

Abstract
Gαs and extra-large Gαs (XLαs) can both transduce receptor activation into intracellular cAMP generation. It is unknown, however, whether these two GNAS-locus products display distinct properties with respect to receptor coupling. Here, we show that XLαs couples to the β2-adrenoceptor more efficiently than Gαs. In transfected human embryonic kidney 293 cells and mouse embryonic fibroblasts null for both Gαs and XLαs (2B2 cells), basal cAMP accumulation mediated by XLαs was higher than that mediated by Gαs. Inverse agonist treatment reduced Gαs-mediated basal activity, whereas its effect was markedly blunted on XLαs-mediated basal activity. Rank order of ligand efficacies regarding cAMP accumulation was the same when the receptor was coupled to XLαs or Gαs. However, ligand-induced and XLαs-mediated cAMP generation was higher than that mediated by Gαs. The relatively high efficiency of XLαs-mediated cAMP generation was conditional, disappearing with increased level of receptor expression or increased efficacy of ligand. Full-agonist responses in XLαs- and Gαs-expressing cells were comparable even at low receptor levels, whereas partial agonist responses became comparable only when the receptor expression was increased (>3 pmol/mg). Radioligand binding studies showed that the high-affinity component in agonist binding to β2-adrenoceptor was more pronounced in cells expressing XLαs than those expressing Gαs. We discuss these findings in the framework of current receptor-G protein activation models and offer an extended ternary complex model that can fully explain our observations.
Heterotrimeric G proteins, consisting of α, β, and γ subunits, constitute a large family of signaling proteins that transmit receptor signals to intracellular effectors. Upon interaction with an active receptor, G proteins undergo a conformational change that results in guanine-nucleotide exchange on the α subunit and dissociation of α and βγ subunits. Dissociated subunits interact with intracellular effectors to modulate their activity. Among others, Gs protein has specifically evolved to transmit receptor signals to the stimulation of adenylyl cyclase that leads to intracellular generation of the second messenger cAMP (for reviews, see Gilman, 1987; Hamm, 1998).
The α subunits of Gs are encoded by the complex GNAS locus on the chromosome 20q13 (Kozasa et al., 1988). This locus generates multiple products through the splicing of different alternative first exons onto a common downstream exon (exon 2). In addition, alternative splicing of exon 3 of Gαs gene results in long and short forms of Gαs protein (Bray et al., 1986). A recently identified product of the GNAS locus is the extra-large αs (XLαs) protein, in which the first exon of Gαs is replaced by the XL-exon that encodes, in rat, 347 instead of 47 amino acids in the amino terminus of Gαs (Kehlenbach et al., 1994). In contrast to Gαs, which is expressed ubiquitously, XLαs is expressed particularly in neuroendocrine tissues (Pasolli et al., 2000) and derived from the paternal allele (Hayward et al., 1998). Polymorphisms affecting the XL-exon have been shown to be associated with prolonged trauma-induced bleeding in humans (Freson et al., 2001). In addition, XLαs knockout mice have shown poor postnatal growth and survival, suggesting an important role for XLαs in postnatal development and adaptation (Plagge et al., 2004). Perinatal defects similar to those in XLαs knockout mice have also been identified in two unrelated children who carried large deletions that comprised the paternal GNAS allele (Geneviève et al., 2005).
XLαs has been shown to interact with Gβγ dimers to form a stable heterotrimeric complex and to undergo cholera toxin-induced ADP-ribosylation. It is activated by GTPγS, and upon binding of GTPγS, undergoes a conformational change similar to Gαs (Klemke et al., 2000). It has also been shown that XLαs couples agonist stimulation of different types of Gαs-coupled receptors to the activation of adenylyl cyclase in transfected cells (Bastepe et al., 2002; Linglart et al., 2006). Finally, the point mutation Q548L in XLαs (equivalent to Q227L in Gαs) results in constitutive adenylyl cyclase stimulation (Klemke et al., 2000). Thus, consistent with the fact that XLαs and Gαs share identical functional domains (except their N termini), XLαs demonstrates Gαs-like properties. Although there have been conflicting reports about its intracellular distribution, the fact that it couples membrane receptors to adenylyl cyclase strongly suggests that XLαs, like Gαs, is also expressed in the plasma membrane (Kehlenbach et al., 1994; Pasolli et al., 2000; Ugur and Jones, 2000; Linglart et al., 2006). However, little is known about the signaling properties of XLαs compared with Gαs. Despite the functional similarities between XLαs and Gαs, their coupling properties to the membrane receptors may diverge because of the difference in their N termini, which has been implicated to be involved in receptor interaction and activation (Fanelli et al., 1999). Apparently, this difference does not result in receptor selectivity, because all the Gs-coupled receptors investigated to date have also been found to be able to couple to XLαs (Bastepe et al., 2002; Linglart et al., 2006). However, the difference between the two proteins may be particularly important in terms of agonist-directed signal trafficking, where different ligands can couple the same receptor to different G proteins with diverging efficacies. Thus, a set of ligands may exhibit different order of efficacy depending on whether a particular receptor is coupled to Gαs or XLαs, which may have a potential pharmacological importance.
In the present study, we therefore investigated signaling properties of XLαs in comparison with GαsL (Gαs long form), by measuring its ability to mediate receptor- and ligand-dependent or -independent activation of adenylyl cyclase. We used human β2-adrenoceptor (βAR) as a prototypical Gs-coupled experimental model. The purpose of the present study was 2-fold: first, to compare the coupling properties of XLαs and Gαs to βAR; and second, to gain further insight into the mechanism of G protein-mediated signaling by using a system in which the same receptor is coupled to two different G proteins as a tool. The latter point is discussed in the framework of the current interpretation of the ternary complex models.
Materials and Methods
Cell Culture. HEK293 cells were grown in DMEM supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), and 10% fetal bovine serum in a humidified atmosphere of 5% CO2 at 37°C. For 2B2 GnasE2-/E2- fibroblasts, where the second exon of Gαs or XLαs was disrupted out, DMEM-F-12 medium supplemented with 5% fetal bovine serum was used, and cells were grown at 33°C.
Plasmid Constructs and Transfections. The point mutation (L519P) present in the original cDNA of XLαs was corrected by replacing the P codon with the L codon at the relevant position by standard site directed mutagenesis techniques on the pcDNA3.1(+) vector (Kehlenbach et al., 1995). cDNAs encoding rat XLαs or rat GαsL were cleaved from the original vectors and reinserted into pcDNA3.1-hygromycin plasmids. cDNA encoding human βAR was inserted into the pcDNA3.1-zeocin plasmid. HEK293 cells and 2B2 cells were transfected with calcium phosphate precipitation (Kingston et al., 1996) and DEAE-dextran methods (Gulick, 1997), respectively. Stable monoclones were selected using appropriate antibiotics. Protein expression levels of the selected clones or transiently transfected cells were determined by radioligand binding or Western blot analysis. HEK293 clones that overexpress βAR at a level of 30 pmol/mg membrane protein were a kind gift of Dr. Tommaso Costa (Istituto Superiore di Sanitá, Rome, Italy). Original cDNAs for human-β2AR, rat-GαsL and rat-XLαs were kind gifts of T. Costa, T. L. Z. Jones (NIH, NIDDK, Bethesda, MD), and W. B. Huttner (Max Plank Institute of Cell Biology and Genetics, Dresden, Germany), respectively.
