscoll M, Jeggo PA. The role of double-strand break repair — insights from human genetics. Nature Rev. Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 17.Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13:458–462. doi: 10.1016/s0962-8924(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 18.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nature Rev. Mol. Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 19.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nature Rev. Mol. Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 20.Mikkelsen RB, Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. 2003;22:5734–5754. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- 21.Robbins ME, Diz DI. Pathogenic role of the renin-angiotensin system in modulating radiation-induced late effects. Int. J. Radiat. Oncol. Biol. Phys. 2006;64:6–12. doi: 10.1016/j.ijrobp.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 22.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling — in control of vascular function. Nature Rev. Mol. Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 23.Weis SM. Vascular permeability in cardiovascular disease and cancer. Curr. Opin. Hematol. 2008;15:243–249. doi: 10.1097/MOH.0b013e3282f97d86. [DOI] [PubMed] [Google Scholar]
- 24.Holthusen H. Erfahrungen über die Verträglichkeitsgrenze für Röntgenstrahlen and deren Nutzanwendung zur Verhütung von Schäden. Strahlentherapie. 1936;57:254–269. in German. [Google Scholar]
- 25.Burnet NG, Johansen J, Turesson I, Nyman J, Peacock JH. Describing patients’ normal tissue reactions: concerning the possibility of individualising radiotherapy dose prescriptions based on potential predictive assays of normal tissue radiosensitivity. Steering Committee of the BioMed2 European Union Concerted Action Programme on the Development of Predictive Tests of Normal Tissue Response to Radiation Therapy. Int. J. Cancer. 1998;79:606–613. doi: 10.1002/(sici)1097-0215(19981218)79:6<606::aid-ijc9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 26.Bentzen SM, Overgaard J. Patient-to-patient variability in the expression of radiation-induced normal tissue injury. Semin. Radiat. Oncol. 1994;4:68–80. doi: 10.1053/SRAO00400068. [DOI] [PubMed] [Google Scholar]
- 27.Holscher T, Bentzen SM, Baumann M. Influence of connective tissue diseases on the expression of radiation side effects: a systematic review. Radiother. Oncol. 2006;78:123–130. doi: 10.1016/j.radonc.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 28.Coles CE, et al. Implementation of breast IMRT via a randomised control trial: a report of the first year’s experience. Radiother. Oncol. 2004;73(Suppl. 1):S15. [Google Scholar]
- 29.Bentzen SM. High-tech in radiation oncology: should there be a ceiling? Int. J. Radiat. Oncol. Biol. Phys. 2004;58:320–330. doi: 10.1016/j.ijrobp.2003.09.057. [DOI] [PubMed] [Google Scholar]
- 30.Turesson I. Individual variation and dose dependency in the progression rate of skin telangiectasia. Int. J. Radiat. Oncol. Biol. Phys. 1990;19:1569–1574. doi: 10.1016/0360-3016(90)90374-s. [DOI] [PubMed] [Google Scholar]
- 31.Bentzen SM, et al. The UK Standardisation of Breast Radiotherapy (START) Trial A of radiotherapy hypofractionation for treatment of early breast cancer: a randomised trial. Lancet Oncol. 2008;9:331–341. doi: 10.1016/S1470-2045(08)70077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burnet NG, Wurm R, Nyman J, Peacock JH. Normal tissue radiosensitivity — how important is it? Clin. Oncol. (R. Coll. Radiol.) 1996;8:25–34. doi: 10.1016/s0936-6555(05)80035-4. [DOI] [PubMed] [Google Scholar]
- 33.Turesson I, Nyman J, Holmberg E, Oden A. Prognostic factors for acute and late skin reactions in radiotherapy patients. Int. J. Radiat. Oncol. Biol. Phys. 1996;36:1065–1075. doi: 10.1016/s0360-3016(96)00426-9. [DOI] [PubMed] [Google Scholar]
- 34.Davidson SE, et al. Short report: a morbidity scoring system for Clinical Oncology practice: questionnaires produced from the LENT SOMA scoring system. Clin. Oncol. (R. Coll. Radiol.) 2002;14:68–69. doi: 10.1053/clon.2001.0029. [DOI] [PubMed] [Google Scholar]
- 35.LENT SOMA tables. Radiother. Oncol. 1995;35:17–60. No authors listed. [PubMed] [Google Scholar]
