

Abstract
Bovine aortic endothelial cells (ECs) respond to nitric oxide (NO) donors
by activating the redox-sensitive NF-E2-related factor 2/antioxidant response
element pathway and up-regulating heme oxygenase (HO)-1 expression. EC
exposure to steady laminar shear stress causes a sustained increase in NO, a
transient increase in reactive oxygen species (ROS), and activation of the
HO-1 gene. Because steady laminar flow increases the mitochondrial superoxide
(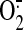 ) production, we
hypothesized that mitochondria-derived ROS play a role in shear-induced HO-1
expression. Flow (10 dynes/cm2, 6 h)-induced expression of HO-1
protein was abolished when BAECs were preincubated and sheared in the presence
of either NG-nitro-l-arginine methyl ester or
N-acetyl-l-cysteine, suggesting that either NO or ROS
up-regulates HO-1. Ebselen and diphenylene iodonium blocked HO-1 expression,
and uric acid had no effect. The mitochondrial electron transport chain
inhibitors, myxothiazol, rotenone, or antimycin A, and the
mitochondria-targeted antioxidant peptide, Szeto-Schiller (SS)-31, which
scavenges
) production, we
hypothesized that mitochondria-derived ROS play a role in shear-induced HO-1
expression. Flow (10 dynes/cm2, 6 h)-induced expression of HO-1
protein was abolished when BAECs were preincubated and sheared in the presence
of either NG-nitro-l-arginine methyl ester or
N-acetyl-l-cysteine, suggesting that either NO or ROS
up-regulates HO-1. Ebselen and diphenylene iodonium blocked HO-1 expression,
and uric acid had no effect. The mitochondrial electron transport chain
inhibitors, myxothiazol, rotenone, or antimycin A, and the
mitochondria-targeted antioxidant peptide, Szeto-Schiller (SS)-31, which
scavenges 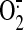 , hydrogen
peroxide (H2O2), peroxynitrite, and hydroxyl radicals,
markedly inhibited the increase in HO-1 expression. These data collectively
suggest that mitochondrial H2O2 mediates the HO-1
induction. MitoSOX and 2′,7′-dichlorofluorescin (DCF) fluorescence
showed that mitochondrial
, hydrogen
peroxide (H2O2), peroxynitrite, and hydroxyl radicals,
markedly inhibited the increase in HO-1 expression. These data collectively
suggest that mitochondrial H2O2 mediates the HO-1
induction. MitoSOX and 2′,7′-dichlorofluorescin (DCF) fluorescence
showed that mitochondrial 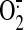 levels and intracellular peroxides, respectively, are higher in sheared ECs
compared with static controls and, in part, dependent on NO. SS-31
significantly inhibited both the shear-induced MitoSOX and DCF fluorescence
signals. Either phosphatidylinositol 3-kinase or mitogen-activated protein
kinase cascade inhibitors blocked the HO-1 induction. In conclusion, under
shear, EC mitochondria-derived H2O2 diffuses to the
cytosol, where it initiates oxidative signaling leading to HO-1 up-regulation
and maintenance of the atheroprotective EC status.
levels and intracellular peroxides, respectively, are higher in sheared ECs
compared with static controls and, in part, dependent on NO. SS-31
significantly inhibited both the shear-induced MitoSOX and DCF fluorescence
signals. Either phosphatidylinositol 3-kinase or mitogen-activated protein
kinase cascade inhibitors blocked the HO-1 induction. In conclusion, under
shear, EC mitochondria-derived H2O2 diffuses to the
cytosol, where it initiates oxidative signaling leading to HO-1 up-regulation
and maintenance of the atheroprotective EC status.
The stress proteins heme oxygenases, which consist of constitutive and inducible isozymes (HO-2 and HO-1, respectively), catalyze the rate-limiting step in the degradation of heme to the bile pigments, biliverdin and bilirubin, eventually releasing iron and carbon monoxide. Because the HO reaction products are known to provide cytoprotection from inflammatory diseases, including atherosclerosis, mechanisms of HO-1 induction have been studied extensively (Ryter et al., 2006). HO-1 is induced by chemicals that produce oxidative stress. The signaling pathways leading to the transcriptional regulation of the HO-1 gene are variable in a cell- and inducer-specific fashion, although all of them involve the participation of protein phosphorylation cascades (Ryter et al., 2006). NF-E2-related factor 2 (Nrf2) is the transcription factor that, upon activation by oxidative stress, translocates to the nucleus, binds to the antioxidant response element (ARE), and activates transcription of phase II genes, including HO-1 (Nguyen et al., 2004). Under physiological conditions, Kelch-like erythroid-derived Cap-N-Collar-Homology-associated protein 1 (Keap1), a suppressor that binds to Nrf2, retains it in the cytoplasm and promotes its proteasomal degradation. Specific cysteine residues of Keap1 are the sensors that recognize inducers of phase II genes. Under oxidative stress, lipid oxidation products, such as the electrophilic lipid 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), may bind to Keap1 and cause alkylation of its critical cysteine residues, rendering Keap1 unable to repress Nrf2. Nrf2 activation may also occur when the inducers or their metabolites stimulate Nrf2 phosphorylation through redox-sensitive protein kinases, which triggers its dissociation from Keap1, its nuclear translocation, and initiation of ARE-dependent transcription (Nguyen et al., 2004).
Cultured EC exposure to steady laminar shear stress causes a sustained
increase in nitric oxide (NO) and a transient one in superoxide
(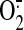 ) production, and the
shear-induced reactive oxygen species (ROS) act as second messengers in the
induction of gene expression (Kuchan and
Frangos, 1994; Chiu et al.,
1997; Yeh et al.,
1999; Han et al.,
2007). Human aortic EC exposure to steady laminar shear stress was
shown to activate HO-1 at the transcriptional level and to involve the
Keap1-Nrf2 pathway and 15d-PGJ2
(Chen et al., 2003;
Hosoya et al., 2005). Although
it is accepted that induction of ARE-regulated genes is a compensatory
response to ROS production (Jones et al.,
2007; Warabi et al.,
2007), more studies are needed to understand the intracellular
mechanisms that regulate this event. HO-1 expression data from sheared human
umbilical ECs (HUVECs) in the presence of ROS scavengers suggested that
xanthine oxidase-, NAD(P)H oxidase-, and mitochondria-derived
) production, and the
shear-induced reactive oxygen species (ROS) act as second messengers in the
induction of gene expression (Kuchan and
Frangos, 1994; Chiu et al.,
1997; Yeh et al.,
1999; Han et al.,
2007). Human aortic EC exposure to steady laminar shear stress was
shown to activate HO-1 at the transcriptional level and to involve the
Keap1-Nrf2 pathway and 15d-PGJ2
(Chen et al., 2003;
Hosoya et al., 2005). Although
it is accepted that induction of ARE-regulated genes is a compensatory
response to ROS production (Jones et al.,
2007; Warabi et al.,
2007), more studies are needed to understand the intracellular
mechanisms that regulate this event. HO-1 expression data from sheared human
umbilical ECs (HUVECs) in the presence of ROS scavengers suggested that
xanthine oxidase-, NAD(P)H oxidase-, and mitochondria-derived
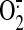 , but not NO, mediates
HO-1 expression (Warabi et al.,
2007). However, agents that release NO induce HO-1 expression in
aortic ECs, possibly via formation of S-nitrosothiols (SNO) in the
regulatory cysteine residues of Keap1
(Motterlini et al., 2002;
Buckley et al., 2003,
2008). We have shown that, in
sheared ECs of either aortic or venous origin, endogenous NO by itself and via
formation of peroxynitrite (ONOO-) in the mitochondria results in
inactivation of the electron transport chain (ETC) and increased
, but not NO, mediates
HO-1 expression (Warabi et al.,
2007). However, agents that release NO induce HO-1 expression in
aortic ECs, possibly via formation of S-nitrosothiols (SNO) in the
regulatory cysteine residues of Keap1
(Motterlini et al., 2002;
Buckley et al., 2003,
2008). We have shown that, in
sheared ECs of either aortic or venous origin, endogenous NO by itself and via
formation of peroxynitrite (ONOO-) in the mitochondria results in
inactivation of the electron transport chain (ETC) and increased
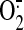 production
(Han et al., 2007;
Jones et al., 2008). Because
steady laminar flow increases
production
(Han et al., 2007;
Jones et al., 2008). Because
steady laminar flow increases
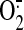 production by the ETC and
oxidative stress mediates HO-1 expression, the purpose of the present study
was to better understand the role of mitochondria-derived ROS in the latter
event and the signaling pathways that regulate it.
production by the ETC and
oxidative stress mediates HO-1 expression, the purpose of the present study
was to better understand the role of mitochondria-derived ROS in the latter
event and the signaling pathways that regulate it.
