

Abstract
Vascular smooth muscle cells have a proliferative phenotype that is important in vascular development, adaptation, and disease. Intracellular calcium handling is thought to play pivotal roles in determining the properties of these cells, and thus previously unrecognized mechanisms for transmembrane calcium movement are of potential interest. An unsolved question is the mechanism of constitutive (passive) calcium leak from the intracellular stores. Studies of other cell types have suggested that the translocon is a calcium leak pathway. Here we investigated the contribution of the translocon in proliferating vascular smooth muscle cells. Calcium leak into the cytoplasm was measured using fura-2, and protein synthesis was measured using radioactive methionine. Puromycin, emetine, and anisomycin are chemicals that inhibit protein synthesis, acting via the translocon; all three agents strongly inhibited protein synthesis in the smooth muscle cells within 1 h. Puromycin, which opens the translocon, evoked a transient increase in cytoplasmic calcium that was similar in amplitude to the calcium rise evoked by thapsigargin. The puromycin effect was abolished by thapsigargin. The treatment of cells for 1 h with emetine or anisomycin, which close the translocon, inhibited the calcium release evoked by puromycin but not the calcium release evoked by extracellular ATP, endothelin-1, or the calcium ionophore ionomycin. Thapsigargin-evoked calcium rises were slightly suppressed by emetine but unaffected by puromycin or anisomycin. The data suggest that the translocon has the capacity to act as a calcium leak pathway in the ribosomal endoplasmic reticulum but that it is normally closed and lacks relevance to physiological calcium leak mechanisms.
Keywords: calcium stores, blood vessels, remodeling
the proliferation and migration of vascular smooth muscle cells (VSMCs) are important in physiology and diseases (3, 25). They are essential for vasculogenesis and physiological adaptation as well as contributing significantly to atherosclerotic plaques, neointimal hyperplasia, in-stent restenoses, and other maladaptive formations of blood vessels (1, 22, 25). As with many cell behaviors, Ca2+ plays pivotal roles. Intriguingly, there are significant Ca2+-handling differences in proliferating compared with contractile VSMCs, including intracellular Ca2+ storage and Ca2+ entry after store depletion (2, 4, 11, 13, 31). Here we address an aspect of the Ca2+ storage system, the Ca2+ leak mechanism from the intracellular stores formed by the sarco(endo)plasmic reticulum.
Generally, in mammalian cells, the Ca2+ content of stores is thought to be governed by Ca2+ uptake driven by sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), counterbalanced by constitutive Ca2+ leak through Ca2+-permeable channels of the endoplasmic reticulum (ER) membrane. A distinction is suggested to exist between Ca2+-leak channels and Ca2+-release channels. The well-recognized Ca2+-release channels are the inositol 1,4,5-trisphosphate (IP3) and ryanodine receptors (19, 35). Although the activity of the release channels is not excluded as a contributor to Ca2+ leak, blocking SERCA with thapsigargin reveals the importance of a constitutive Ca2+ leak in the absence of elevated IP3 concentrations (28, 29). The elevated Ca2+ concentration upon the application of thapsigargin may secondarily stimulate the ryanodine receptors, but the ryanodine receptors are not thought to be the leak mechanism (10, 17, 32). Consequently, there have been investigations to identify whether separate proteins might account for the constitutive Ca2+ leak. Two candidates are the translocon complex and presenilin-1 (9, 23, 24, 30, 32). Here we focus on the translocon.
The translocon is an ER membrane protein complex mediating the translocation of nascent polypeptide into the ER. It, therefore, has a critical role in the synthesis and topological organization of transmembrane proteins (33). Various polypeptide synthesis inhibitors act by modifying the activity of the translocon. One inhibitor is puromycin, which opens the translocon by mimicking aminoacyl tRNA, releasing the nascent polypeptide chain (26). The antibiotic anisomycin and anti-protozoal agent emetine, by contrast, close the translocon by inhibiting the peptidyl transferase (9, 24, 26).