- 36.LENT SOMA scales for all anatomic sites. Int. J. Radiat. Oncol. Biol. Phys. 1995;31:1049–1091. doi: 10.1016/0360-3016(95)90159-0. No authors listed. [DOI] [PubMed] [Google Scholar]
- 37.Chassagne D, et al. A glossary for reporting complications of treatment in gynecological cancers. Radiother. Oncol. 1993;26:195–202. doi: 10.1016/0167-8140(93)90260-f. [DOI] [PubMed] [Google Scholar]
- 38.Litwin MS, et al. The UCLA Prostate Cancer Index: development, reliability, and validity of a health-related quality of life measure. Med. Care. 1998;36:1002–1012. doi: 10.1097/00005650-199807000-00007. [DOI] [PubMed] [Google Scholar]
- 39.Price SJ, et al. Early radiotherapy dose response and lack of hypersensitivity effect in normal brain tissue: a sequential dynamic susceptibility imaging study of cerebral perfusion. Clin. Oncol. (R. Coll. Radiol.) 2007;19:577–587. doi: 10.1016/j.clon.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 40.Giotopoulos G, et al. The late radiotherapy normal tissue injury phenotypes of telangiectasia, fibrosis and atrophy in breast cancer patients have distinct genotype-dependent causes. Br. J. Cancer. 2007;96:1001–1007. doi: 10.1038/sj.bjc.6603637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johansson S, Svensson H, Denekamp J. Timescale of evolution of late radiation injury after postoperative radiotherapy of breast cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2000;48:745–750. doi: 10.1016/s0360-3016(00)00674-x. [DOI] [PubMed] [Google Scholar]
- 42.Bentzen SM, Vaeth M, Pedersen DE, Overgaard J. Why actuarial estimates should be used in reporting late normal-tissue effects of cancer treatment ... now! Int. J. Radiat. Oncol. Biol. Phys. 1995;32:1531–1534. doi: 10.1016/0360-3016(95)00262-W. [DOI] [PubMed] [Google Scholar]
- 43.Caplan RJ, Pajak TF, Cox JD. Analysis of the probability and risk of cause-specific failure. Int. J. Radiat. Oncol. Biol. Phys. 1994;29:1183–1186. doi: 10.1016/0360-3016(94)90416-2. [DOI] [PubMed] [Google Scholar]
- 44.Haie-Meder C, et al. Analysis of complications in a prospective randomized trial comparing two brachytherapy low dose rates in cervical carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 1994;29:953–960. doi: 10.1016/0360-3016(94)90388-3. [DOI] [PubMed] [Google Scholar]
- 45.Peters LJ, Withers HR, Brown BW. Complicating issues in complication reporting. Int. J. Radiat. Oncol. Biol. Phys. 1995;31:1349–1351. doi: 10.1016/0360-3016(95)00041-V. [DOI] [PubMed] [Google Scholar]
- 46.Taylor AM, et al. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature. 1975;258:427–429. doi: 10.1038/258427a0. [DOI] [PubMed] [Google Scholar]
- 47.Woods WG, Byrne TD, Kim TH. Sensitivity of cultured cells to gamma radiation in a patient exhibiting marked in vivo radiation sensitivity. Cancer. 1988;62:2341–2345. doi: 10.1002/1097-0142(19881201)62:11<2341::aid-cncr2820621114>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 48.Little JB, Nove J. Sensitivity of human diploid fibroblast cell strains from various genetic disorders to acute and protracted radiation exposure. Radiat. Res. 1990;123:87–92. [PubMed] [Google Scholar]
- 49.Loeffler JS, Harris JR, Dahlberg WK, Little JB. In vitro radiosensitivity of human diploid fibroblasts derived from women with unusually sensitive clinical responses to definitive radiation therapy for breast cancer. Radiat. Res. 1990;121:227–231. [PubMed] [Google Scholar]
- 50.Plowman PN, Bridges BA, Arlett CF, Hinney A, Kingston JE. An instance of clinical radiation morbidity and cellular radiosensitivity, not associated with ataxia-telangiectasia. Br. J. Radiol. 1990;63:624–628. doi: 10.1259/0007-1285-63-752-624. [DOI] [PubMed] [Google Scholar]
- 51.Alter BP. Radiosensitivity in Fanconi’s anemia patients. Radiother. Oncol. 2002;62:345–347. doi: 10.1016/s0167-8140(01)00474-1. [DOI] [PubMed] [Google Scholar]
- 52.Rogers PB, Plowman PN, Harris SJ, Arlett CF. Four radiation hypersensitivity cases and their implications for clinical radiotherapy. Radiother. Oncol. 2000;57:143–154. doi: 10.1016/s0167-8140(00)00249-8. [DOI] [PubMed] [Google Scholar]