Immunoblots and SDS-Polyacrylamide Gel Electrophoresis. Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA) by using standard procedures. Proteins were detected by custom-designed polyclonal antibody (produced by Pacific Immunology Corp., Ramona, CA) raised against the C-terminal decapeptide [NH2-(Cys)-Arg-Met-His-Leu-Arg-Gln-Tyr-Glu-Leu-Leu] of Gαs (and XLαs), and enhanced chemiluminescence as described by the manufacturer (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Densitometric analysis of blots was carried out by using an image analysis system (Diana version 1.6; Aida version 2.43; Raytest, Straubenhardt, Germany).
Membrane Preparations and Receptor Binding Assays. Cells were pelleted at 200 g for 5 min at room temperature, resuspended in homogenization buffer [5 mM Tris-HCl, pH 7.4, and protease inhibitor mixture (Roche Diagnostics, Mannheim, Germany)], and homogenized by passing the suspension (10–15 times) through a 26-gauge syringe tip on ice. The homogenate was centrifuged at 450g for 10 min at 4°C, and the resulting supernatant was centrifuged at 100,000g for 30 min at 4°C (Optima LE-80K ultracentrifuge; Beckman Coulter, Fullerton, CA). The pellet was resuspended in a buffer containing 50 mM Tris-HCl, pH 7.4, 0.3 mg/ml dithiothreitol, 5 mM MgCl2, and protease inhibitor mixture and then repelleted by centrifugation at 100,000g for 30 min at 4°C. The final pellet was suspended in 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, protease inhibitor mixture, and 25% sucrose at a protein concentration of approximately 2 mg/ml and stored at -80°C.
In saturation binding assays, 0.5 to 1 μg of membrane protein was incubated with [125I]iodocyanopindolol (100,000 dpm/well) in a total volume of 100 μl of buffer (100 mM KCl, 10 mM MgCl2, and 50 mM Tris-HCl, pH 7.4) for 2 h at 37°C in 96-well plates. The reaction was stopped by rapid filtration through a Whatman GFB glass fiber filter (Whatman, Maidstone, UK) by using a cell harvester (Skatron Instruments, Lier, Norway). Radioactivity on the filters was counted by using a scintillation counter (MicroBeta 1450 Trilux; PerkinElmer Wallac, Turku, Finland). Nonspecific binding was determined in the presence of 1 μM cyanopindolol. Competition binding assays were conducted similarly except that varying concentrations of indicated competitor ligands and 20,000 cpm/well [125I]iodocyanopindolol were used in the presence or absence of GTPγS (1 μM) or GPD (100 μM) + AlF (20 μM NaCl/10 mM NaF) at a final buffer volume of 200 μl. Nucleotide-induced shift in agonist binding curves was found to be more complete with GDP + AlF than with GTPγS. Therefore, we presented the results of the experiments in which GDP + AlF were used. Binding curves were analyzed by nonlinear regression of a four-parameter logistic equation or numerical solution of multisite binding equation in the presence of multiple ligands by means of an in-house Excel routine (Microsoft, Redmond, WA). Binding curves obtained in parallel experiments in the presence or absence of guanine nucleotide were analyzed by sharing receptor concentration and affinity values among binding curves. The effect of parameter sharing was tested by using F statistics.
Determination cAMP Accumulation. Cells were seeded in 96-well plates at a density of 5 to 10 × 103 cells/well 24 h before the experiment. Two hours before the assay, cells were washed once with serum-free DMEM. Assays were conducted in a total volume of 100 μl at 37°C for 5 min. After incubating the cells with the receptor ligands for 5 min at room temperature, cAMP assay was initiated by adding 1 mM isobutylmethylxanthine and terminated by adding 100 μl of 0.2 N HCl. cAMP accumulation was determined by a radioimmunoassay as described previously (Ugur and Onaran, 1997).
Immunocytochemistry and Confocal Microscopy. Cells, grown on glass coverslips, were washed three times with phosphate-buffered saline (PBS) and fixed with 2% paraformaldehyde (w/v) in PBS for 20 min. After permeabilization with 0.1% Triton X-100 in PBS (v/v) for 15 min and blocking for 15 min with 1% bovine serum albumin in PBS (w/v), cells were incubated with an antibody raised against carboxyl terminus decapeptide of Gαs (see Immunoblots and SDS-Polyacrylamide Gel Electrophoresis for the specification of the antibody) at a dilution of 1:500 for 1 h, washed with PBS, and then incubated with Cy3-conjugated anti-rabbit antibody (1:1000) (Zymax, San Luis Obispo, CA) for 1 h at room temperature. Cells were then washed three times with PBS and once with distilled water, mounted with Immu-Mount reagent (Shandon, Waltham, MA), and visualized by the use of a confocal microscope (TSC SP5; Leica, Wetzlar, Germany).
Modeling and Numerical Simulations. To explain the experimental observations on a quantitative basis we used a modified ternary complex model as schematized in Fig. 1. In the classical ternary complex models, the G protein activation has been considered implicitly as equivalent to the amount of receptor-G protein complex formed, regardless of whether the receptor activation is considered explicitly or not. In the present case however, the G protein activation is considered explicitly as a binary process. Thus, the model given in Fig. 1 is a new interpretation of the well known ternary complex model that has been widely used to explain ligand behavior in different contexts. The reason for such a modification is discussed under Discussion.
Fig. 1.

Description of the equilibrium model where the ternary complex model was extended to include G protein activation (G-G* transition) explicitly. Left, meanings of the parameters are given schematically: Three binding partners are designated as H, R, and G for ligand, receptor, and G proteins, respectively. Binding sites on the proteins are shown as black circles. Activation of G protein is symbolized as a gray area in the G protein, in which the activation reaction (*↔0) takes place. Three reaction constants (K, M, and L) for relevant binding (or isomerization) reactions, and three allosteric couplings (α, β, and γ) that links these reactions are indicated on the picture. Among the allosteric factors, γ is a second-order effect that is transmitted between ligand binding and G protein activation reactions once the RG complex is formed. Note that ligand efficacy in the model comprises of the mixed effects of α and γ. Right, equilibrium reactions and corresponding equilibrium constants are shown. Elementary reactions are indicated with thick lines.