Our study was conducted on bovine aortic ECs (BAECs) exposed to 10
dynes/cm2 for 6 h. To examine whether the shear-induced increase in
HO-1 protein expression depends on mitochondrial function, and NO is involved,
ECs were preincubated and sheared in the presence of the NO synthase (NOS)
inhibitor, NG-nitro-l-arginine methyl ester
(l-NAME); ROS scavengers, such as
N-acetyl-l-cysteine (NAC), ebselen, and uric acid (UA); or
mitochondrial ETC inhibitors, such as rotenone, myxothiazol, and antimycin A.
To delineate the role of mitochondrial ROS, the effect of a
mitochondria-targeted antioxidant peptide, Szeto-Schiller (SS)-31
(d-Arg-2′,6′-dimethyltyrosine-Lys-Phe-NH2),
which exhibits 1000-fold mitochondrial accumulation and scavenges
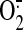 , hydrogen peroxide
(H2O2), ONOO-, and hydroxyl radicals
(OH.) (Szeto, 2006,
2008), was examined. SS-31
belongs to a family of aromatic cationic tetrapeptides that owe their ROS
scavenging activity to the phenolic group on tyrosine or dimethyltyrosine
(replacement with phenylalanine resulted in a nonscavenging analog, SS-20).
Tyrosine-containing SS peptides were shown to reduce mitochondrial ROS levels
in epithelial and neuronal cells exposed to ETC inhibitors and to protect from
mitochondrial permeability transition and cytotoxicity
(Zhao et al., 2004; Szeto,
2004,
2008). Our results confirmed a
major role of the mitochondria-derived H2O2, resulting
from dismutation of
, hydrogen peroxide
(H2O2), ONOO-, and hydroxyl radicals
(OH.) (Szeto, 2006,
2008), was examined. SS-31
belongs to a family of aromatic cationic tetrapeptides that owe their ROS
scavenging activity to the phenolic group on tyrosine or dimethyltyrosine
(replacement with phenylalanine resulted in a nonscavenging analog, SS-20).
Tyrosine-containing SS peptides were shown to reduce mitochondrial ROS levels
in epithelial and neuronal cells exposed to ETC inhibitors and to protect from
mitochondrial permeability transition and cytotoxicity
(Zhao et al., 2004; Szeto,
2004,
2008). Our results confirmed a
major role of the mitochondria-derived H2O2, resulting
from dismutation of 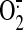 produced by the ETC, in the shear-induced up-regulation of HO-1
expression.
produced by the ETC, in the shear-induced up-regulation of HO-1
expression.
Materials and Methods
EC Culture. BAECs were purchased from Cambrex (East Rutherford, NJ) and cultured in Dulbecco's modified Eagle's medium (low glucose with l-glutamine and sodium bicarbonate) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA) in a humidified atmosphere of 95% air-5% CO2. ECs (passages 3-9) were seeded onto glass slides (75 × 38 mm; Thermo Fisher Scientific, Waltham, MA) that were sterilized, air-dried, and coated with a 0.5% gelatin-subbing solution containing 0.05% potassium chromium sulfate (Sigma-Aldrich, St. Louis, MO) and were used within 24 h upon confluence.
EC Exposure to Shear Stress and Antioxidants. ECs were serum-starved overnight in Dulbecco's modified Eagle's medium with 0.5% fetal bovine serum and antibiotics and exposed to steady (gravity-driven) laminar shear stress of 10 dynes/cm2 (in the low range of human arterial shear stresses) for either 30 min or 6 h in the same medium. In brief, three glass slides with EC monolayers were assembled side-by-side into a parallel-plate flow chamber, and the chamber was connected at both ends to a reservoir forming a flow loop, as described previously (Frangos et al., 1985; Han et al., 2007). A schematic diagram of a flow loop is provided in Supplemental Fig. 1. Flow rate through the chamber was monitored by an ultrasonic flow sensor (Transonic Systems Inc., Ithaca, NY). Recirculating medium was constantly exposed to a countercurrent flow of a sterile-filtered gas mixture (95% air-5% CO2) that was warmed and humidified by bubbling through water. The temperature of the entire system was kept at 37°C, and medium temperature was monitored real-time by an inline sensor (World Precision Instruments, Inc., Sarasota, FL). For static controls, ECs were placed in tissue culture incubators for the same time period as the sheared monolayers.
Some monolayers were preincubated with the NOS inhibitor, l-NAME (1 mM for 4 h); the nonspecific antioxidant and glutathione (GSH) precursor, NAC (10 mM for 30 min); the GSH peroxidase mimetic and ONOO- scavenger, ebselen (5 μM for 1 h); the flavoprotein inhibitor, diphenylene iodonium (DPI; 20 μM for 30 min), which inhibits the NAD(P)H oxidase, NOS, xanthine oxidase, cytochrome P450 reductase, and the mitochondrial ETC flavoproteins (including the flavoprotein subcomplex of complex I); the ONOO- scavenger, UA (100 μM for 30 min); the inhibitors of ETC complex III, myxothiazol and antimycin A (each at 10 μM for 30 min); and the inhibitor of ETC complex I, rotenone (2 μM for 1 h), and either subjected to shear or left static in medium containing the same concentration of the respective drug (all from Sigma-Aldrich). Some ECs were preincubated with either the mitochondria-targeted antioxidant peptide SS-31 (1, 10, or 100 nM for 1 h) or the nonscavenging SS-20 and subjected to shear with the same concentration of drug (a gift from Dr. Szeto, Cornell University) (Zhao et al., 2004). Last, some ECs were preincubated with the phosphatidylinositol 3-kinase (PI3K) inhibitor, wortmannin (500 nM for 1 h); the mitogen-activated protein kinase (MAPK) kinase (MEK) 1 inhibitor, PD98059 (25 μM for 1 h), which blocks phosphorylation and activation of extracellular signal-regulated kinase (ERK) 1/2; or the p38 MAPK inhibitor, SB202190 (20 μM for 1 h), and subjected to shear with the same concentration of drug (all from Sigma-Aldrich). Cell viability was determined by trypan blue exclusion at the end of treatments, and it was ≥90%. Concentrations of tested compounds were chosen based on their efficacy in inhibiting intracellular signaling pathways and/or Nrf2 nuclear translocation/HO-1 expression in either ECs or epithelial cells treated with either oxidized lipids or NO donors (Buckley et al., 2003; Iles et al., 2005; Watanabe et al., 2006). Stock solutions of NAC, UA, and SS peptides were made with distilled water and diluted in phosphate-buffered saline (PBS) before mixing with the incubation/perfusion medium. Stock solutions of all other compounds were made with dimethyl sulfoxide (DMSO) and diluted in PBS, but the final DMSO concentration was never higher than 0.03% (v/v). Static controls were treated with DMSO at the same concentration as sheared ECs.
Western Blot Analysis. ECs were scraped into ice-cold lysis buffer (0.06 M Tris·HCl, 2% SDS, and 5% glycerol, pH 6.8). Cell extracts were homogenized by sonication, boiled for 5 min, and centrifuged at 10,000 rpm for 10 min at 4°C to remove the insoluble material. Protein concentration in cell lysates was determined using the bicinchoninic acid assay (Pierce Chemical, Rockford, IL). Supernatant containing 30 μg of protein was mixed with 2× sample buffer and applied in duplicate on 4 to 20% Tris·HCl gels (Bio-Rad, Hercules, CA) for protein separation by SDS-polyacrylamide gel electrophoresis at 100-V constant voltage. Proteins were electrophoretically transferred at 100-V constant voltage at room temperature for 1 h onto nitrocellulose membranes (Bio-Rad). After incubation in blocking solution (10 mM Tris·HCl, 0.2% nonfat milk, 100 mM NaCl, and 0.1% Tween 20, pH 7.5), duplicate membranes were hybridized with primary antibodies against HO-1 (Assay Designs, Ann Arbor, MI) and β-actin (Sigma-Aldrich), respectively. Membrane-bound primary antibodies were detected using secondary antibodies conjugated with alkaline phosphatase. Immunoblots were developed on films using the enhanced chemiluminescence method (Bio-Rad) and analyzed by densitometry.