There has been interest in whether the translocon might confer not only a polypeptide pathway across the ER membrane but also a pathway for ions and small molecules, particularly when the polypeptide chain releases from the translocon (10, 14, 16, 17). In this regard it can be envisaged that ion permeability, and particularly Ca2+ permeability, of the translocon has the potential to be a mechanism linking protein synthesis to ER Ca2+, regulating protein synthesis by changing the Ca2+ content of the ER (27). Biochemical studies of the translocon have suggested that leak is prevented by the BiP protein sealing the translocon at the moment of polypeptide release (12, 13). However, functional studies have led to the opposite conclusion, suggesting that the translocon is a major pathway for Ca2+ leak in cell types including a prostate cancer cell line (9, 32) and salivary gland cells (24). Here we investigated whether the translocon contributes to Ca2+ leak in proliferating VSMCs. The translocon must be present in VSMCs but has not been previously investigated as a contributor to Ca2+ handling. While we are interested in the general principles for VSMCs, we specifically focused on VSMCs from the human saphenous vein because this blood vessel is a common coronary artery bypass graft that is prone to failure because of neointimal hyperplasia (1).
METHODS
Cells.
Freshly discarded human saphenous vein VSMCs were obtained with informed consent from patients undergoing open heart surgery in the General Infirmary at Leeds. Approval was granted by the Leeds Teaching Hospitals Local Research Ethics Committee. As previously described (34), VSMCs were grown in Dulbecco's modified Eagle's medium (DMEM) containing GlutaMAX-1, supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin (GIBCO) at 37°C in a 5% CO2-95% room air incubator. VSMCs were used for experiments at passage 2–5. For measurement of protein synthesis, the VSMCs were replated at 70–80% confluence in six-well plates (Sarstedt) for 24–36 h before experimentation. For Ca2+ measurements, VSMCs were replated on Nunclon Delta Microwel plates (96 well) at 70–80% confluence for 24–36 h before experimentation.
Chemicals and ionic solutions.
General salts and puromycin, anisomycin, emetine, thapsigargin, ionomycin, ATP, endothelin-1 were from Sigma. Radioactive [35S]methionine was from MP Biomedicals Europe. The puromycin stock solution was 200 mM in water, stored at −20°C. The emetine stock solution was 40 mM in water, stored at 4°C. The anisomycin stock solution was in dimethylsulfoxide at 200 mM, stored at 4°C. Standard bath solution (SBS) contained (in mM) 130 NaCl, 5 KCl, 1.2 MgCl2, 1.5 CaCl2, 8 d-glucose, and 10 HEPES; osmolarity was adjusted to 290–300 mosmol/l with mannitol (pH 7.4). Ca2+-free SBS was SBS without the addition of CaCl2.
Protein synthesis measurement.
VSMCs were preincubated for 1 h with cysteine and methionine-deprived DMEM and then incubated for a further 1 h in DMEM supplemented with radioactive [35S]methionine and puromycin, emetine, anisomycin, or vehicle at 37°C. VSMCs were scraped, collected, lightly centrifuged, and washed three times with phosphate-buffered saline. VSMCs were then treated with 2% sodium dodecyl sulfate and proteins precipitated with 10% trichloroacetic acid, using bovine serum albumin as a carrier. Pellets were dissolved in 0.5 M NaOH for 1 h. Radioactive emission was determined in a liquid scintillation counter.
Ca2+ measurement.