In the present scheme, three unconditional affinity constants, K, M, and L, govern ligand-receptor binding, receptor-G protein binding, and state transition of the G protein, respectively. Three allosteric constants, α, β and γ, depict the coupling between the following processes: 1) ligand binding to the receptor and receptor binding to the G protein (α), 2) receptor binding to G protein and G protein activation (β), and 3) ligand binding to receptor-G protein complex and G protein activation (γ). See the left panel of Fig. 1 for a schematic presentation of the affinity and allosteric constants. All these constants can be defined as follows:
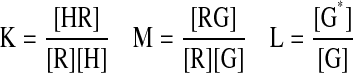 |
 |
Combining the definitions of the above-mentioned reaction constants with the conservation equations for the three components H, R, and G yields the following equations for the corresponding free species:
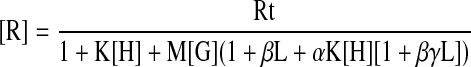 |
(1) |
 |
(2) |
 |
(3) |
Rt, Ht, and Gt in eqs. 1, 2, 3 signify the total concentrations of the corresponding components. Given the reaction constants and total concentrations of the three components, we calculated free concentrations of the three components by solving eqs. 1, 2, 3 numerically using an algorithm that has been described previously (Costa et al., 1992; Onaran et al., 1993). This algorithm has been proved to converge to a unique solution vector for the free species (Pradines et al., 2001). Once the concentrations of the free species are thus obtained, the concentrations of all the other species can be readily calculated by using the definitions of the reaction constants given above. See Results for the choice of parameter values.
Other Procedures. The number of living cells was determined by methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay as described by the manufacturer (Sigma-Aldrich, Taufkirchen, Germany). Protein concentration in the membrane preparations was determined by Bradford assay using bovine serum albumin as standard (Bradford, 1976).
All standard reagents (e.g., buffers, salts, detergents) were from Sigma-Aldrich or Thermo Fisher Scientific at appropriate purity. βAR ligands were from Tocris Bioscience (Bristol, UK). Guanine nucleotides were from Roche Diagnostics. [125I]iodocyanopindolol and [125I]NaI were purchased from Amersham Biosciences (Chalfont St. Giles, Buckinghamshire, UK). β-Adrenoceptor ligands were obtained from the following suppliers: Tocris Biosciences for clenbuterol, cimaterol, procaterol hydrochloride, dobutamine hydrochloride, pronethalol hydrochloride, (S)-(-)-propranolol hydrochloride, sotalol hydrochloride, ICI-188,551 hydrochloride, cyanopindolol hemifumarate, (S)-(-)-pindolol, and ICI-89,406; and Sigma-Aldrich for (-)-isoproterenol hydrochloride, (-)-adrenalin, (-)-alprenolol hydrochloride, timolol maleate, and terbutaline hemisulfate.
Results
Expression and Cellular Localization of Gαs and XLαs Proteins. Transfection of HEK293 or 2B2 cells with GαsL or XLαs resulted in considerable overexpression of the proteins in the membrane fractions (Fig. 2A). Note that HEK293 cells endogenously express the long (52-kDa) and the short (45-kDa) forms of Gαs but not XLαs (94 kDa), whereas the untransfected 2B2 mouse embryonic fibroblasts express neither Gαs nor XLαs because of homozygous ablation of GNAS exon 2. Expression levels of GαsL or XLαs were similar in the 2B2 clones selected for further experiments (Fig. 2B). Because the similarity of the expression levels has a critical importance for the interpretation of data presented in the following sections, we gave the details of the measurement procedure in Supplemental Fig. 1. cAMP response to agonist stimulation in these clones was restored by GαsL or XLαs expression (Fig. 2C).
Fig. 2.

Expression of GαsL or XLαs in HEK293 or 2B2 cells. A, representative Western blots of membrane preparations from transiently transfected HEK293 or permanently cloned 2B2 cells as indicated in the picture; φ, G, and X signify no transfection, transfection with GαsL or XLαs, respectively. B, densitometric analysis of 2B2 cell clones that were used throughout the present experiments. Mean values (bars) were calculated from four independent membrane preparations (for each clone) by using three serial dilutions (5, 2.5, and 1.25 μg/lane) for each preparation. Lines on the bars represent S.E.M. No significant differences were found between GαsL and XLαs signals in these clones (p > 0.05, as assessed by Student's t test). C, intracellular cAMP accumulation in untransfected cells, or in GαsL or XLαs expressing 2B2 cell clones measured in the presence of 0.1 mM isoproterenol. cAMP response were normalized to the number of living cells in the wells as assessed by parallel MTT assays. The numerical value of the MTT signal was ∼0.3 on average. Response in transfected cells is presented as average response as we observed no significant difference between GαsL or XLαs expressing clones. D, cellular distribution of GαsL or XLαs in HEK293 cells that overexpress βAR. Cells were transfected with the cDNA of GαsL or XLαs (as indicated in the picture), and confocal images were obtained after immunostaining by using an antibody against the carboxyl terminus of Gαs (the same antibody was used in the immunoblotting experiments). Each picture consists of a collage of cell images chosen intentionally to show that the distribution pattern of both GαsL and XLαs exhibits a cell-to-cell variation. Scale bars, 10 μm.
We used confocal microscopy and transiently transfected HEK293 cells to determine subcellular localization of XLαs or GαsL. Endogenous Gαs in HEK293 cells did not produce a detectable fluorescence signal in untransfected cells, allowing us to distinguish XLαs (or additional GαsL) signal in the transfected HEK293 cells by using an antibody against the common carboxyl terminus of Gαs and XLαs. Localization pattern of the proteins differed considerably among cells for both XLαs and GαsL (Fig. 2D): cell membrane, diffuse cytoplasmic, and perinuclear staining were all evident in both cases. Thus, in HEK293 cells we were unable to diagnose any obvious difference between the distribution patterns of GαsL and XLαs as opposed to what has been reported previously (Kehlenbach et al., 1994; Uǧur and Jones 2000; Linglart et al., 2006). Despite the diffuse cytoplasmic staining in some cells we found no XLαs in soluble fractions of the cell homogenates, which indicated that XLαs was mostly associated with membranes. Unlike XLαs, a small fraction of Gαs could be found in the soluble fractions (data not shown).
Despite the obvious expression of GαsL or XLαs (Fig. 2, A–C) in stably transfected 2B2 clones, we failed to obtain a good quality immunostaining for GαsL or XLαs in these cells because of a high background signal that resulted apparently from the nonspecific interactions of the fluorescent antibodies with some constituents of the 2B2 cells.