Fluorescence Detection of Mitochondrial
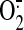 and Intracellular
Peroxides. MitoSOX red (514-nm excitation/585-nm emission) and
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; 488-nm
excitation/525-nm emission) were used to detect mitochondrial
and Intracellular
Peroxides. MitoSOX red (514-nm excitation/585-nm emission) and
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; 488-nm
excitation/525-nm emission) were used to detect mitochondrial
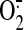 and intracellular
peroxides, respectively, and 4′,6-diamidino-2-phenylindole (DAPI; 359-nm
excitation/461-nm emission) to label cell nuclei (Invitrogen, Carlsbad, CA).
DCFH-DA is converted to DCFH inside the cell, and on exposure to either
H2O2 or ONOO-, it is oxidized to the green
fluorescent product 2′,7′-dichlorofluorescin (DCF)
(Tarpey et al., 2004). MitoSOX
Red is a mitochondria-targeted form of dihydroethidium that is relatively
specific for
and intracellular
peroxides, respectively, and 4′,6-diamidino-2-phenylindole (DAPI; 359-nm
excitation/461-nm emission) to label cell nuclei (Invitrogen, Carlsbad, CA).
DCFH-DA is converted to DCFH inside the cell, and on exposure to either
H2O2 or ONOO-, it is oxidized to the green
fluorescent product 2′,7′-dichlorofluorescin (DCF)
(Tarpey et al., 2004). MitoSOX
Red is a mitochondria-targeted form of dihydroethidium that is relatively
specific for 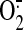 and
undergoes oxidation to form the DNA-binding red fluorophore ethidium bromide
(Tarpey et al., 2004). Before
shear exposure, EC monolayers were incubated with either DCFH-DA (10 μM) or
MitoSOX red (5 μM) and DAPI (1 μM) for 10 min in the incubator. At the
end of shear (30 min), ECs were washed with PBS, mounted with Fluoromount-G
(Southern Biotechnology Associates, Birmingham, AL), and images (62×)
were obtained by confocal microscopy (Zeiss LSM 510; Carl Zeiss Inc.,
Thornwood, NY) and overlaid using LSM software. Digital images from three
fields of view were collected per experiment and corrected for
autofluorescence, and background fluorescence was excluded from calculations
by thresholding. The mean fluorescence intensity per image was calculated and
averaged over the three images, using MetaMorph software (Molecular Devices,
Sunnyvale, CA).
and
undergoes oxidation to form the DNA-binding red fluorophore ethidium bromide
(Tarpey et al., 2004). Before
shear exposure, EC monolayers were incubated with either DCFH-DA (10 μM) or
MitoSOX red (5 μM) and DAPI (1 μM) for 10 min in the incubator. At the
end of shear (30 min), ECs were washed with PBS, mounted with Fluoromount-G
(Southern Biotechnology Associates, Birmingham, AL), and images (62×)
were obtained by confocal microscopy (Zeiss LSM 510; Carl Zeiss Inc.,
Thornwood, NY) and overlaid using LSM software. Digital images from three
fields of view were collected per experiment and corrected for
autofluorescence, and background fluorescence was excluded from calculations
by thresholding. The mean fluorescence intensity per image was calculated and
averaged over the three images, using MetaMorph software (Molecular Devices,
Sunnyvale, CA).
Statistical Analysis. Both HO-1/β-actin protein expression and fluorescence data were normalized to corresponding static controls and expressed as mean ± S.E.M. of n = 3 independent experiments. Significant differences among treatments were determined by using one-way analysis of variance followed by Bonferroni's test for pair-wise comparisons. P values < 0.05 were considered significant.
Results
NO and ROS Are Responsible for the HO-1 Induction by Shear Stress. BAEC exposure to steady laminar shear stress (10 dynes/cm2, 6 h) caused a 6-fold increase in HO-1 protein expression (Fig. 1). To investigate the role of shear-induced NO and ROS in HO-1 induction, ECs were preincubated and sheared in the presence of the NOS inhibitor l-NAME (1 mM) and the antioxidant and GSH precursor NAC (10 mM), respectively. Although NAC was slightly more potent, either l-NAME or NAC abolished the shear-induced increase in HO-1 expression, suggesting that both NO and ROS play important roles in HO-1 induction (Fig. 1).
Fig. 1.

Effect of l-NAME and NAC on shear-induced HO-1 expression. BAECs were preincubated and sheared (10 dynes/cm2, 6 h) in the presence of either l-NAME (1 mM) or NAC (10 mM), and HO-1 protein expression was measured as described under Materials and Methods. Representative immunoblot for HO-1 (32 kDa) and loading control β-actin (42 kDa) and densitometric analysis for levels of HO-1 protein relative to β-actin in static or sheared ECs in presence or absence of l-NAME and NAC are shown. Values are mean ± S.E.M. *, significantly different from static control group; P ≤ 0.05; n = 3. †, significantly different from shear group; P ≤ 0.05; n = 3.
H2O2 Mediates the HO-1 Induction, and
Mitochondrial Function Is Required. To determine the particular ROS that
mediates the increase in HO-1 protein expression, ECs were preincubated and
sheared in the presence of either the GSH peroxidase mimetic and
ONOO- scavenger ebselen (5 μM), the flavoprotein inhibitor DPI
(20 μM), or the ONOO- scavenger UA (100 μM). Ebselen
abolished the HO-1 induction, suggesting that either
H2O2 (or its metabolite OH.) or
ONOO- is involved (Fig.
2A). DPI also abolished the HO-1 induction, suggesting that the
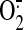 produced by NAD(P)H
oxidase, NOS, the mitochondrial ETC, and other ROS sources, which contain
flavoproteins, is upstream in the redox signaling pathways that regulate HO-1
expression (Fig. 2A). UA did
not affect the shear-induced response; hence, ONOO- is not involved
(Fig. 2A). These data
collectively suggest that H2O2 (and/or secondary
OH.), dismutated from flavoprotein-derived
produced by NAD(P)H
oxidase, NOS, the mitochondrial ETC, and other ROS sources, which contain
flavoproteins, is upstream in the redox signaling pathways that regulate HO-1
expression (Fig. 2A). UA did
not affect the shear-induced response; hence, ONOO- is not involved
(Fig. 2A). These data
collectively suggest that H2O2 (and/or secondary
OH.), dismutated from flavoprotein-derived
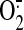 , plays a role in HO-1
induction.
, plays a role in HO-1
induction.
Fig. 2.

Effect of selective antioxidants and ETC inhibitors on shear-induced HO-1 expression. ECs were preincubated and sheared in the presence of ebselen (5 μM), DPI (20 μM), UA (100 μM), myxothiazol (10 μM), rotenone (2 μM), or antimycin A (10 μM), and HO-1 protein expression was measured as before. A, representative immunoblot for HO-1 and β-actin and densitometric analysis for HO-1 protein levels relative to β-actin in static or sheared ECs in the presence or absence of antioxidants ebselen, DPI, and UA (because they had no effect on HO-1 expression in static ECs, only their effects on sheared ECs are shown). B, same as in A but in the presence or absence of ETC inhibitors myxothiazol, rotenone, and antimycin A. Values are mean ± S.E.M. *, significantly different from static control group; P ≤ 0.05; n = 3. †, significantly different from shear group; P ≤ 0.05; n = 3.
When ECs were preincubated and sheared in the presence of the ETC complex III inhibitors myxothiazol and antimycin A (10 μM), myxothiazol abolished and antimycin A significantly inhibited the shear-induced increase in HO-1 expression (Fig. 2B). The ETC complex I inhibitor rotenone (2 μM) also significantly blocked the shear-induced response (Fig. 2B). These data suggest that the activities of complexes I and III of the mitochondrial ETC contribute to the pathways leading to HO-1 induction.