VSMC Ca2+ measurements were made on a FlexStation II384 (Molecular Devices), which is a real-time fluorimeter suitable for multiwell live-cell assays. Our experiments used 96-well plates, enabling direct multiwell comparisons of test and control conditions on the same batch of cells. The fluorescence indicator fura-2 acetoxymethyl ester (AM) (Invitrogen) was used to measure changes in intracellular ionized Ca2+ concentration. The VSMCs were incubated with 2 μM fura-2 AM and 0.1% pluronic acid in SBS for 1 h at 37°C. VSMCs were washed two to three times in SBS and then treated with agents (protein synthesis inhibitors) at 37°C for another 1 h in a Ca2+-free SBS. Once loaded with fura-2, VSMCs were excited with 340- and 380-nm light, and the emission was collected at 510 nm. Changes in intracellular ionized Ca2+ concentration are indicated as the change in the fura-2 fluorescence ratio for the two excitation wavelengths. Measurements were made in Ca2+-free SBS at room temperature (21 ± 2°C), unless indicated otherwise.
Data analysis.
Data are expressed as means ± SE, where n is the number of independent experiments and N is the number of wells used in multi-well plates. In each case, Ca2+ responses were determined from the average of five data points at the peak of the response. A Student's t-test was used to compare control and test data sets, which were from paired experiments. A statistically significant difference is indicated by P < 0.05. Origin software was used for data analysis.
RESULTS
Effectiveness of translocon pharmacology.
Studies of the acute functional impact of the translocon on Ca2+ signaling depend on pharmacological agents that alter the activity of the translocon. These agents are protein synthesis inhibitors. We selected three agents (puromycin, emetine, and anisomycin) and confirmed that each is an inhibitor of protein synthesis in VSMCs under the conditions necessary for the study of Ca2+ leak. Protein synthesis was quantified by measuring [35S]methionine incorporation. Within 1 h, each agent strongly inhibited protein synthesis (Fig. 1). The same 1-h incubation time was used for subsequent Ca2+ measurement experiments, except when acute responses to puromycin were studied.
Fig. 1.

Chemical inhibition of protein synthesis. Radioactive methionine incorporation in vascular smooth muscle cells (VSMCs) treated for 1 h with vehicle control, puromycin (200 μM), anisomycin (200 μM), or emetine (40 μM) (n = 3 independent experiments; N = 8 wells; P < 0.05 for each inhibitor relative to control). cpm, Counts per minute.
Puromycin-evoked Ca2+ release.
An application of puromycin to VSMCs loaded with fura-2 evoked a transient rise in cytoplasmic Ca2+ (Fig. 2A, and Table 1); the effect was similar at 37°C (n = 3; supplemental Fig. 1; note: supplemental material may be found posted with the online version of this article). The effect of puromycin occurred in the absence of extracellular Ca2+ (Fig. 2A) and was lost in VSMCs pretreated with thapsigargin (Fig. 2B, and Table 1), which depletes intracellular Ca2+ stores. Emetine and anisomycin inhibited the puromycin-evoked Ca2+ signal (Fig. 3, and Table 1). The data suggest that puromycin evokes Ca2+ release via the translocon.
Fig. 2.

Puromycin-evoked Ca2+ release. All graphs show measurement of intracellular Ca2+ (Ca
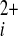 ) from VSMCs in the absence of extracellular Ca2+ (0 Ca2+). Data in A and B are each from single paired-independent experiments. A: effect of extracellular application of puromycin (200 μM; a) or vehicle control (b). B: effect of extracellular application of puromycin (200 μM) after pretreatment with vehicle control (a) or 1 μM thapsigargin (b) for 0.5 h at room temperature. ΔF, change in fluorescence; T, time.
) from VSMCs in the absence of extracellular Ca2+ (0 Ca2+). Data in A and B are each from single paired-independent experiments. A: effect of extracellular application of puromycin (200 μM; a) or vehicle control (b). B: effect of extracellular application of puromycin (200 μM) after pretreatment with vehicle control (a) or 1 μM thapsigargin (b) for 0.5 h at room temperature. ΔF, change in fluorescence; T, time.
Table 1.