Stimulation of Adenylyl Cyclase Activity. We measured cAMP production in the presence or absence of βAR ligands in intact HEK293 cells cotransfected with βAR and XLαs or GαsL. As shown in Fig. 3A, cells transfected with XLαs showed higher basal and agonist stimulated cAMP accumulation than those transfected with Gαs, although the expression levels of each G protein α-subunit and the βAR were comparable. In GαsL-transfected cells, inverse agonists timolol and ICI-118,551 reduced the basal cAMP levels. In contrast, the basal cAMP level in XLαs-transfected cells was not responsive to these inverse agonists (Fig. 3A). Thus, we asked whether the elevated basal XLαs activity was independent of receptor coupling. Increasing the level of βAR expression resulted in an increase of basal cAMP accumulation in both GαsL- and XLαs-transfected cells, demonstrating that the high basal activity of XLαs is associated, at least partly, with the receptor. At the high receptor expression levels, the basal cAMP level in XLαs-transfected cells remained insensitive to inverse agonists (Fig. 3A, right).
Fig. 3.

Cyclic AMP responses in HEK293 (A) or 2B2 (B) cells in the absence or presence of saturating concentrations of indicated ligands. The level of βAR expression in each cell type is indicated in the picture. GαsL and XLαs expression levels were comparable in these cells as explained in Fig. 2B. cAMP response were normalized to the number of living cells in the wells as assessed by parallel MTT assays. The numerical value of the MTT signal was ∼0.3 on average. Results are mean + S.E.M. values of 10 (A) or eight (B) independent quadruplicate experiments.
One interpretation for this result might be that XLαs was partially or completely unable to distinguish between the inverse agonist-bound and the empty receptor conformations. However, a more comprehensive scenario can also explain this observation, along with the observations presented below, by a different mechanism (see Discussion).
To observe pure XLαs response to βAR and to avoid possible interference of endogenous Gαs, we used the Gαs-deficient 2B2 cells. As expected, cAMP production of these cells was insensitive to β-adrenergic stimulation. Transfection of these cells with XLαs or GαsL restored the cAMP response, because a small amount of βAR (∼100 fmol/mg) is expressed endogenously (Fig. 2C). We nevertheless stably overexpressed βAR along with XLαs or Gαs for examining basal receptor activity, which was otherwise undetectable. Selected clonal cells expressed comparable levels of GαsL and XLαs (Fig. 2B). At ∼1 pmol/mg βAR, the basal cAMP level in XLαs-transfected cells was unaltered in response to inverse agonists (Fig. 3B, left). When the receptor expression level was increased to ∼5 pmol/mg, an inverse agonist effect of ICI-118,551 emerged in XLαs-transfected cells as well (Fig. 3B, right), but the magnitude of this effect was small compared with that observed in GαsL-transfected cells; ICI-118,551-induced inhibition of basal activity was 65% in GαsL but was 30% in XLαs-transfected cells. These results confirmed the above-mentioned observation that XLαs-transfected cells were more resistant to inverse agonist effects than Gαs-transfected cells.
The observation that inverse agonist-induced responses of GαsL and XLαs were different from one another suggested that variation in receptor state that can be induced by different ligands, is also perceived differently by these two proteins. Thus, we systematically screened a set of βAR ligands with a broad spectrum of efficacy for their ability to stimulate cAMP accumulation in GαsL- or XLαs-transfected cells. Figure 4, A and B, shows cAMP responses of GαsL- or XLαs-transfected 2B2 cells that overexpress βAR at a level of ∼1or ∼5 pmol/mg. Overall, relative intrinsic activity of each ligand observed in XLαs-transfected cells was comparable with that in GαsL-expressing cells. Although the basal cAMP accumulation was relatively high in XLαs-transfected cells (consistent with results presented above), the difference between XLαs and GαsL was less evident upon agonist stimulation. At ∼1 pmol/mg receptor expression, maximal cAMP accumulation was similar in XLαs- and GαsL-transfected cells for strong, but not for partial, agonists (Fig. 4A). When the receptor expression was increased to ∼5 pmol/mg, the similarity between XLαs- and Gαs-transfected cells in terms of maximal cAMP accumulation was also observed for the partial agonist dobutamine (Fig. 4B). We were unable to further increase the receptor expression in 2B2 cells. Thus, to address the question as to whether higher receptor expression levels would result in similar levels of XLαs- and Gαs-mediated maximal cAMP accumulation even in response to agonists with lower efficacy than dobutamine, we used HEK293 cell clones that express βAR at a level of 30 pmol/mg. At this receptor expression level, the difference between XLαs- and GαsL-mediated cAMP accumulation responses to agonists was no longer detectable: even those agents with very low intrinsic activity, starting from propranolol, which displayed a weak agonistic effect at this level of receptor expression, were able to stimulate XLαs and GαsL equally well (Fig. 4C). This phenomenon is best seen when the responses are shown on normalized scales (Fig. 4D). In Fig. 4D, three categories of ligands are identifiable: 1) ICI-118,551, timolol, and sotalol, for which XLαs is more efficient than GαsL in mediating cAMP production; 2) from propranolol to pindolol, for which XLαs and GαsL are equally efficient; and 3) from partial agonist dobutamine to full agonist clenbuterol, for which overexpression of XLαs or Gαs did not increase the response any more than that obtained in HEK293 cells that overexpress βAR only. Together, these results suggested that XLαs is intrinsically more efficient than GαsL in mediating receptor-induced cAMP accumulation, but this phenomenon is observed when the efficacy of ligand and/or the receptor expression level is relatively low.
Fig. 4.

Cyclic AMP responses in the absence or presence of saturating concentrations of indicated ligands in the cells that express comparable amount of GαsL or XLαs but varying level of βAR: 2B2 cells expressing 1 pmol/mg βAR (A), 2B2 cells expressing 5 pmol/mg βAR (B), and HEK293 cells expressing 30 pmol/mg βAR (C). Ligand concentrations were 10-6 or 10-4 M depending on ligand's affinity to βAR. Results are mean + S.E.M. values of three to four independent quadruplicate experiment. cAMP response were normalized to the number of living cells in the wells as assessed by parallel MTT assays. Significant differences in A to C are shown with the asterisk (*), as assessed by Student's test. Two cases were considered as noise: marginal difference (p = 0.06) in sotalol in C and significant difference in clenbuterol in A (p < 0.05). D, increase in cAMP response upon transfection of HEK293 cells (that express 30 pmol/mg βAR) with GαsL or XLαs was given as -fold increase in cAMP response relative to vector transfected cells that express the same amount of βAR in the absence or presence of indicated ligands. The plot is constructed by dividing the data given in C by the response observed in vector transfected cells. Three groups of ligands are indicated in the picture (see text). E, cAMP responses in 2B2 cells that express GαsL were plotted against cAMP responses in 2B2 cells that express XLαs in the absence or presence of following ligands that include inverse agonists and very weak partial agonists: 1, ICI-118,551; 2, timolol; 3, sotalol; 4, propranolol; 5, ICI-89,406; 6, no ligand; 7, pronethalol; 8, cyanopindolol; 9, pindolol; and 10, alprenolol (data are from B). Data are fitted with two straight lines having different slopes. Slopes of the lines differ by a factor of 3.