Mitochondrial H2O2 Is Responsible for the Increase
in HO-1 Expression. To investigate the role of mitochondrial ROS, ECs were
preincubated and sheared in the presence of either the mitochondria-targeted
antioxidant peptide SS-31 (1, 10, or 100 nM) or its nonscavenging analog
SS-20. SS-31 displayed a biphasic dose-response relationship, where it
abolished HO-1 induction at the middle dose (10 nM) and afforded a significant
inhibition at 1 nM and a modest one at 100 nM
(Fig. 3). SS-20 had no effect
at either dose tested (only 10 nM is shown;
Fig. 3). Because SS-31 is known
to scavenge mitochondrial
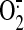 ,
H2O2, and ONOO- and, to a lesser extent,
OH. (Szeto, 2008),
and H2O2 can freely diffuse to the cytosol
(Cadenas, 2004), our data
suggest that the mitochondrial H2O2 plays an important
role in shear-induced HO-1 up-regulation.
,
H2O2, and ONOO- and, to a lesser extent,
OH. (Szeto, 2008),
and H2O2 can freely diffuse to the cytosol
(Cadenas, 2004), our data
suggest that the mitochondrial H2O2 plays an important
role in shear-induced HO-1 up-regulation.
Fig. 3.

Effect of mitochondria-targeted antioxidant peptides on shear-induced HO-1 expression. ECs were preincubated and sheared in the presence of either SS-31 (1, 10, or 100 nM) or its nonscavenging analog SS-20. Representative immunoblot for HO-1 and β-actin and densitometric analysis for HO-1 protein levels relative to β-actin in static or sheared ECs in the presence or absence of SS-31 and SS-20 are shown. Because both SS peptides had no effect on HO-1 expression in static ECs, only their effects on sheared ECs are included. Because SS-20 had no effect on HO-1 expression in sheared ECs at either concentration tested, only 10 nM is shown. Values are mean ± S.E.M. *, significantly different from static control group; P ≤ 0.05; n = 3. †, significantly different from shear group; P ≤ 0.05; n = 3.
Mitochondrial H2O2 Is a Major Source of
Intracellular ROS in Sheared ECs. MitoSOX red fluorescence was
significantly higher in sheared (30 min) ECs compared with static controls,
suggesting a shear-induced increase in
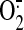 production by the ETC
(Fig. 4, A and B). Rotenone and
antimycin A enhanced the MitoSOX red fluorescence in static ECs, as expected
(Turrens, 2003), but
significantly inhibited the increase in fluorescence in sheared ECs
(Fig. 4, A and B). This is
probably because shear exposure inhibits complex I and III activities by
∼80 and ∼70%, respectively (Han et
al., 2007), and, at least in the case of complex III, decreased
complex activity reduces the enzyme-mediated
production by the ETC
(Fig. 4, A and B). Rotenone and
antimycin A enhanced the MitoSOX red fluorescence in static ECs, as expected
(Turrens, 2003), but
significantly inhibited the increase in fluorescence in sheared ECs
(Fig. 4, A and B). This is
probably because shear exposure inhibits complex I and III activities by
∼80 and ∼70%, respectively (Han et
al., 2007), and, at least in the case of complex III, decreased
complex activity reduces the enzyme-mediated
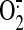 production
(Chen et al., 2006).
l-NAME did not affect the fluorescence signal in static ECs but
significantly inhibited the increase in fluorescence in sheared ECs,
suggesting NO involvement in the shear-induced mitochondrial
production
(Chen et al., 2006).
l-NAME did not affect the fluorescence signal in static ECs but
significantly inhibited the increase in fluorescence in sheared ECs,
suggesting NO involvement in the shear-induced mitochondrial
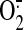 production
(Fig. 4, A and B). At each
concentration tested, SS-31 significantly inhibited the MitoSOX red
fluorescence in sheared ECs (10 and 100 nM totally abolished the signal; only
1 and 10 nM are shown), whereas SS-20 had no effect (only 10 nM is shown;
Fig. 4, A and B).
production
(Fig. 4, A and B). At each
concentration tested, SS-31 significantly inhibited the MitoSOX red
fluorescence in sheared ECs (10 and 100 nM totally abolished the signal; only
1 and 10 nM are shown), whereas SS-20 had no effect (only 10 nM is shown;
Fig. 4, A and B).
Fig. 4.

Effect of ETC inhibitors and selective antioxidants on mitochondrial
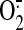 production. ECs were
preincubated with mitoSOX red (5 μM) and DAPI (1 μM) and with rotenone
(2 μM), antimycin A (10 μM), l-NAME (1 mM), or SS-31 or SS-20
(1, 10, or 100 nM) and sheared (30 min) in the presence of the respective
drug. Digital images (62×) of mitoSOX and DAPI fluorescence were
obtained by confocal microscopy and overlaid. A, representative digital images
from ECs exposed to different treatments. Because SS-31 at either 10 or 100 nM
totally abolished the shear-induced fluorescence signal, only 1 and 10 nM are
shown. Because SS-20 had no effect at either concentration tested, only 10 nM
is shown. B, using digital image processing, the mean mitoSOX fluorescence
intensity per image was calculated, averaged over three fields of view per
experiment, and then averaged over three independent experiments. Normalized
(to static controls) mitoSOX fluorescence values are mean ± S.E.M.
*, significantly different from static control group; P ≤ 0.05;
n = 3. †, significantly different from shear group; P
≤ 0.05; n = 3.
production. ECs were
preincubated with mitoSOX red (5 μM) and DAPI (1 μM) and with rotenone
(2 μM), antimycin A (10 μM), l-NAME (1 mM), or SS-31 or SS-20
(1, 10, or 100 nM) and sheared (30 min) in the presence of the respective
drug. Digital images (62×) of mitoSOX and DAPI fluorescence were
obtained by confocal microscopy and overlaid. A, representative digital images
from ECs exposed to different treatments. Because SS-31 at either 10 or 100 nM
totally abolished the shear-induced fluorescence signal, only 1 and 10 nM are
shown. Because SS-20 had no effect at either concentration tested, only 10 nM
is shown. B, using digital image processing, the mean mitoSOX fluorescence
intensity per image was calculated, averaged over three fields of view per
experiment, and then averaged over three independent experiments. Normalized
(to static controls) mitoSOX fluorescence values are mean ± S.E.M.
*, significantly different from static control group; P ≤ 0.05;
n = 3. †, significantly different from shear group; P
≤ 0.05; n = 3.
When ECs were pretreated with ebselen, rotenone, l-NAME, SS-31,
or SS-20 and with DCFH-DA and then left static (30 min) in the presence of the
respective drug, DCF fluorescence was not affected in any case, except in the
case of rotenone, which caused a significant increase (only rotenone is shown;
Fig. 5, A and B). When ECs were
pretreated as before and then sheared (30 min) in the presence of the
respective drug, the shear-induced increase in DCF fluorescence was abolished
by ebselen, significantly inhibited by rotenone, l-NAME, or SS-31
(1, 10, or 100 nM), and not affected by SS-20 (only 10 nM is shown;
Fig. 5, A and B). Although
SS-31 at 1 nM was slightly less potent in blocking both the shear-induced
MitoSOX red and DCF signals, the effect was not significantly different from
that observed by SS-31 at either 10 or 100 nM (Figs.
4 and
5). Based on the fact that
SS-31 scavenges mitochondrial
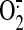 ,
H2O2, ONOO, and OH., the DCF signal reports
intracellular H2O2 and ONOO-, and
mitochondrial H2O2 freely diffuses to the cytosol
(Cadenas, 2004), the data
suggest that mitochondrial H2O2 is a major part of the
intracellular peroxide pool.
,
H2O2, ONOO, and OH., the DCF signal reports
intracellular H2O2 and ONOO-, and
mitochondrial H2O2 freely diffuses to the cytosol
(Cadenas, 2004), the data
suggest that mitochondrial H2O2 is a major part of the
intracellular peroxide pool.
Fig. 5.