Summary of Ca2+ measurement data
| Control Response, ΔF Ratio | Test Response, ΔF Ratio | Inhibition, % | P Value | n | N | |
|---|---|---|---|---|---|---|
| Puromycin | ||||||
| Thapsigargin | 0.313±0.061 | 0.019±0.002 | 93.9 | 0* | 3 | 60 |
| Emetine | 0.306±0.014 | 0.119±0.037 | 61.2 | 0.0070* | 3 | 32 |
| Anisomycin | 0.226±0.012 | 0.080±0.048 | 64.6 | 0.0079* | 3 | 24 |
| Thapsigargin | ||||||
| Emetine | 0.341±0.072 | 0.278±0.058 | 18.5 | 0.0012* | 5 | 36 |
| Puromycin | 0.274±0.062 | 0.294±0.068 | −7.3 | 0.5829 | 5 | 28 |
| Anisomycin (200 μM) | 0.262±0.048 | 0.273±0.032 | −4.2 | 0.4307 | 3 | 24 |
| Anisomycin | 0.211±0.086 | 0.197±0.079 | 6.5 | 0.1416 | 3 | 36 |
| Anisomycin (37°C) | 0.244±0.017 | 0.249±0.030 | −2.0 | 0.8963 | 5 | 76 |
| Anisomycin (1.5 mM Ca2+, 37°C) | 0.183±0.038 | 0.155±0.054 | 15.5 | 0.6823 | 4 | 48 |
| Anisomycin (10 mM Ca2+, 37°C) | 0.166±0.040 | 0.150±0.031 | 9.8 | 0.7599 | 4 | 48 |
| Ionomycin | ||||||
| Emetine | 0.669±0.065 | 0.608±0.065 | 9.2 | 0.0967 | 3 | 18 |
| Puromycin | 0.654±0.046 | 0.681±0.046 | −4.1 | 0.7671 | 3 | 18 |
| Anisomycin | 1.264±0.289 | 1.148±0.233 | 9.2 | 0.0732 | 3 | 40 |
| Anisomycin (37°C) | 0.502±0.087 | 0.383±0.077 | 23.7 | 0.0725 | 4 | 60 |
| ATP | ||||||
| Emetine | 0.214±0.081 | 0.207±0.060 | 3.3 | 0.7546 | 4 | 64 |
| Anisomycin | 0.226±0.077 | 0.235±0.095 | −3.2 | 0.8792 | 3 | 24 |
| Puromycin | 0.323±0.028 | 0.253±0.087 | 21.8 | 0.6235 | 3 | 24 |
| Endothelin-1 | ||||||
| Emetine | 0.504±0.186 | 0.466±0.058 | 7.5 | 0.6530 | 3 | 44 |
Values are means ± SE; n, number of independent experiments; N, number of wells of the 96-well plate. Puromycin, thapsigargin, emetine, ionomycin, ATP, and endothelin-1 were used at 200, 3, 40, 5, 100 and 0.1 μM, respectively. Anisomycin was used at 400 μM unless indicated. Experiments were performed in the absence of extracellular Ca2+ and at room temperature, unless at 37°C as indicated. Preincubation with inhibitors was in the absence of Ca2+ for 1 h unless the preincubation included 1.5 or 10 mM Ca2+ where indicated. ΔF, change in fluorescence.
P < 0.05.
Fig. 3.

Inhibition of puromycin-evoked Ca2+ release by translocon inhibitors. All graphs show measurement of intracellular Ca2+ from VSMCs in the absence of extracellular Ca2+ (0 Ca2+). Data in A and B are each from single paired-independent experiments. A: effect of puromycin (200 μM) after pretreatment with vehicle control (a) or 40 μM emetine (b) for 1 h. B: effect of puromycin (200 μM) after pretreatment with vehicle control (a) or 400 μM anisomycin (b) for 1 h.
Constitutive Ca2+ leak.