A more detailed analysis of the response pattern of weak and inverse agonists is given in Fig. 4E for intermediate level of receptor expression, in which the discrepancy between GαsL and XLαs was best seen. In Fig. 4E, maximal cAMP accumulations in the presence of indicated ligands in GαsL-transfected 2B2 cells are plotted against those that were obtained in XLαs-transfected cells, in which it is evident that XLαs mediates (partial) agonist responses better than GαsL but is relatively insensitive to inverse agonist-induced inhibition of basal activity as assessed by the slopes of the lines in Fig. 4E.
Ligand Binding. The above-mentioned observation that XLαs mediates βAR signaling more efficiently than GαsL (at least conditionally) suggests that XLαs couples better to βAR and/or stimulates adenylyl cyclase more efficiently than does GαsL. Although the findings presented above (Fig. 4) are consistent with the former possibility, more direct evidence for this possibility could be obtained by using a ligand binding strategy; the efficiency of receptor-G protein coupling should affect agonist binding pattern, whereas that of G protein-effector coupling is not expected to have a consequence on ligand binding. Therefore, we analyzed agonist binding affinity of βAR in 2B2 cells expressing XLαs or GαsL in the presence or absence of guanine nucleotides. This setting can be considered as an experimental paradigm in the framework of ternary complex-like models to reveal the efficiency of receptor-G protein coupling. As shown in Fig. 5 and Table 1, in the presence of GDP + AlF, isoproterenol bound to βAR with low affinity both in XLαs- and GαsL-expressing cell membranes. In the absence of the guanine nucleotide, the binding isotherm fit to a double-site binding model in both cases. In fact, the low-affinity values estimated from the double-site fit in the absence of the guanine nucleotide were consistent with those estimated in the presence of the guanine nucleotide. Increasing the receptor expression reduced the proportion of high-affinity binding sites and nucleotide-induced shift without affecting the binding affinities (Table 1), which is consistent with the predictions of the ternary complex model. The proportion of high-affinity binding sites was higher in XLαs-expressing cells than in GαsL-expressing cells, and the nucleotide-induced shifts in isoproterenol binding were more pronounced in membrane preparations from XLαs-expressing cells. Finally, binding affinity for high-affinity binding sites was higher in XLαs-expressing than in GαsL-expressing cells. We obtained similar results in HEK293 cells that overexpress βAR and XLαs or GαsL (Supplemental Fig. 2). Combined, these results show that βAR-G protein coupling is more efficient in the case of XLαs than GαsL.
Fig. 5.

Competition binding curves ([125I]-iodocyanopindolol versus isoproterenol) obtained in the membranes from 2B2 cell clones that express comparable amount of GαsL or XLαs and indicated amounts of βAR. Binding curves were obtained in the absence or presence of GDP (10 μM) +AlCl3 (20 μM) + NaF (10 mM) as indicated in the picture. Binding of hot ligand is given as a fraction of total receptor (Rt) in each case. Solid curves are nonlinear regressions of numerically solved competition binding equations for two binding sites. Areas between binding curves obtained in the absence or presence of nucleotide (ΔAUC) are given in the pictures as a measure of nucleotide-induced shift in binding. See Table 1 for estimated parameters. Data are mean ± S.E.M. values of three independent quadruplicate experiments.
TABLE 1.
Estimated parameters of isoproterenol binding eurves in 2B2 cells that express β AR receptors at two different levels, and XLαs or GαsL at similar levels Parameters are estimated by the regression of numerical solution of competition binding equations assuming a single dissociation constant of 40 pM for [125I]iodocyanopindolol. Affinities for isoproterenol were estimated simultaneously by sharing the parameters between two curves obtained in the presence or absence of GDP; AlF (100 μ M; 10 mM). This procedure did not cause a significant worsening of residual variance compared with independent estimation of the parameters (p > 0.05 as assessed by F statistics). Percentage of contributions of high- and low-affinity components are indicated as RH and RL and corresponding affinities as KH and KL, respectively (see Fig. 5 for the biding curves).
|
Addition
|
Parameter
|
Low βAR
|
High βAR
|
||
|---|---|---|---|---|---|
| GαsL | XLαs | GαsL | XLαs | ||
| log(KL) | 6.3 ± 0.06 | 6.5 ± 0.06 | 6.3 ± 0.05 | 6.4 ± 0.03 | |
| log(KH) | 8.1 ± 0.12 | 8.9 ± 0.05 | 8.2 ± 0.05 | 8.7 ± 0.05 | |
| GDP + AlF | RL (%) | 97 ± 0.2 | 91 ± 3 | 95 ± 1 | 90 ± 4 |
| RH (%) | 3 ± 0.2 | 9 ± 3 | 5 ± 1 | 10 ± 4 | |
| No nucleotide | RL (%) | 59 ± 5 | 49 ± 5 | 78 ± 3 | 59 ± 3 |
| RH (%) | 41 ± 5 | 51 ± 5 | 22 ± 3 | 41 ± 3 | |
| βAR (pmol/mg) | 0.4 ± 0.02 | 1.0 ± 0.04 | 4.9 ± 0.53 | 3.5 ± 0.22 | |
Numerical Simulations. Numerical simulations that are made by using the scheme in Fig. 1 are shown in Fig. 6. Gαs and XLαs are simulated as possessing different affinities for the receptor (M = 5 × 109 M-1 for Gαs and M = 3 × 1011 M-1 for XLαs). Ligands were simulated as follows: inverse agonist (α< 1, γ= 1), neutral ligand (α= 1, γ= 1), and agonist (α> 1, γ>1). For agonism, values of α and γ were chosen equal for simplicity and changed together to simulate partial and full agonists. The value of γ for inverse agonists was set to 1 (see below). For a particular ligand type, values of α and γ were set constant when simulating different types of G proteins. The following parameters were constant for different G protein and ligand types: β= 20, K = 107 M-1; [Gtotal] = 10-10 M; and L = [G*]/[G] = 0.01, yielding a very low spontaneous G protein activity in the absence of receptor intervention. The first row of Fig. 6A shows the predicted G protein activation depending on receptor density in the absence or saturating presence of three kinds of ligands: an inverse agonist, a partial agonist, or a full agonist. Note that the activation induced by the partial agonist differs for the two G proteins only at low receptor concentrations ([Rt] ≅10-10 M). This difference disappears at high receptor concentrations ([Rt] ≅10-9 M), and the activation of the two G proteins by the partial agonist becomes equal at a level below the maximal response of the system (compare the saturation levels obtained in the case of partial and full agonist in the first row of Fig. 6A). This prediction is consistent with the experimental observations presented in Fig. 4, A–C. The second row of Fig. 6A shows the formation of RG complex, which can be considered as what the traditional interpretation of the ternary complex would predict for activation in corresponding situations. In the latter case, as opposed to the former case, the system is predicted to be maximally active when the difference between the two G proteins disappears (compare the saturation levels obtained in the case of partial and full agonist in the second row of Fig. 6A), which was not the case experimentally. The assumption that inverse agonists, as opposed to agonists, do not affect G protein activation directly (γ = 1) leads to the discontinuity observed in the experiments (compare Figs. 6B and 4E). Interestingly, the present model also predicts that the inverse agonist effect should have a maximum (more pronounced for Gαs than XLαs) depending on receptor expression, and this was observed experimentally (Fig. 6, C and D). Although not demonstrated numerically in the present report, the entire scenario is compatible with the binding patterns observed in Fig. 5.