Effect of ETC inhibitors and selective antioxidants on intracellular peroxide levels. ECs were preincubated with DCFH-DA (10 μM) and DAPI (1 μM) and with ebselen (5 μM), rotenone (2 μM), l-NAME (1 mM), or SS-31 or SS-20 (1, 10, or 100 nM) and sheared (30 min) in the presence of the respective drug. Digital images (62×) of DCF and DAPI fluorescence were obtained by confocal microscopy and overlaid. A, representative digital images from ECs exposed to different treatments. From all drugs tested on static ECs, only rotenone changed the DCF fluorescence signal (only rotenone is included). Because SS-20 had no effect on the fluorescence in sheared ECs at either concentration tested, only 10 nM is shown. B, using digital image processing, the mean DCF fluorescence intensity per image was calculated, averaged over three fields of view per experiment, and then averaged over three independent experiments. Normalized (to static controls) DCF fluorescence values are mean ± S.E.M. *, significantly different from static control group; P ≤ 0.05; n = 3. †, significantly different from shear group; P ≤ 0.05; n = 3.
PI3K and MAPK Are Involved in the Increase in HO-1 Expression. To investigate the involvement of protein phosphorylation cascades in the shear-induced HO-1 up-regulation, ECs were preincubated and sheared in the presence of the PI3K inhibitor wortmannin (500 nM), the MEK1 inhibitor PD98059 (25 μM; also an ERK1/2 inhibitor because it inhibits its phosphorylation by MEK1), or the p38 MAPK inhibitor SB202190 (20 μM). Either wortmannin or PD98059 abolished the shear-induced increase in HO-1 expression, and SB202190 had no effect (Fig. 6), suggesting that PI3K and ERK1/2 are signaling molecules in HO-1 induction.
Fig. 6.

Effect of protein phosphorylation cascade inhibitors on shear-induced HO-1 expression. ECs were preincubated and sheared in the presence of wortmannin (500 nM), PD98059 (25 μM), or SB202190 (20 μM). Representative immunoblot for HO-1 and β-actin and densitometric analysis for HO-1 protein levels relative to β-actin in static or sheared ECs in the presence or absence of wortmannin, PD98059, and SB202190 are shown. Because they all had no effect on HO-1 expression in static ECs, only their effects on sheared ECs are included. Values are mean ± S.E.M. *, significantly different from static control group; P ≤ 0.05; n = 3. †, significantly different from shear group; P ≤ 0.05; n = 3.
Discussion
The present study provides the first evidence that the mitochondria-derived
H2O2 plays an important role in the intracellular
signaling pathways, leading to shear-induced up-regulation of the
cytoprotective HO-1 in cultured BAECs. The shear-induced increase in HO-1
protein expression was abolished by saturating concentrations of either
l-NAME or NAC, suggesting that both NO and ROS mediate the HO-1
up-regulation. Based on the effects of the antioxidants ebselen, DPI, and UA
on HO-1 expression, it was concluded that H2O2 (and/or
its product OH.), dismutated from flavoprotein-derived
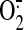 , is essential for HO-1
induction. The ETC inhibitors myxothiazol, rotenone, and antimycin A at the
concentrations tested either abolished or significantly inhibited the
shear-induced increase in HO-1 expression, suggesting that mitochondrial
function is required for this event. Based on the effect of the
mitochondria-targeted antioxidant peptide, SS-31, which scavenges
, is essential for HO-1
induction. The ETC inhibitors myxothiazol, rotenone, and antimycin A at the
concentrations tested either abolished or significantly inhibited the
shear-induced increase in HO-1 expression, suggesting that mitochondrial
function is required for this event. Based on the effect of the
mitochondria-targeted antioxidant peptide, SS-31, which scavenges
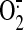 ,
H2O2, ONOO-, and OH.
(Szeto, 2008), on the HO-1
expression, and because it is known that mitochondrial
H2O2 freely diffuses to the cytosol
(
,
H2O2, ONOO-, and OH.
(Szeto, 2008), on the HO-1
expression, and because it is known that mitochondrial
H2O2 freely diffuses to the cytosol
(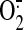 is dismutated to
H2O2 by superoxide dismutase within the matrix and
intermembrane space; ONOO- and OH. react with
intramitochondrial targets) (Cadenas,
2004), it was concluded that the mitochondria-derived
H2O2 is primarily responsible for the shear-induced HO-1
up-regulation. The essential role of mitochondrial H2O2
in HO-1 expression was demonstrated previously in human hepatoma cells exposed
to metabolic stress by glucose deprivation
(Chang et al., 2003). In
vascular ECs, the functional significance of mitochondrial ROS has started
receiving attention only in recent years
(Zhang and Gutterman, 2007).
Mitochondria-derived H2O2 was found responsible for the
flow-induced vasodilation in human coronary arterioles
(Liu et al., 2003).
Furthermore, it was shown that mitochondria-derived ROS are required for the
cyclic strain-induced increases in vascular cell adhesion molecule-1
expression via nuclear factor-κB (Ali
et al., 2004). To our knowledge, before this study, none has
demonstrated a specific role for mitochondrial ROS in sheared ECs.
is dismutated to
H2O2 by superoxide dismutase within the matrix and
intermembrane space; ONOO- and OH. react with
intramitochondrial targets) (Cadenas,
2004), it was concluded that the mitochondria-derived
H2O2 is primarily responsible for the shear-induced HO-1
up-regulation. The essential role of mitochondrial H2O2
in HO-1 expression was demonstrated previously in human hepatoma cells exposed
to metabolic stress by glucose deprivation
(Chang et al., 2003). In
vascular ECs, the functional significance of mitochondrial ROS has started
receiving attention only in recent years
(Zhang and Gutterman, 2007).
Mitochondria-derived H2O2 was found responsible for the
flow-induced vasodilation in human coronary arterioles
(Liu et al., 2003).
Furthermore, it was shown that mitochondria-derived ROS are required for the
cyclic strain-induced increases in vascular cell adhesion molecule-1
expression via nuclear factor-κB (Ali
et al., 2004). To our knowledge, before this study, none has
demonstrated a specific role for mitochondrial ROS in sheared ECs.
Our finding that NO is an important mediator in shear-induced HO-1
up-regulation in BAECs agrees with studies where diverse NO donors were shown
to induce HO-1 expression and activity in aortic ECs
(Motterlini et al., 2002). To
be specific, BAEC exposure to the NO donor spermine NONOate (500 μM,
generating 4 μM NO/min) led to increases in Nrf2 nuclear translocation and
HO-1 protein expression that were blocked by EC pretreatment with either NAC
or MAPK pathway inhibitors (Buckley et al.,
2003). Furthermore, NO was recently shown to cause thiol-dependent
modulation of Keap1, possibly via S-nitrosylation, leading to loss of
its ability to negatively regulate Nrf2
(Buckley et al., 2008). NO can
modify thiol residues to produce SNO via interaction with: 1) oxidants, such
as 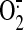 , and formation of
higher oxides of nitrogen, such as nitrogen dioxide, dinitrogen trioxide, and
ONOO- (probably not the main pathway because UA did not block the
shear-induced HO-1 up-regulation); 2) reduced thiol followed by electron
abstraction; or 3) metal centers, such as iron, either operating as catalysts
or through the generation of nitrosonium cation
(Gow et al., 2004). Shear
stress increases the amount of S-nitrosylated proteins in ECs, but
both the basal and the shear-induced levels of SNO differ in HUVECs compared
with human aortic ECs (BAECs were not examined)
(Hoffmann et al., 2003), which
may account for the fact that NO does not play a role in HO-1 induction in
sheared HUVECs (Dai et al.,
2007; Warabi et al.,
2007).
, and formation of
higher oxides of nitrogen, such as nitrogen dioxide, dinitrogen trioxide, and
ONOO- (probably not the main pathway because UA did not block the
shear-induced HO-1 up-regulation); 2) reduced thiol followed by electron
abstraction; or 3) metal centers, such as iron, either operating as catalysts
or through the generation of nitrosonium cation
(Gow et al., 2004). Shear
stress increases the amount of S-nitrosylated proteins in ECs, but
both the basal and the shear-induced levels of SNO differ in HUVECs compared
with human aortic ECs (BAECs were not examined)
(Hoffmann et al., 2003), which
may account for the fact that NO does not play a role in HO-1 induction in
sheared HUVECs (Dai et al.,
2007; Warabi et al.,
2007).