Because the translocon is capable of conferring Ca2+ leak, we explored whether it is relevant to the constitutive Ca2+ leak that is evident when the SERCA of the store is blocked by thapsigargin, applied to VSMCs in the absence of extracellular Ca2+. Although there was a small inhibitory effect of emetine on the thapsigargin response (Table 1), puromycin had no effect (Table 1) and anisomycin had no effect, even at the high concentration of 0.4 mM (Fig. 4A, and Table 1). An independent method for investigating constitutive Ca2+ leak is to measure the total amount of Ca2+ releasable from intracellular stores because the total content of the stores will be greater if the constitutive Ca2+ leak is inhibited. To determine the total Ca2+ content, we applied ionomycin, which permeabilizes the membrane of the stores to Ca2+, causing rapid Ca2+ leak. Ionomycin evoked a large Ca2+ transient in the absence of extracellular Ca2+, which was not affected by emetine (Fig. 4B) or anisomycin (Table 1).
Fig. 4.

Lack of effect of translocon inhibitors on constitutive Ca2+ leak. All graphs show measurement of intracellular Ca2+ from VSMCs in the absence of extracellular Ca2+ (0 Ca2+). Data in A and B are each from single paired-independent experiments. A: effect of thapsigargin (3 μM) after pretreatment with vehicle control (a) or 400 μM anisomycin (b) for 1 h. B: effect of ionomycin (5 μM) after pretreatment with vehicle control (a) or 40 μM emetine (b) for 1 h.
The above experiments were performed at room temperature and after a 1-h preincubation in Ca2+-free medium (to avoid contamination from Ca2+-entry signals). Although Ca2+ leak occurred, it was conceivable that the conditions were biased against the contribution of the translocon. However, a repetition of the experiments at 37°C and the preincubation in Ca2+-containing medium revealed no effects of anisomycin on the thapsigargin and ionomycin responses (Table 1). In one set of experiments, we increased the Ca2+ concentration in the preincubation medium to 10 mM in an effort to overload the Ca2+ stores (20), but again there was no effect of anisomycin (Table 1).
The data suggest that the translocon has no role in the constitutive Ca2+ leak mechanism.
Ca2+ release evoked by physiological agonists.
Two physiological agonists that evoke Ca2+ release in saphenous vein VSMCs are endothelin-1 and ATP (5, 7). Both agonists act through G protein-coupled receptors that induce the production of IP3. Neither endothelin-1 nor ATP responses were affected by emetine or anisomycin (Fig. 5, and Table 1). Therefore, the data suggest that the Ca2+ release evoked by physiological agonists does not involve the translocon.
Fig. 5.

Lack of effect of translocon inhibitors on Ca2+ release evoked by physiological agonists. All graphs show measurement of intracellular Ca2+ from VSMCs in the absence of extracellular Ca2+ (0 Ca2+). Data in A and B are each from single paired-independent experiments. A: effect of endothelin-1 (100 nM) after pretreatment with vehicle control (a) or 40 μM emetine (b) for 1 h. B: effect of ATP (100 μM) after pretreatment with vehicle control (a) or 400 μM anisomycin (b) for 1 h.
DISCUSSION
The study shows a striking Ca2+-release event when VSMCs are exposed to puromycin. The event is suggested to take place as a result of the opening of the translocon because it is suppressed by the translocon inhibitors anisomycin and emetine. Importantly, studies of both the puromycin Ca2+-release signal and direct measurements of protein synthesis enabled us to confirm that anisomycin and emetine inhibit the translocon under our experimental conditions. Under these conditions, passive Ca2+ leak and Ca2+ release evoked by physiological agonists were not affected by translocon inhibition. Therefore, although Ca2+ leak via the translocon is possible in VSMCs, the translocon is not normally a passive Ca2+ leak mechanism of these cells and does not act as a Ca2+ pathway influencing responses to endothelin-1 or ATP. Therefore, the data suggest that there is translocon closure to Ca2+ leak unless there is a premature release of the nascent polypeptide chain caused by an agent such as puromycin.