Fig. 6.

Numerical simulations by using the model given in Fig. 1. A, G protein activation (i.e., G* formation) in the presence or absence of saturating concentrations of indicated type of ligands is given in the first row as a fraction of total G (i.e., [G* + RG* + HRG*]/[total G]), depending on receptor concentration. Fractional formation of RG complex in corresponding situations is given in the second row as indicated in the picture (see text for parameter values). B, G protein activity in the presence (or absence) of inverse agonists and weak partial agonists were simulated for XLαs or Gαs according to the model described in Fig. 1 at a constant receptor concentration (Rtotal = 2 × 10-10 M). Activity (i.e., total G*/total G) calculated for Gαs was plotted against the one that was calculated for XLαs in the presence of different ligands (represented by each dot in the picture). XLαs and Gαs were simulated as in A (i.e., with different M values for R). Ligands were simulated as follows: Inverse agonists (α= 0.125, 0.25, and 0.5; γ= 1), neutral antagonist (α= 1; γ= 1), and partial agonists (α=γ= 1.15, 1.32, 1.52, 1.75, 2.01, and 2.31). Values of all other parameters are the same as in A. The position of neutral ligand is indicated with two dotted lines in the picture. Simulated results were in good agreement with the experimental data given in Fig. 4E. C, relative response of an inverse agonist (i.e., activity in the presence of ligand/basal activity) was simulated for XLαs or Gαs at varying receptor concentrations as indicated in the picture. XLαs and Gαs were simulated as in A and B. Inverse agonist was simulated with the parameters α= 0.1 and γ= 1. D, experimentally observed relative response to inverse agonist ICI-118,551 (i.e., cyclase activity in the presence of ICI-118,551/basal cyclase activity) at three different receptor concentrations in XLαs-orGαsL-expressing cells as indicated in the picture. Data in the picture were calculated from the data given Fig. 4, A to C, for ICI-118,551. The simulation in C is in good agreement with the experimental data.
Discussion
We investigated receptor coupling properties of Gαs and its variant XLαs, revealing that XLαs couples βAR signaling to adenylyl cyclase more efficiently than GαsL. The difference between XLαs and GαsL was apparently because of the difference between coupling abilities of these G proteins to the receptor.
Gαs is required for numerous agonist responses. Unlike Gαs, which is ubiquitous, XLαs seems to be more abundant in neuroendocrine tissues and brain (Kehlenbach et al., 1994; Pasolli et al., 2000), although XLαs transcript has been detected in many different tissues (Hayward et al., 1998; Plagge et al., 2004). The phenotypes observed from mice in which either XLαs or Gαs is knocked out alone suggest that these two proteins have markedly different physiological roles (Plagge et al., 2004; Chen et al., 2005; Germain-Lee et al., 2005). Although the unique cellular roles of XLαs remain to be defined, it has been clearly shown that XLαs can mediate cyclase stimulation in response to receptor activation (Klemke et al., 2000; Bastepe et al., 2002; Linglart et al., 2006). Our results now verify these findings and suggest furthermore that XLαs may be an important contributor of cAMP signaling, even in tissues where XLαs levels are markedly lower than Gαs levels. Consistent with this prediction, XLαs mRNA is markedly lower than Gαs mRNA in growth plate chondrocytes, but XLαs ablation together with the ablation of one Gαs copy (paternal Gnas exon 2 disruption) results in a more severe phenotype, i.e., premature chondrocyte hypertrophy, than the ablation of one Gαs copy alone (maternal Gnas exon 2 disruption) (Bastepe et al., 2004). cAMP signaling is involved in a majority of cellular responses, and the superiority of XLαs over Gαs in terms of receptor coupling and cAMP generation may thus have important implications in physiology and diseases. Naturally occurring GNAS mutations, with the exception of those located in exon 1, are predicted to affect not only Gαs but also XLαs. The changes in XLαs activity can be involved in the pathogenesis of diseases caused by these mutations, such as various endocrine and nonendocrine tumors (activating) or Albright's hereditary osteodystrophy (inactivating).
In a series of studies, divergent signaling abilities of GαsL and GαsS (splice variants of Gαs) have been reported, in which the difference was attributed to a higher rate of dissociation of GDP from GαsL than GαsS (Seifert et al., 1998; Wenzel-Seifert et al., 2001, 2002). In these studies, receptor-G protein fusions were used to obtain 1:1 stoichiometry of receptor/G protein, which provided a good model for investigating the coupling efficiency between receptor and G protein. In the present work, however, βAR-XLαs fusion protein did not function in 2B2 cells, but it mediated agonist-induced cyclase activation in HEK293 cells (Supplemental Figs. 3 and 4). Interactions of receptor or G protein in the fusion protein with their nonfused partners in the cell membrane have been reported (Burt et al., 1998; Molinari et al., 2003). Hence, the observed discrepancy between HEK293 and 2B2 cells that express βAR-XLαs fusion protein can be interpreted as follows: the receptor in the fusion construct interacts fruitfully with endogenously expressed Gαs proteins, which is present in HEK293 but not in 2B2 cells. Hence, no response is observed in 2B2 cells as no intra- or interfusion interaction can actually occur between receptor and XLαs (Supplemental Fig. 3 for a schematic representation of the idea). Therefore, we were unable to compare XLαs and GαsL in the fusion model. Thus, receptor/G protein stoichiometry was variable (but controlled) in our experiments; this variability, in contrast, eventually proved to be an advantage for the present case (see below).