- 53.Leong T, Borg M, McKay M. Clinical and cellular radiosensitivity in inherited human syndromes. Clin. Oncol. (R. Coll. Radiol.) 2004;16:206–209. doi: 10.1016/j.clon.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 54.Burnet NG, et al. Prediction of normal-tissue tolerance to radiotherapy from in-vitro cellular radiation sensitivity. Lancet. 1992;339:1570–1571. doi: 10.1016/0140-6736(92)91833-t. [DOI] [PubMed] [Google Scholar]
- 55.Geara FB, Peters LJ, Ang KK, Wike JL, Brock WA. Prospective comparison of in vitro normal cell radiosensitivity and normal tissue reactions in radiotherapy patients. Int. J. Radiat. Oncol. Biol. Phys. 1993;27:1173–1179. doi: 10.1016/0360-3016(93)90540-c. [DOI] [PubMed] [Google Scholar]
- 56.Burnet NG, et al. The relationship between cellular radiation sensitivity and tissue response may provide the basis for individualising radiotherapy schedules. Radiother. Oncol. 1994;33:228–238. doi: 10.1016/0167-8140(94)90358-1. [DOI] [PubMed] [Google Scholar]
- 57.Johansen J, Bentzen SM, Overgaard J, Overgaard M. Relationship between the in vitro radiosensitivity of skin fibroblasts and the expression of subcutaneous fibrosis, telangiectasia, and skin erythema after radiotherapy. Radiother. Oncol. 1996;40:101–109. doi: 10.1016/0167-8140(96)01777-x. [DOI] [PubMed] [Google Scholar]
- 58.West CM, et al. Lymphocyte radiosensitivity is a significant prognostic factor for morbidity in carcinoma of the cervix. Int. J. Radiat. Oncol. Biol. Phys. 2001;51:10–15. doi: 10.1016/s0360-3016(01)01575-9. [DOI] [PubMed] [Google Scholar]
- 59.Russell NS, et al. Low predictive value of intrinsic fibroblast radiosensitivity for fibrosis development following radiotherapy for breast cancer. Int. J. Radiat. Biol. 1998;73:661–670. doi: 10.1080/095530098141915. [DOI] [PubMed] [Google Scholar]
- 60.Peacock J, et al. Cellular radiosensitivity and complication risk after curative radiotherapy. Radiother. Oncol. 2000;55:173–178. doi: 10.1016/s0167-8140(00)00173-0. [DOI] [PubMed] [Google Scholar]
- 61.Dikomey E, Borgmann K, Peacock J, Jung H. Why recent studies relating normal tissue response to individual radiosensitivity might have failed and how new studies should be performed. Int. J. Radiat. Oncol. Biol. Phys. 2003;56:1194–1200. doi: 10.1016/s0360-3016(03)00188-3. [DOI] [PubMed] [Google Scholar]
- 62.Dickson J, Magee B, Stewart A, West CM. Relationship between residual radiation-induced DNA double-strand breaks in cultured fibroblasts and late radiation reactions: a comparison of training and validation cohorts of breast cancer patients. Radiother. Oncol. 2002;62:321–326. doi: 10.1016/s0167-8140(01)00432-7. [DOI] [PubMed] [Google Scholar]
- 63.Ozsahin M, et al. CD4 and CD8 T-lymphocyte apoptosis can predict radiation-induced late toxicity: a prospective study in 399 patients. Clin. Cancer Res. 2005;11:7426–7433. doi: 10.1158/1078-0432.CCR-04-2634. [DOI] [PubMed] [Google Scholar]
- 64.West CM, Elliott RM, Burnet NG. The genomics revolution and radiotherapy. Clin. Oncol. (R. Coll. Radiol.) 2007;19:470–480. doi: 10.1016/j.clon.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 65.Kruse JJ, Stewart FA. Gene expression arrays as a tool to unravel mechanisms of normal tissue radiation injury and prediction of response. World J. Gastroenterol. 2007;13:2669–2674. doi: 10.3748/wjg.v13.i19.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sonis S, et al. Gene expression changes in peripheral blood cells provide insight into the biological mechanisms associated with regimen-related toxicities in patients being treated for head and neck cancers. Oral Oncol. 2007;43:289–300. doi: 10.1016/j.oraloncology.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 67.Rieger KE, et al. Toxicity from radiation therapy associated with abnormal transcriptional responses to DNA damage. Proc. Natl Acad. Sci. USA. 2004;101:6635–6640. doi: 10.1073/pnas.0307761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Badie C, et al. Aberrant CDKN1A transcriptional response associates with abnormal sensitivity to radiation treatment. Br. J. Cancer. 