MitoSOX fluorescence responded to ETC inhibitors, and, based on the effect
of l-NAME on the shear-induced increase in fluorescence, it was
concluded that NO is, at least in part, responsible for mitochondrial
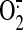 production in sheared
BAECs. Utilization of excess NO by mitochondria is known to involve ubiquinol
oxidation that increases the
production in sheared
BAECs. Utilization of excess NO by mitochondria is known to involve ubiquinol
oxidation that increases the
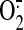 production rate and
formation of ONOO- that, via inhibition of the ETC at multiple
sites, amplifies the
production rate and
formation of ONOO- that, via inhibition of the ETC at multiple
sites, amplifies the 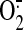 generation (Boveris and Cadenas,
2000). SS-31, at each dose tested, markedly inhibited the
shear-induced increase in both DCF and MitoSOX fluorescence. Because it is
known that DCF responds to intracellular peroxides, SS-31 scavenges
mitochondrial
generation (Boveris and Cadenas,
2000). SS-31, at each dose tested, markedly inhibited the
shear-induced increase in both DCF and MitoSOX fluorescence. Because it is
known that DCF responds to intracellular peroxides, SS-31 scavenges
mitochondrial 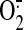 ,
H2O2, ONOO-, and OH., and only
H2O2 freely diffuses through membranes, it was concluded
that the mitochondria are the major source of H2O2 in
sheared BAECs. Because NO is responsible for mitochondrial
,
H2O2, ONOO-, and OH., and only
H2O2 freely diffuses through membranes, it was concluded
that the mitochondria are the major source of H2O2 in
sheared BAECs. Because NO is responsible for mitochondrial
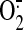 generation and
mitochondria-derived H2O2 is a major part of
intracellular ROS, inhibition of the endothelial NOS also significantly
decreased the shear-induced DCF signal. However, several groups, including
ours, have shown that EC exposure to steady laminar flow causes a transient
increase in
generation and
mitochondria-derived H2O2 is a major part of
intracellular ROS, inhibition of the endothelial NOS also significantly
decreased the shear-induced DCF signal. However, several groups, including
ours, have shown that EC exposure to steady laminar flow causes a transient
increase in 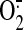 levels that
originates from the plasma membrane-bound NAD(P)H oxidase
(De Keulenaer et al., 1998;
Yeh et al., 1999;
Duerrschmidt et al., 2006).
Because a significant portion of the
levels that
originates from the plasma membrane-bound NAD(P)H oxidase
(De Keulenaer et al., 1998;
Yeh et al., 1999;
Duerrschmidt et al., 2006).
Because a significant portion of the
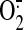 produced by NAD(P)H
oxidase diffuses to the extracellular space
(Barbacanne et al., 2000), and
shear-induced
produced by NAD(P)H
oxidase diffuses to the extracellular space
(Barbacanne et al., 2000), and
shear-induced 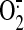 generation
was measured using the cytochrome c assay in media samples and was
blocked by the NAD(P)H oxidase inhibitor gp91ds-tat
(Duerrschmidt et al., 2006),
it may be that, depending on the method for ROS detection, the contribution of
mitochondrial ROS is less apparent. It may also be that the relative
contribution of the two ROS sources, NAD(P)H oxidase and mitochondria, changes
with time during shear. Last, it is possible that l-NAME inhibits
the fluorescence signals because shear-induced NOS activation results in
generation
was measured using the cytochrome c assay in media samples and was
blocked by the NAD(P)H oxidase inhibitor gp91ds-tat
(Duerrschmidt et al., 2006),
it may be that, depending on the method for ROS detection, the contribution of
mitochondrial ROS is less apparent. It may also be that the relative
contribution of the two ROS sources, NAD(P)H oxidase and mitochondria, changes
with time during shear. Last, it is possible that l-NAME inhibits
the fluorescence signals because shear-induced NOS activation results in
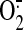 production because of NOS
uncoupling, as was shown in sheared pulmonary artery ECs
(Mata-Greenwood et al., 2006).
It is worth noting that SS-31 demonstrated a biphasic dose-response
relationship in the inhibition of HO-1 induction, but this was not the case
with its effect on the MitoSOX and DCF signals, suggesting that there may be
H2O2-independent pathways leading to shear-induced HO-1
up-regulation. These may depend exclusively on NO because theoretically, NO
can modify thiols (in Keap1) independently of its interaction with
oxidants.
production because of NOS
uncoupling, as was shown in sheared pulmonary artery ECs
(Mata-Greenwood et al., 2006).
It is worth noting that SS-31 demonstrated a biphasic dose-response
relationship in the inhibition of HO-1 induction, but this was not the case
with its effect on the MitoSOX and DCF signals, suggesting that there may be
H2O2-independent pathways leading to shear-induced HO-1
up-regulation. These may depend exclusively on NO because theoretically, NO
can modify thiols (in Keap1) independently of its interaction with
oxidants.
Studies have implicated a major role for protein phosphorylation-dependent
signaling cascades in HO-1 induction by different agents
(Ryter et al., 2006). Under
our experimental conditions, PI3K and ERK1/2, but not p38 MAPK, were involved
in HO-1 up-regulation. Although we did not examine the role of lipid oxidation
products, 15d-PGJ2 is known to be essential for steady laminar flow
to activate the Keap1-Nrf2 pathway (Hosoya
et al., 2005). 15d-PGJ2 can bind directly to and modify
thiols in Keap1 leading to its dissociation from Nrf2 and transactivation of
ARE-regulated genes (Levonen et al.,
2004; Hosoya et al.,
2005). In ECs exposed to lipid oxidation products, it was shown
that they primarily localize to the mitochondria
(Landar et al., 2006), where
they form protein adducts and increase the mitochondrial Ca2+
uptake, leading to 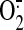 generation by the ETC (Watanabe et al.,
2006). In these ECs, either inhibitors of oxidative
phosphorylation (rotenone, antimycin A) or mitochondria-targeted vitamin E
inhibited ERK1/2 activation and HO-1 induction, providing important evidence
that the mitochondrial ROS mediate HO-1 expression via ERK1/2
(Watanabe et al., 2006).
generation by the ETC (Watanabe et al.,
2006). In these ECs, either inhibitors of oxidative
phosphorylation (rotenone, antimycin A) or mitochondria-targeted vitamin E
inhibited ERK1/2 activation and HO-1 induction, providing important evidence
that the mitochondrial ROS mediate HO-1 expression via ERK1/2
(Watanabe et al., 2006).
In summary, the present study, in combination with work by others, allows
us to delineate the pathways that lead to Keap1-Nrf2 dissociation, Nrf2
nuclear translocation, and Nrf2-mediated HO-1 expression in BAECs exposed to
steady laminar flow (Fig. 7).
Shear-induced NO increases the mitochondrial
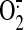 production, which is
dismutated to H2O2, and the excess
H2O2 escapes to the cytosol
(Cadenas, 2004).
H2O2 forms lipid oxidation products
(Zmijewski et al., 2005) that
can activate protein kinases, such as PI3K (associated with the
serine/threonine kinase Akt) and ERK1/2
(Watanabe et al., 2006),
resulting in Nrf2 phosphorylation and its dissociation from Keap1
(Ryter et al., 2006;
Salazar et al., 2006). Lipid
oxidation products also cause alkylation of critical cysteine residues of
Keap1, leading to its dissociation from Nrf2
(Levonen et al., 2004), and
can localize to the mitochondria, where they further increase
production, which is
dismutated to H2O2, and the excess
H2O2 escapes to the cytosol
(Cadenas, 2004).
H2O2 forms lipid oxidation products
(Zmijewski et al., 2005) that
can activate protein kinases, such as PI3K (associated with the
serine/threonine kinase Akt) and ERK1/2
(Watanabe et al., 2006),
resulting in Nrf2 phosphorylation and its dissociation from Keap1
(Ryter et al., 2006;
Salazar et al., 2006). Lipid
oxidation products also cause alkylation of critical cysteine residues of
Keap1, leading to its dissociation from Nrf2
(Levonen et al., 2004), and
can localize to the mitochondria, where they further increase
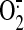 production
(Landar et al., 2006). Last,
NO can directly contribute to Nrf2 activation by modifying the critical
cysteine residues of Keap1, possibly via S-nitrosylation
(Buckley et al., 2008).
However, flow in the arterial circulation, although still laminar, is
unsteady. Flow is pulsatile in the straight portions of the arteries and
oscillatory in curvatures and bifurcations, which are the areas prone to
atherosclerotic lesion formation
(Chatzizisis et al., 2007).