Strikingly, the observed difference between XLαs- and GαsL was conditional. It disappeared with increasing ligand efficacy, threshold of which was dependent on the expression level of the receptor (Fig. 4). At first sight, this observation implies a saturation effect in the receptor-G protein coupling. However, cyclase activities mediated by XLαs or GαsL became comparable at a level far below the full agonist-induced maximal cyclase activity observable in the cells (Fig. 4, B and C). Hence, this pattern requires further considerations in the framework of the interpretation of ternary complex models that have been used successfully to explain ligand behavior. What is not compatible with this framework is the following: in a system where a receptor couples with different efficiencies to two different G proteins, which in turn, transmit the receptor signal to a unique effector with the same efficiency, the models predict that the ligand-induced effector activations that are mediated by these two G proteins cannot be equal when the stimulated response is below the maximal response of the system. Hence, the models, in their currently interpreted forms, cannot predict the observed equalization of Gαs- and XLαs-mediated responses below the maximal level of adenylyl cyclase activation that is achievable in the presence of full βAR agonists. This “defect”, which is not an intrinsic property of the ternary complex models, stems from the assumption that receptor-bound G protein is fully active and can easily be eliminated by assuming that formation of receptor-G protein complex is not necessarily equivalent to full activation of G protein and that agonist binding to receptor can have further “activating” effects on the G protein via conformational changes when receptor and G proteins are bound to each other. Such a scenario has already been suggested for rhodopsin-Gt interaction (Fanelli and Dell'Orco, 2005). Consequences of this assumption become obvious when we introduce G protein activation into the ternary complex model as a simple two-state process (G-active or G-inactive; Fig. 1). Inclusion of a G protein activation step in the ternary interaction scheme inevitably divides the ligand efficacy into two parts: ability of ligand to modify R-G binding (governed by the allosteric constant α in Fig. 1) and to modify G protein activation (i.e., G-G* transition) in the RG complex (governed by the allosteric constant γ in Fig. 1). On the basis of these fundamentals, the observed behavior of XLαs and Gαs in mediating βAR signal can be explained almost entirely by making the following additional assumptions (Fig. 6); XLαs and GαsL differ only in their unconditional tendency to bind receptor (MXLαs > MGαsL); efficacy of ligands on βAR does not depend on the identity of G protein to which the receptor is coupled (at least in the case of XLαs and GαsL); spontaneous tendency of RG complex to get activated is relatively low (L << 1 and 1 <β); efficacy of neutral or agonist ligands are evenly distributed over α and γ (for the sake of simplicity); and finally, inverse agonists have no direct effect on G protein activation (i.e., γ= 1) but reduces RG binding (i.e., α< 1). The last assumption is rather speculative but required to explain the inverse agonist effect on βAR-XLαs coupling compared with GαsL. In this framework, the basic mechanism and the scenario that explains the advantage of XLαs over GαsL depending on ligand efficacy and receptor expression can be stated as follows. The origin of difference between the two G proteins is their diverging affinities for the receptor, which is operative (and observable) only when RG interaction is not saturated. The saturation depends on the combined effect of receptor expression and ligand efficacy. Once RG saturation occurs, the difference between XLαs and GαsL disappears, because ligands are assumed to have γ values that do not depend on the kind of G protein. However, even at this saturation point weak agonists do not necessarily induce maximal response of the system (i.e., G-G* conversion in RG complex may not be complete upon agonist binding depending on the γ value of the agonist) (Fig. 6). This scenario also supports a long debated idea that inactive receptor and G protein tend to form an RG complex that does not necessarily lead to G protein activation, which actually occurs upon activation of receptor in the complex. Accordingly, agonist-induced (or spontaneous) receptor activation directly transmits an activating conformational signal to a precoupled G protein without necessarily affecting the stability of the RG complex. Several lines of indirect evidence support this statement (e.g., Fanelli and Dell'Orco, 2005).
In summary, the present study showed that βAR can couple to XLαs better than Gαs and that the rank order of ligand efficacies does not change when it is coupled to XLαs. Thus, despite the differences observed between Gαs and XLαs signaling, no ligand-dependent divergence is predicted to occur between XLαs and Gαs in transmitting βAR signal to adenylyl cyclase. Nevertheless, βAR signaling becomes relatively resistant to inverse agonists when the receptor is coupled to XLαs, which may have pharmacological implications depending on the distribution of βAR-XLαs coupling in the body. This may justify further studies on βAR-XLαs coupling (or receptor-XLαs coupling in general) in physiological integrity. Finally, the experimental system where the two kinds of G proteins are coupled to the same receptor with different efficiencies enabled us to reevaluate the interpretation of the ternary complex model, especially when it is used to explain or understand G protein activation.
Supplementary Material
This work was supported in part by the Turkish Scientific and Technical Research Council [Grants 104s472, 107s086]; the Ankara University Research Fund [Grant BAP 2002 0809 088]; the Ankara University Biotechnology Institute [Grant 103]; and the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant R01-DK073911].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.108.149989.
ABBREVIATIONS: XLαs, extra-large Gαs; GTPγS, guanosine 5′-O-(3-thio)triphosphate; GαsL,Gαs long form; βAR, β2-adrenoceptor; HEK, human embryonic kidney; DMEM, Dulbecco's modified Eagle's medium; PBS, phosphate-buffered saline; MTT, methylthiazolyldiphenyl-tetrazolium bromide; ICI-188,551; (±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol; ICI-89,406, N-(2-((3-(2-cyanophenoxy)-2-hydroxypropyl)amino)ethyl)-N′-phenylurea.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
References
- Bastepe M, Gunes Y, Perez-Villamil B, Hunzelman J, Weinstein LS, and Jüppner H (2002) Receptor mediated adenylyl cyclase activation through XLαs, the extra large variant of the stimulatory G protein α-subunit. Mol Endocrinol 16 1912-1919. [DOI] [PubMed] [Google Scholar]
- Bastepe M, Weinstein LS, Ogata N, Kawaguchi H, Jüppner H, Kronenberg HM, and Chung UI (2004) Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc Natl Acad Sci U S A 101 14794-14799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72 248-254. [DOI] [PubMed] [Google Scholar]