Relatively rapid puromycin-evoked release of ER Ca2+ has been previously suggested based on direct measurements of luminal Ca2+ or other ER permeability assays (14, 17, 24, 32). Our observation of a cytoplasmic Ca2+-release signal is consistent with these previous results, as well as the rapid effect of puromycin on protein synthesis (18). We were, however, surprised to observe that the release signal is comparable in amplitude and time course to signals evoked by other agents including thapsigargin and ATP. Superficially, the similarity suggests a release from a common intracellular Ca2+ store. However, although thapsigargin prevented the puromycin response, puromycin failed to inhibit the thapsigargin response. Therefore, the data suggest that the translocon is contained in ER elements that depend on Ca2+ uptake via SERCA but that it is absent from other SERCA-dependent elements. The latter translocon-independent elements may be sub-plasma membrane Ca2+ stores involved in IP3-dependent Ca2+ signaling, a conclusion that is consistent with the statistically insignificant effect of puromycin on the ATP response. That thapsigargin depletes the puromycin-sensitive store yet evokes puromycin-resistant Ca2+ leak tells us that Ca2+ leak from the translocon-containing ER (ribosomal ER) also does not occur through the translocon.
Our findings suggest that the translocon is tightly sealed against Ca2+ flux in VSMCs, even when the cells are proliferating and actively synthesizing new proteins. The question arises, therefore, as to whether leakiness of the translocon is different in VSMCs compared with the LNCaP prostate cancer cell line or salivary gland cells where other investigators have concluded that the translocon is a physiological Ca2+ leak mechanism (9, 24). Ong et al. (24) indirectly observed Ca2+ release in response to a translocon opener, an effect that was strongly inhibited by emetine. However, thapsigargin-evoked Ca2+ release was only slightly affected by emetine. We observed almost exactly the same result with emetine, but our data additionally show that emetine lacks specificity for the translocon because its effect on the thapsigargin response was not mimicked by anisomycin or puromycin. Flourakis et al. (9) observed that 200 μM anisomycin caused about 30% inhibition of Ca2+ release evoked by thapsigargin in LNCaP cells; unfortunately, using the same protocol, we have not reproduced this result in VSMCs (Table 1) or LNCaP cells (M. S. Amer, unpublished data). Our interpretation of the observations is that we cannot yet be certain there are major differences in leakiness of the translocon to Ca2+ in different cell types.
To the best of our knowledge, there is no physiological or pathological situation that leads to a premature release of the nascent polypeptide chain. Therefore, it seems appropriate to conclude that the translocon is normally efficiently closed to Ca2+ leak. Nevertheless, the translocon can readily be made leaky to Ca2+ by puromycin. Puromycin is used therapeutically as an antiprotozoal agent and has been compared with cycloheximide as a potential pharmacological approach to stabilizing atherosclerotic plaques (6). Intriguingly, the latter study showed that puromycin, but not cycloheximide, evoked VSMC apoptosis. Our data may help to explain this differential effect because puromycin causes Ca2+ release in VSMCs, whereas cycloheximide would be expected to have no effect on Ca2+ release because it is a translocon inhibitor.
Our data suggest that the translocon does not explain the constitutive (passive) leak of Ca2+ from intracellular stores of VSMCs. Consequently, the molecular mechanism of Ca2+ leak in these cells remains unclear. It is important to consider the potential contribution of ryanodine receptors, even though such a contribution appears absent in some other cell types (10, 17, 32). Significantly, we have observed no effect of 0.1 mM ryanodine on the thapsigargin response of human saphenous vein VSMCs, and an activator of ryanodine receptors (10 mM caffeine) failed to evoke Ca2+ release, even following efforts to overload the Ca2+ stores using 10 mM Ca2+ in the extracellular medium (supplemental Figs. 2 and 3). The same caffeine was shown to be effective as a Ca2+-release agent on ventricular myocytes (supplemental Fig. 4). Such observations do not encourage a further consideration of ryanodine receptors as the leak mechanism in VSMCs. We have no data on the roles of IP3 receptors in the leak mechanism, but IP3 receptor inhibitors (2-aminoethoxydiphenylborate and xestospongin C) either enhanced or had no effect on passive Ca2+ leak from stores of the A7r5 VSMC line (8, 21), arguing against IP3 receptors playing a role.