2008;98:1845–1851. doi: 10.1038/sj.bjc.6604381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quarmby S, et al. Differential expression of cytokine genes in fibroblasts derived from skin biopsies of patients who developed minimal or severe normal tissue damage after radiotherapy. Radiat. Res. 2002;157:243–248. doi: 10.1667/0033-7587(2002)157[0243:deocgi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 70.Svensson JP, et al. Analysis of gene expression using gene sets discriminates cancer patients with and without late radiation toxicity. PLoS Med. 2006;3:e422. doi: 10.1371/journal.pmed.0030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodningen OK, Borresen-Dale AL, Alsner J, Hastie T, Overgaard J. Radiation-induced gene expression in human subcutaneous fibroblasts is predictive of radiation-induced fibrosis. Radiother. Oncol. 2008;86:314–320. doi: 10.1016/j.radonc.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 72.Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nature Rev. Genet. 2007;8:639–646. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- 73.Stranger BE, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hurles ME, Dermitzakis ET, Tyler-Smith C. The functional impact of structural variation in humans. Trends Genet. 2008;24:238–245. doi: 10.1016/j.tig.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mulero-Navarro S, Esteller M. Epigenetic biomarkers for human cancer: the time is now. Crit. Rev. Oncol. Hematol. 2008;68:1–11. doi: 10.1016/j.critrevonc.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 76.Quarmby S, et al. Association of transforming growth factor beta-1 single nucleotide polymorphisms with radiation-induced damage to normal tissues in breast cancer patients. Int. J. Radiat. Biol. 2003;79:137–143. [PubMed] [Google Scholar]
- 77.Andreassen CN, Alsner J, Overgaard M, Overgaard J. Prediction of normal tissue radiosensitivity from polymorphisms in candidate genes. Radiother. Oncol. 2003;69:127–135. doi: 10.1016/j.radonc.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 78.Andreassen CN, et al. TGFB1 polymorphisms are associated with risk of late normal tissue complications in the breast after radiotherapy for early breast cancer. Radiother. Oncol. 2005;75:18–21. doi: 10.1016/j.radonc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 79.Andreassen CN, Alsner J, Overgaard M, Sorensen FB, Overgaard J. Risk of radiation-induced subcutaneous fibrosis in relation to single nucleotide polymorphisms in TGFB1, SOD2, XRCC1, XRCC3, APEX and ATM — a study based on DNA from formalin fixed paraffin embedded tissue samples. Int. J. Radiat. Biol. 2006;82:577–586. doi: 10.1080/09553000600876637. [DOI] [PubMed] [Google Scholar]
- 80.Baumann M, Holscher T, Begg AC. Towards genetic prediction of radiation responses: ESTRO’s GENEPI project. Radiother. Oncol. 2003;69:121–125. doi: 10.1016/j.radonc.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 81.Burnet NG, Elliott RM, Dunning A, West CM. Radiosensitivity, radiogenomics and RAPPER. Clin. Oncol. (R. Coll. Radiol.) 2006;18:525–528. doi: 10.1016/j.clon.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 82.Chanock SJ, et al. Replicating genotype-phenotype associations. Nature. 2007;447:655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 83.Easton DF, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Visscher PM, Andrew T, Nyholt DR. Genome-wide association studies of quantitative traits with related individuals: little (power) lost but much to be gained. Eur. J. Hum. Genet. 2008;16:387–390. doi: 10.1038/sj.ejhg.5201990. [DOI] [PubMed] [Google Scholar]
- 85.Ma L, Runesha HB, Dvorkin D, Garbe JR, Da Y. Parallel and serial computing tools for testing single-locus and epistatic SNP effects of quantitative traits in genome-wide association studies. BMC Bioinformat. 2008;9:315. doi: 10.1186/1471-2105-9-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chambers JC, et al. Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nature Genet. 2008;40:716–718. doi: 10.1038/ng.156. [DOI] [PubMed] [Google Scholar]