- Bray P, Carter A, Simons C, Guo V, Puckett C, Kamholz J, Spiegel A, and Nirenberg M (1986) Human clones for four species of Gsα signal transduction protein. Proc Natl Acad Sci U S A 83 8893-9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt AR, Sautel M, Wilson MA, Rees S, Wise A, and Milligan G (1998) Agonist occupation of an α2a-adrenoreceptor-Gi1α fusion protein results in activation of both receptor-linked and endogenous Gi proteins: comparisons of their contributions to GTPase activity and signal transduction and analysis of receptor-G protein activation stoichiometry. J Biol Chem 273 10367-10375. [DOI] [PubMed] [Google Scholar]
- Chen M, Gavrilova O, Liu J, Xie T, Deng C, Nguyen AT, Nackers LM, Lorenzo J, Shen L, and Weinstein LS (2005) Alternative Gnas gene products have opposite effects on glucose and lipid metabolism. Proc Natl Acad Sci U S A 102 7386-7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa T, Ogino Y, Munson PJ, Onaran HO, and Rodbard D (1992) Drug efficacy at guanine nucleotide-binding regulatory protein-linked receptors: thermodynamic interpretation of negative antagonism and of receptor activity in the absence of ligand. Mol Pharmacol 41 549-560. [PubMed] [Google Scholar]
- Fanelli F and Dell'Orco D (2005) Rhodopsin activation follows precoupling with transducin: inferences from computational analysis. Biochemistry 44 14695-14700. [DOI] [PubMed] [Google Scholar]
- Fanelli F, Menziani C, Scheer A, Cotecchia S, and De Benedetti PG (1999) Theoretical study of the electrostatically driven step of receptor-G protein recognition. Proteins 37 145-156. [PubMed] [Google Scholar]
- Freson K, Hoylaerts MF, Jaeken J, Eyssen M, Arnout J, Vermylen J, and Van Geet C (2001) Genetic variation of the extra-large stimulatory G protein alpha-subunit leads to Gs hyperfunction in platelets and is a risk factor for bleeding. Thromb Haemost 86 733-738. [PubMed] [Google Scholar]
- Germain-Lee EL, Schwindinger W, Crane JL, Zewdu R, Zweifel LS, Wand G, Huso DL, Saji M, Ringel MD, and Levine MA (2005) A mouse model of Albright Hereditary osteodystrophy generated by targeted disruption of exon 1 of the Gnas gene. Endocrinology 146 4697-4709. [DOI] [PubMed] [Google Scholar]
- Geneviève D, Sanlaville D, Faivre L, Kottler ML, Jambou M, Gosset P, Boustani-Samara D, Pinto G, Ozilou C, Abeguilé G, et al. (2005) Paternal deletion of the GNAS imprinted locus (including Gnasxl) in two girls presenting with severe pre- and post-natal growth retardation and intractable feeding difficulties. Eur J Hum Genet 13 1033-1039. [DOI] [PubMed] [Google Scholar]
- Gilman AG (1987) G proteins: transducers of receptor-generated signals. Annu Rev Biochem 56 615-649. [DOI] [PubMed] [Google Scholar]
- Gulick T (1997) Transfection using DEAE-dextran, in Current Protocols in Molecular Biology, pp 9.2.1-9.2.10. John Wiley & Sons, Inc., New York. [DOI] [PubMed]
- Hamm HE (1998) The many faces of G protein signaling. J Biol Chem 273 669-672. [DOI] [PubMed] [Google Scholar]
- Hayward BE, Kamiya M, Strain L, Moran V, Campbell R, Hayashizaki Y, and Bonthron DT (1998) The human GNAS 1 gene is imprinted and encodes distinct paternally and biallelically expressed G proteins. Proc Natl Acad Sci U S A 95 10038-10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehlenbach RH, Matthey J, and Huttner WB (1994) XLαs is a new type of G protein. Nature 372 804-809. [DOI] [PubMed] [Google Scholar]
- Kehlenbach RH, Matthey J, and Huttner WB (1995) XLαs is a new type of G protein. Nature 375 253. [DOI] [PubMed] [Google Scholar]
- Kingston RE, Chen CA, Okayama H, and Rose JK (1996) Transfection of DNA into eukaryotic cells, in Current Protocols in Molecular Biology, pp. 9.1.1-9.1.11, John Wiley & Sons, Inc., New York.
- Klemke M, Pasolli HA, Kehlenbach RH, Offermanns S, Schultz G, and Huttner WB (2000) Characterization of the extra-large G protein α-subunit XLαs. II. Signal transduction properties. J Biol Chem 275 33633-33640. [DOI] [PubMed] [Google Scholar]
- Kozasa T, Itoh H, Tsukamoto T, and Kaziro Y (1988) Isolation and characterization of the human Gsα gene. Proc Natl Acad Sci U S A 85 2081-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linglart A, Mahon MJ, Kerachian MA, Berlach DM, Hendy GN, Jüppner H, and Bastepe M (2006) Coding GNAS mutations leading to hormone resistance impair in vitro agonist- and cholera toxin-induced adenosine cyclic 3′,5′-monophosphate formation mediated by human XLαs. Endocrinology 147 2253-2262. [DOI] [PubMed] [Google Scholar]
- Molinari P, Ambrosio C, Riitano D, Sbraccia M, Grò MC, and Costa T (2003) Promiscuous coupling at receptor-Gα fusion proteins: the receptor of one covalent complex interacts with the α-subunit of another. J Biol Chem 278 15778-15788. [DOI] [PubMed] [Google Scholar]
- Onaran HO, Costa T, and Rodbard D (1993) βγ-Subunits of guanine nucleotide-binding proteins and regulation of spontaneous receptor activity: thermodynamic model for the interaction between receptors and guanine nucleotide-binding protein subunits. Mol Pharmacol 43 245-256. [PubMed] [Google Scholar]
- Pasolli HA, Klemke M, Kehlenbach RH, Wang Y, and Huttner WB (2000) Characterization of the extra-large G protein α-subunit XLαs. I. Tissue distribution and subcellular localization. J Biol Chem 275 33622-33632. [DOI] [PubMed] [Google Scholar]
- Plagge A, Gordon E, Dean W, Boiani R, Cinti S, Peters J, and Kelsey G (2004) The imprinted signaling protein XLαs is required for postnatal adaptation to feeding. Nat Genet 36 818-826. [DOI] [PubMed] [Google Scholar]
- Pradines JR, Hasty J, and Pakdaman K (2001) Complex ligand-protein systems: a globally convergent iterative method for the n×m case. J Math Biol 43 313-324. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K, Lee TW, Gether U, Sanders-Bush E, and Kobilka BK (1998) Different effects of Gsα splice variants on β2-adrenoreceptor-mediated signaling. The β2-adrenoreceptor coupled to the long splice variant of Gsα has properties of a constitutively active receptor. J Biol Chem 273 5109-5116. [PubMed] [Google Scholar]
- Ugur O and Onaran HO (1997) Allosteric equilibrium model explains steady-state coupling of beta-adrenergic receptors to adenylate cyclase in turkey erythrocyte membranes. Biochem J 323 765-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugur O and Jones TL (2000) A proline-rich region and nearby cysteine residues target XLαs to the Golgi complex region. Mol Biol Cell 11 1421-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel-Seifert K, Kelley MT, Buschauer A, and Seifert R (2001) Similar apparent constitutive activity of human histamine h2-receptor fused to long and short splice variants of Gsα. J Pharmacol Exp Ther 299 1013-1020. [PubMed] [Google Scholar]
- Wenzel-Seifert K, Liu HY, and Seifert R (2002) Similarities and differences in the coupling of human β1- and β2-ARs to Gsα splice variants. Biochem Pharmacol 64 9-20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.