In conclusion, our observations are not consistent with the translocon acting as a physiological Ca2+ leak pathway or with Ca2+ permeability of the translocon serving as a mechanism linking Ca2+ handling to protein synthesis. The data are consistent with the translocon having the capacity to enable Ca2+ leak from ribosomal ER but, nevertheless, suggest that the translocon is not the normal Ca2+ leak mechanism of the ribosomal ER. The capacity for translocon-dependent Ca2+ leak is not apparent in the subplasma membrane Ca2+ stores (non-ribosomal ER) that enables IP3-dependent Ca2+ release. We emphasize that our study focused on VSMCs and so does not exclude differential Ca2+ leakiness of the translocon in other cell types and contexts. Furthermore, pharmacologically, the translocon can be made leaky to Ca2+, which may explain the functional differences in effects of protein synthesis inhibitors that either open or close the translocon.
GRANTS
This study was supported by the Wellcome Trust and a scholarship from the Egyptian Ministry of Higher Education (to M. S. Amer).
Supplementary Material
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Angelini GD, Jeremy JY. Towards the treatment of saphenous vein bypass graft failure—a perspective of the Bristol Heart Institute. Biorheology 39: 491–499, 2002. [PubMed] [Google Scholar]
- 2.Beech DJ Ion channel switching and activation in smooth-muscle cells of occlusive vascular diseases. Biochem Soc Trans 35: 890–894, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Berk BC Vascular smooth muscle growth: autocrine growth mechanisms. Physiol Rev 81: 999–1030, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Berra-Romani R, Mazzocco-Spezzia A, Pulina MV, Golovina VA. Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am J Physiol Cell Physiol 295: C779–C790, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borna C, Wang L, Gudbjartsson T, Karlsson L, Jern S, Malmsjo M, Erlinge D. Contractions in human coronary bypass vessels stimulated by extracellular nucleotides. Ann Thorac Surg 76: 50–57, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Croons V, Martinet W, Herman AG, De Meyer GR. Differential effect of the protein synthesis inhibitors puromycin and cycloheximide on vascular smooth muscle cell viability. J Pharmacol Exp Ther 325: 824–832, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Davenport AP, Maguire JJ. The endothelin system in human saphenous vein graft disease. Curr Opin Pharmacol 1: 176–182, 2001. [DOI] [PubMed] [Google Scholar]
- 8.De Smet P, Parys JB, Callewaert G, Weidema AF, Hill E, De Smedt H, Erneux C, Sorrentino V, Missiaen L. Xestospongin C is an equally potent inhibitor of the inositol 1,4,5-trisphosphate receptor and the endoplasmic-reticulum Ca2+ pumps. Cell Calcium 26: 9–13, 1999. [DOI] [PubMed] [Google Scholar]
- 9.Flourakis M, Van Coppenolle F, Lehen'kyi V, Beck B, Skryma R, Prevarskaya N. Passive calcium leak via translocon is a first step for iPLA2-pathway regulated store operated channels activation. FASEB J 20: 1215–1217, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Giunti R, Gamberucci A, Fulceri R, Banhegyi G, Benedetti A. Both translocon and a cation channel are involved in the passive Ca2+ leak from the endoplasmic reticulum: a mechanistic study on rat liver microsomes. Arch Biochem Biophys 462: 115–121, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Golovina VA Cell proliferation is associated with enhanced capacitative Ca2+ entry in human arterial myocytes. Am J Physiol Cell Physiol 277: C343–C349, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Haigh NG, Johnson AE. A new role for BiP: closing the aqueous translocon pore during protein integration into the ER membrane. J Cell Biol 156: 261–270, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamman BD, Hendershot LM, Johnson AE. BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell 92: 747–758, 1998. [DOI] [PubMed] [Google Scholar]