- 87.Frayling TM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu YJ, et al. Genome-wide association scans identified CTNNBL1 as a novel gene for obesity. Hum. Mol. Genet. 2008;17:1803–1813. doi: 10.1093/hmg/ddn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scuteri A, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weedon MN, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nature Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sanna S, et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nature Genet. 2008;40:198–203. doi: 10.1038/ng.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lettre G, et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nature Genet. 2008;40:584–591. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sandhu MS, et al. LDL-cholesterol concentrations: a genome-wide association study. Lancet. 2008;371:483–491. doi: 10.1016/S0140-6736(08)60208-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kooner JS, et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nature Genet. 2008;40:149–151. doi: 10.1038/ng.2007.61. [DOI] [PubMed] [Google Scholar]
- 95.Kathiresan S, et al. A genome-wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Med. Genet. 2007;8(Suppl. 1):S17. doi: 10.1186/1471-2350-8-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saxena R, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 97.Freimer N, Sabatti C. The human phenome project. Nature Genet. 2003;34:15–21. doi: 10.1038/ng0503-15. [DOI] [PubMed] [Google Scholar]
- 98.Flint J, Munafo MR. The endophenotype concept in psychiatric genetics. Psychol. Med. 2007;37:163–180. doi: 10.1017/S0033291706008750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Newton-Cheh C, et al. Genome-wide association study of electrocardiographic and heart rate variability traits: the Framingham Heart Study. BMC Med. Genet. 2007;8(Suppl. 1):S7. doi: 10.1186/1471-2350-8-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mohlke KL, Boehnke M, Abecasis GR. Metabolic and cardiovascular traits: an abundance of recently identified common genetic variants. Hum. Mol. Genet. 2008;17:R102–R108. doi: 10.1093/hmg/ddn275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feijen M, Gerritsen J, Postma DS. Genetics of allergic disease. Br. Med. Bull. 2000;56:894–907. doi: 10.1258/0007142001903580. [DOI] [PubMed] [Google Scholar]
- 102.Scott D. Chromosomal radiosensitivity and low penetrance predisposition to cancer. Cytogenet. Genome Res. 2004;104:365–370. doi: 10.1159/000077517. [DOI] [PubMed] [Google Scholar]
- 103.Roberts SA, et al. Heritability of cellular radiosensitivity: a marker of low-penetrance predisposition genes in breast cancer? Am. J. Hum. Genet. 1999;65:784–794. doi: 10.1086/302544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu X, et al. Mutagen sensitivity has high heritability: evidence from a twin study. Cancer Res. 2006;66:5993–5996. doi: 10.1158/0008-5472.CAN-06-1007. [DOI] [PubMed] [Google Scholar]
- 105.Borgmann K, et al. Genetic determination of chromosomal radiosensitivities in G0- and G2-phase human lymphocytes. Radiother. Oncol. 2007;83:196–202. doi: 10.1016/j.radonc.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 106.Finnon P, et al. Evidence for significant heritability of apoptotic and cell cycle responses to ionising radiation. Hum. Genet. 2008;123:485–493. doi: 10.1007/s00439-008-0500-1. [DOI] [PubMed] [Google Scholar]
- 107.Curwen GB, et al. G2 chromosomal radiosensitivity in Danish survivors of childhood and adolescent cancer and their offspring. Br. J. Cancer. 2005;93:1038–1045. doi: 10.1038/sj.bjc.6602807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schmitz A, Bayer J, Dechamps N, Goldin L, Thomas G. Heritability of susceptibility to ionizing radiation-induced apoptosis of human lymphocyte subpopulations. Int. J. Radiat. Oncol. Biol. Phys. 2007;68:1169–1177. doi: 10.1016/j.ijrobp.2007.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. N. Engl. J. Med. 2008;358:2796–2803. doi: 10.1056/NEJMsa0708739. [DOI] [PubMed] [Google Scholar]
- 110.Janssens AC, van Duijn CM. Genome-based prediction of common diseases: advances and prospects. Hum. Mol. Genet. 2008;17:R166–R173. doi: 10.1093/hmg/ddn250. [DOI] [PubMed] [Google Scholar]
- 111.Suit H. The Gray Lecture 2001: coming technical advances in radiation oncology. Int. J. Radiat. Oncol. Biol. Phys. 2002;53:798–809. doi: 10.1016/s0360-3016(02)02851-1. [DOI] [PubMed] [Google Scholar]