Steady laminar and pulsatile laminar flows (considered
“atheroprotective”) induce EC HO-1 up-regulation, whereas
oscillatory laminar flow (considered “atheroprone”) does not
(Hosoya et al., 2005;
Dai et al., 2007). In
oscillatory flow, there is less bioavailable NO because of down-regulation of
endothelial NOS expression and increased
production
(Landar et al., 2006). Last,
NO can directly contribute to Nrf2 activation by modifying the critical
cysteine residues of Keap1, possibly via S-nitrosylation
(Buckley et al., 2008).
However, flow in the arterial circulation, although still laminar, is
unsteady. Flow is pulsatile in the straight portions of the arteries and
oscillatory in curvatures and bifurcations, which are the areas prone to
atherosclerotic lesion formation
(Chatzizisis et al., 2007).
Steady laminar and pulsatile laminar flows (considered
“atheroprotective”) induce EC HO-1 up-regulation, whereas
oscillatory laminar flow (considered “atheroprone”) does not
(Hosoya et al., 2005;
Dai et al., 2007). In
oscillatory flow, there is less bioavailable NO because of down-regulation of
endothelial NOS expression and increased
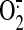 generation by the NAD(P)H
oxidase (Hsiai et al., 2007).
According to the present study, less NO may be, at least in part, responsible
for decreased Nrf2 nuclear translocation and suppressed HO-1 expression in
areas of the vasculature that are exposed to oscillatory flow. Suppressed
expression of phase II genes contributes to the proinflammatory phenotype
observed in those areas and leads to atherogenesis.
generation by the NAD(P)H
oxidase (Hsiai et al., 2007).
According to the present study, less NO may be, at least in part, responsible
for decreased Nrf2 nuclear translocation and suppressed HO-1 expression in
areas of the vasculature that are exposed to oscillatory flow. Suppressed
expression of phase II genes contributes to the proinflammatory phenotype
observed in those areas and leads to atherogenesis.
Fig. 7.

A proposed scheme for HO-1 induction in BAECs exposed to steady laminar
flow. Shear-induced NO, produced by the endothelial NOS, diffuses to the
mitochondria where, by inactivating the ETC, increases
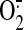 production. Excess
H2O2 diffuses to the cytosol, where it forms reactive
lipid products, which can activate protein kinases, such as PI3K/Akt and
ERK1/2, leading to Nrf2 phosphorylation and its release from Keap1. Lipid
oxidation products can directly modify Keap1 thiols by alkylation of critical
cysteine residues, inhibiting its activity and contributing to the
stabilization of Nrf2. Lipid oxidations products may preferentially localize
to the mitochondria resulting in further increases in
production. Excess
H2O2 diffuses to the cytosol, where it forms reactive
lipid products, which can activate protein kinases, such as PI3K/Akt and
ERK1/2, leading to Nrf2 phosphorylation and its release from Keap1. Lipid
oxidation products can directly modify Keap1 thiols by alkylation of critical
cysteine residues, inhibiting its activity and contributing to the
stabilization of Nrf2. Lipid oxidations products may preferentially localize
to the mitochondria resulting in further increases in
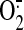 production. NO can
directly modify Keap1 thiols by, for example, forming SNO. Stabilized Nrf2
translocates to the nucleus, where it binds to the ARE and initiates gene
transcription of phase II genes, including HO-1. Sites of inhibition of the
pathway by l-NAME, SS-31, ebselen, NAC, wortmannin, and PD98059 are
shown.
production. NO can
directly modify Keap1 thiols by, for example, forming SNO. Stabilized Nrf2
translocates to the nucleus, where it binds to the ARE and initiates gene
transcription of phase II genes, including HO-1. Sites of inhibition of the
pathway by l-NAME, SS-31, ebselen, NAC, wortmannin, and PD98059 are
shown.
Supplementary Material
Acknowledgments
We thank Dr. Yeong-Renn Chen (Davis Heart and Lung Research Institute, Ohio State University, Columbus, OH) for a critical review of the manuscript. We also thank Dr. Charles I. Jones III for technical assistance and the Confocal Microscopy Core at the Davis Heart and Lung Research Institute for use of facilities.
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL91417, HL38324, HL63744, HL65608]; the National Institutes of Health National Institute on Drug Abuse [Grant DA08924]; and the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK73595].
Patent applications have been filed by Cornell Research Foundation Inc. for the technology (SS peptides) described in this article. H.H.S. is the inventor. Cornell Research Foundation Inc., on behalf of Cornell University, has licensed the technology for further research and development to a commercial enterprise in which Cornell Research Foundation Inc. and H.H.S. have financial interests.
doi:10.1124/jpet.108.145557.
ABBREVIATIONS: HO, heme oxygenase; Nrf2, NF-E2-related factor 2;
ARE, antioxidant response element; Keap1, Kelch-like erythroid-derived
Cap-N-Collar-Homology-associated protein 1; 15d-PGJ2,
15-deoxy-Δ12,14-prostaglandin J2; EC, endothelial
cell; NO, nitric oxide; 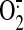 ,
superoxide; ROS, reactive oxygen species; HUVEC, human umbilical vein EC; SNO,
S-nitrosothiol; ONOO-, peroxynitrite; ETC, electron transport
chain; BAEC, bovine aortic EC; NOS, NO synthase; l-NAME,
NG-nitro-l-arginine methyl ester; NAC,
N-acetyl-l-cysteine; UA, uric acid; SS, Szeto-Schiller;
H2O2, hydrogen peroxide; OH., hydroxyl
radical; GSH, glutathione; DPI, diphenylene iodonium; PI3K,
phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein kinase; MEK,
mitogen-activated protein kinase kinase; PD98059,
2′-amino-3′-methoxyflavone; ERK, extracellular signal-regulated
kinase; SB202190,
4-[4-(4-fluorophenyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]phenol; PBS,
phosphate-buffered saline; DMSO, dimethyl sulfoxide; DCFH-DA,
2′,7′-dichlorodihydrofluorescein diacetate; DAPI,
4′,6-diamidino-2-phenylindole; DCF,
2′,7′-dichlorofluorescin.