- 14.Heritage D, Wonderlin WF. Translocon pores in the endoplasmic reticulum are permeable to a neutral, polar molecule. J Biol Chem 276: 22655–22662, 2001. [DOI] [PubMed] [Google Scholar]
- 15.House SJ, Potier M, Bisaillon J, Singer HA, Trebak M. The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease. Pflügers Arch 456: 769–785, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lizak B, Czegle I, Csala M, Benedetti A, Mandl J, Banhegyi G. Translocon pores in the endoplasmic reticulum are permeable to small anions. Am J Physiol Cell Physiol 291: C511–C517, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Lomax RB, Camello C, Van Coppenolle F, Petersen OH, Tepikin AV. Basal and physiological Ca2+ leak from the endoplasmic reticulum of pancreatic acinar cells. Second messenger-activated channels and translocons. J Biol Chem 277: 26479–26485, 2002. [DOI] [PubMed] [Google Scholar]
- 18.MacInnes JW, Luttges MW. Interactive effects of cycloheximide and puromycin in altering brain polyribosomes and neutral and behavioural responses to electroshock in mice. J Neurochem 21: 775–781, 1973. [DOI] [PubMed] [Google Scholar]
- 19.Mikoshiba K IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem 102: 1426–1446, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Mironneau J, Coussin F, Jeyakumar LH, Fleischer S, Mironneau C, Macrez N. Contribution of ryanodine receptor subtype 3 to Ca2+ responses in Ca2+-overloaded cultured rat portal vein myocytes. J Biol Chem 276: 11257–11264, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Missiaen L, Callewaert G, De Smedt H, Parys JB. 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell Calcium 29: 111–116, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Mitra AK, Agrawal DK. In stent restenosis: bane of the stent era. J Clin Pathol 59: 232–239, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest 117: 1230–1239, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ong HL, Liu X, Sharma A, Hegde RS, Ambudkar IS. Intracellular Ca2+ release via the ER translocon activates store-operated calcium entry. Pflügers Arch 453: 797–808, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Pestka S Inhibitors of ribosome functions. Annu Rev Microbiol 25: 487–562, 1971. [DOI] [PubMed] [Google Scholar]
- 27.Roy A, Wonderlin WF. The permeability of the endoplasmic reticulum is dynamically coupled to protein synthesis. J Biol Chem 278: 4397–4403, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA 87: 2466–2470, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci 19: 131–135, 1998. [DOI] [PubMed] [Google Scholar]
- 30.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell 126: 981–993, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallot O, Combettes L, Jourdon P, Inamo J, Marty I, Claret M, Lompre AM. Intracellular Ca2+ handling in vascular smooth muscle cells is affected by proliferation. Arterioscler Thromb Vasc Biol 20: 1225–1235, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Van Coppenolle F, Vanden Abeele F, Slomianny C, Flourakis M, Hesketh J, Dewailly E, Prevarskaya N. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J Cell Sci 117: 4135–4142, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Walter P, Lingappa VR. Mechanism of protein translocation across the endoplasmic reticulum membrane. Annu Rev Cell Biol 2: 499–516, 1986. [DOI] [PubMed] [Google Scholar]
- 34.Xu SZ, Muraki K, Zeng F, Li J, Sukumar P, Shah S, Dedman AM, Flemming PK, McHugh D, Naylor J, Cheong A, Bateson AN, Munsch CM, Porter KE, Beech DJ. A sphingosine-1-phosphate-activated calcium channel controlling vascular smooth muscle cell motility. Circ Res 98: 1381–1389, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem 76: 367–385, 2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.