- 112.Horiot JC, et al. Hyperfractionation versus conventional fractionation in oropharyngeal carcinoma: final analysis of a randomized trial of the EORTC cooperative group of radiotherapy. Radiother. Oncol. 1992;25:231–241. doi: 10.1016/0167-8140(92)90242-m. [DOI] [PubMed] [Google Scholar]
- 113.Clarke M, et al. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;366:2087–2106. doi: 10.1016/S0140-6736(05)67887-7. [DOI] [PubMed] [Google Scholar]
- 114.Nyman J. Normal Skin Reactions in Radiotherapy: Proliferation, Progression and Prognostic Factors. Univ. Gothenburg; 1995. Thesis. [Google Scholar]
- 115.West CM, Hendry JH. Intrinsic radiosensitivity as a predictor of patient response to radiotherapy. BJR Suppl. 1992;24:146–152. [PubMed] [Google Scholar]
- 116.West CM, et al. The intrinsic radiosensitivity of normal and tumour cells. Int. J. Radiat. Biol. 1998;73:409–413. doi: 10.1080/095530098142248. [DOI] [PubMed] [Google Scholar]
- 117.Hart RM, Kimler BF, Evans RG, Park CH. Radiotherapeutic management of medulloblastoma in a pediatric patient with ataxia telangiectasia. Int. J. Radiat. Oncol. Biol. Phys. 1987;13:1237–1240. doi: 10.1016/0360-3016(87)90200-8. [DOI] [PubMed] [Google Scholar]
- 118.Agren A, Brahme A, Turesson I. Optimization of uncomplicated control for head and neck tumors. Int. J. Radiat. Oncol. Biol. Phys. 1990;19:1077–1085. doi: 10.1016/0360-3016(90)90037-k. [DOI] [PubMed] [Google Scholar]
- 119.Abraham J, Earl HM, Pharoah PD, Caldas C. Pharmacogenetics of cancer chemotherapy. Biochim. Biophys. Acta. 2006;1766:168–183. doi: 10.1016/j.bbcan.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 120.Radiotherapy for cancer. Acta Oncol. 1996;35(Suppl. 6):75–83. No authors listed. [PubMed] [Google Scholar]
- 121.Hopewell JW, Trott KR. Volume effects in radiobiology as applied to radiotherapy. Radiother. Oncol. 2000;56:283–288. doi: 10.1016/s0167-8140(00)00236-x. [DOI] [PubMed] [Google Scholar]
- 122.Thomas DC, Haile RW, Duggan D. Recent developments in genomewide association scans: a workshop summary and review. Am. J. Hum. Genet. 2005;77:337–345. doi: 10.1086/432962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Eeles RA, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nature Genet. 2008;40:316–332. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 125.Tomlinson I, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nature Genet. 2007;39:984–988. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- 126.Broderick P, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nature Genet. 2007;39:1315–1317. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- 127.Lettre G, Rioux JD. Autoimmune diseases: insights from genome-wide association studies. Hum. Mol. Genet. 2008;17:R116–121. doi: 10.1093/hmg/ddn246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kato N, et al. High-density association study and nomination of susceptibility genes for hypertension in the Japanese National Project. Hum. Mol. Genet. 2008;17:617–627. doi: 10.1093/hmg/ddm335. [DOI] [PubMed] [Google Scholar]
- 129.Levy D, et al. Framingham Heart Study 100K Project: genome-wide associations for blood pressure and arterial stiffness. BMC Med. Genet. 2007;8(Suppl. 1):S3. doi: 10.1186/1471-2350-8-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Turesson I, Thames HD. Repair capacity and kinetics of human skin during fractionated radiotherapy: erythema, desquamation, and telangiectasia after 3 and 5 year’s follow-up. Radiother. Oncol. 1989;15:169–188. doi: 10.1016/0167-8140(89)90131-x. [DOI] [PubMed] [Google Scholar]
- 131.Schultheiss TE. The radiation dose-response of the human spinal cord. Int. J. Radiat. Oncol. Biol. Phys. 2008;71:1455–1459. doi: 10.1016/j.ijrobp.2007.11.075. [DOI] [PubMed] [Google Scholar]
- 132.Bentzen SM, Overgaard M. Relationship between early and late normal-tissue injury after postmastectomy radiotherapy. Radiother. Oncol. 1991;20:159–165. doi: 10.1016/0167-8140(91)90092-u. [DOI] [PubMed] [Google Scholar]
- 133.Bentzen SM, Overgaard M, Overgaard J. Clinical correlations between late normal tissue endpoints after radiotherapy: implications for predictive assays of radiosensitivity. Eur. J. Cancer. 1993;29A:1373–1376. doi: 10.1016/0959-8049(93)90004-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.