,
superoxide; ROS, reactive oxygen species; HUVEC, human umbilical vein EC; SNO,
S-nitrosothiol; ONOO-, peroxynitrite; ETC, electron transport
chain; BAEC, bovine aortic EC; NOS, NO synthase; l-NAME,
NG-nitro-l-arginine methyl ester; NAC,
N-acetyl-l-cysteine; UA, uric acid; SS, Szeto-Schiller;
H2O2, hydrogen peroxide; OH., hydroxyl
radical; GSH, glutathione; DPI, diphenylene iodonium; PI3K,
phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein kinase; MEK,
mitogen-activated protein kinase kinase; PD98059,
2′-amino-3′-methoxyflavone; ERK, extracellular signal-regulated
kinase; SB202190,
4-[4-(4-fluorophenyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]phenol; PBS,
phosphate-buffered saline; DMSO, dimethyl sulfoxide; DCFH-DA,
2′,7′-dichlorodihydrofluorescein diacetate; DAPI,
4′,6-diamidino-2-phenylindole; DCF,
2′,7′-dichlorofluorescin.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
References
- Ali MH, Pearlstein DP, Mathieu CE, and Schumacker PT (2004) Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechano-transduction. Am J Physiol Lung Cell Mol Physiol 287 L486-L496. [DOI] [PubMed] [Google Scholar]
- Barbacanne MA, Souchard JP, Darblade B, Iliou JP, Nepveu F, Pipy B, Bayard F, and Arnal JF (2000) Detection of superoxide anion released extracellularly by endothelial cells using cytochrome c reduction, ESR, fluorescence and lucigenin-enhanced chemiluminescence techniques. Free Radic Biol Med 29 388-396. [DOI] [PubMed] [Google Scholar]
- Boveris A and Cadenas E (2000) Mitochondrial production of hydrogen peroxide regulation by nitric oxide and the role of ubisemiquinone. IUBMB Life 50 245-250. [DOI] [PubMed] [Google Scholar]
- Buckley BJ, Li S, and Whorton AR (2008) Keap1 modification and nuclear accumulation in response to S-nitrosocysteine. Free Radic Biol Med 44 692-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley BJ, Marshall ZM, and Whorton AR (2003) Nitric oxide stimulates Nrf2 nuclear translocation in vascular endothelium. Biochem Biophys Res Commun 307 973-979. [DOI] [PubMed] [Google Scholar]
- Cadenas E (2004) Mitochondrial free radical production and cell signaling. Mol Aspects Med 25 17-26. [DOI] [PubMed] [Google Scholar]
- Chang SH, Garcia J, Melendez JA, Kilberg MS, and Agarwal A (2003) Haem oxygenase 1 gene induction by glucose deprivation is mediated by reactive oxygen species via the mitochondrial electron-transport chain. Biochem J 371 877-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, and Stone PH (2007) Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol 49 2379-2393. [DOI] [PubMed] [Google Scholar]
- Chen XL, Varner SE, Rao AS, Grey JY, Thomas S, Cook CK, Wasserman MA, Medford RM, Jaiswal AK, and Kunsch C (2003) Laminar flow induction of antioxidant response element-mediated genes in endothelial cells: a novel anti-inflammatory mechanism. J Biol Chem 278 703-711. [DOI] [PubMed] [Google Scholar]
- Chen YR, Chen CL, Yeh A, Liu X, and Zweier JL (2006) Direct and indirect roles of cytochrome b in the mediation of superoxide generation and NO catabolism by mitochondrial succinate-cytochrome c reductase. J Biol Chem 281 13159-13168. [DOI] [PubMed] [Google Scholar]
- Chiu JJ, Wung BS, Shyy JY, Hsieh HJ, and Wang DL (1997) Reactive oxygen species are involved in shear stress-induced intercellular adhesion molecule-1 expression in endothelial cells. Arterioscler Thromb Vasc Biol 17 3570-3577. [DOI] [PubMed] [Google Scholar]
- Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, and Gimbrone MA Jr (2007) Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res 101 723-733. [DOI] [PubMed] [Google Scholar]
- De Keulenaer GW, Chappell DC, Ishizaka N, Nerem RM, Alexander RW, and Griendling KK (1998) Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: role of a superoxide-producing NADH oxidase. Circ Res 82 1094-1101. [DOI] [PubMed] [Google Scholar]
- Duerrschmidt N, Stielow C, Muller G, Pagano PJ, and Morawietz H (2006) NO-mediated regulation of NAD(P)H oxidase by laminar shear stress in human endothelial cells. J Physiol 576 557-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangos JA, Eskin SG, McIntire LV, and Ives CL (1985) Flow effects on prostacyclin production in cultured human endothelial cells. Science 227 1477-1479. [DOI] [PubMed] [Google Scholar]
- Gow AJ, Farkouh CR, Munson DA, Posencheg MA, and Ischiropoulos H (2004) Biological significance of nitric oxide-mediated protein modifications. Am J Physiol Lung Cell Mol Physiol 287 L262-L268. [DOI] [PubMed] [Google Scholar]
- Han Z, Chen YR, Jones CI 3rd, Meenakshisundaram G, Zweier JL, and Alevriadou BR (2007) Shear-induced reactive nitrogen species inhibit mitochondrial respiratory complex activities in cultured vascular endothelial cells. Am J Physiol Cell Physiol 292 C1103-C1112. [DOI] [PubMed] [Google Scholar]
- Hoffmann J, Dimmeler S, and Haendeler J (2003) Shear stress increases the amount of S-nitrosylated molecules in endothelial cells: important role for signal transduction. FEBS Lett 551 153-158. [DOI] [PubMed] [Google Scholar]
- Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K, Warabi E, Noguchi N, Itoh K, and Yamamoto M (2005) Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem 280 27244-27250. [DOI] [PubMed] [Google Scholar]
- Hsiai TK, Hwang J, Barr ML, Correa A, Hamilton R, Alavi M, Rouhanizadeh M, Cadenas E, and Hazen SL (2007) Hemodynamics influences vascular peroxynitrite formation: implication for low-density lipoprotein apo-B-100 nitration. Free Radic Biol Med 42 519-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iles KE, Dickinson DA, Wigley AF, Welty NE, Blank V, and Forman HJ (2005) HNE increases HO-1 through activation of the ERK pathway in pulmonary epithelial cells. Free Radic Biol Med 39 355-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CI 3rd, Han Z, Presley T, Varadharaj S, Zweier JL, Ilangovan G, and Alevriadou BR (2008) Endothelial cell respiration is affected by the oxygen tension during shear exposure: role of mitochondrial peroxynitrite. Am J Physiol Cell Physiol 295 C180-C191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CI 3rd, Zhu H, Martin SF, Han Z, Li Y, and Alevriadou BR (2007) Regulation of antioxidants and phase 2 enzymes by shear-induced reactive oxygen species in endothelial cells. Ann Biomed Eng 35 683-693. [DOI] [PubMed] [Google Scholar]
- Kuchan MJ and Frangos JA (1994) Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am J Physiol 266 C628-C636. [DOI] [PubMed] [Google Scholar]
- Landar A, Zmijewski JW, Dickinson DA, Le Goffe C, Johnson MS, Milne GL, Zanoni G, Vidari G, Morrow JD, and Darley-Usmar VM (2006) Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am J Physiol Heart Circ Physiol 290 H1777-H1787. [DOI] [PubMed] [Google Scholar]
- Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, and Darley-Usmar VM (2004) Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J 378 373-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, and Gutterman DD (2003) Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res 93 573-580. [DOI] [PubMed] [Google Scholar]
- Mata-Greenwood E, Jenkins C, Farrow KN, Konduri GG, Russell JA, Lakshmin-rusimha S, Black SM, and Steinhorn RH (2006) eNOS function is developmentally regulated: uncoupling of eNOS occurs postnatally. Am J Physiol Lung Cell Mol Physiol 290 L232-L241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motterlini R, Green CJ, and Foresti R (2002) Regulation of heme oxygenase-1 by redox signals involving nitric oxide. Antioxid Redox Signal 4 615-624. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Yang CS, and Pickett CB (2004) The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic Biol Med 37 433-441. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Alam J, and Choi AM (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86 583-650. [DOI] [PubMed] [Google Scholar]
- Salazar M, Rojo AI, Velasco D, de Sagarra RM, and Cuadrado A (2006) Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem 281 14841-14851. [DOI] [PubMed] [Google Scholar]
- Szeto HH (2006), inventor; Cornell Research Foundation, Inc., assignee. Methods for reducing oxidative damages. U.S. Patent 20060084606. 2006 January 20.
- Szeto HH, Schiller P, and Zhao K (2004), inventors; Cornell Research Foundation, Inc., and Institut de Recherches Cliniques de Montreal, assignees. Methods for preventing mitochondrial permeability transition. U.S. Patent 20040248808. 2004 December 9.
- Szeto HH (2008) Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal 10 601-619. [DOI] [PubMed] [Google Scholar]
- Tarpey MM, Wink DA, and Grisham MB (2004) Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol 286 R431-R444. [DOI] [PubMed] [Google Scholar]
- Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552 335-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warabi E, Takabe W, Minami T, Inoue K, Itoh K, Yamamoto M, Ishii T, Kodama T, and Noguchi N (2007) Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: role of reactive oxygen/nitrogen species. Free Radic Biol Med 42 260-269. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Zmijewski JW, Takabe W, Umezu-Goto M, Le Goffe C, Sekine A, Landar A, Watanabe A, Aoki J, Arai H, et al. (2006) Activation of mitogen-activated protein kinases by lysophosphatidylcholine-induced mitochondrial reactive oxygen species generation in endothelial cells. Am J Pathol 168 1737-1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh LH, Park YJ, Hansalia RJ, Ahmed IS, Deshpande SS, Goldschmidt-Clermont PJ, Irani K, and Alevriadou BR (1999) Shear-induced tyrosine phosphorylation in endothelial cells requires Rac1-dependent production of ROS. Am J Physiol 276 C838-C847. [DOI] [PubMed] [Google Scholar]
- Zhang DX and Gutterman DD (2007) Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol 292 H2023-H2031. [DOI] [PubMed] [Google Scholar]
- Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, and Szeto HH (2004) Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279 34682-34690. [DOI] [PubMed] [Google Scholar]
- Zmijewski JW, Landar A, Watanabe N, Dickinson DA, Noguchi N, and Darley-Usmar VM (2005) Cell signalling by oxidized lipids and the role of reactive oxygen species in the endothelium. Biochem Soc Trans 33 1385-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.