

Abstract
Fibroblast growth factor (FGF) 1 and FGF-2 are prototypic members of the FGF family, which to date comprises at least 18 members. Surprisingly, even though FGF-1 and FGF-2 share more than 80% sequence similarity and an identical structural fold, these two growth factors are biologically very different. FGF-1 and FGF-2 differ in their ability to bind isoforms of the FGF receptor family as well as the heparin-like glycosaminoglycan (HLGAG) component of proteoglycans on the cell surface to initiate signaling in different cell types. Herein, we provide evidence for one mechanism by which these two proteins could differ biologically. Previously, it has been noted that FGF-1 and FGF-2 can oligomerize in the presence of HLGAGs. Therefore, we investigated whether FGF-1 and FGF-2 oligomerize by the same mechanism or by a different one. Through a combination of matrix-assisted laser desorption ionization mass spectrometry and chemical crosslinking, we show here that, under identical conditions, FGF-1 and FGF-2 differ in the degree and kind of oligomerization. Furthermore, an extensive analysis of FGF-1 and FGF-2 uncomplexed and HLGAG complexed crystal structures enables us to readily explain why FGF-2 forms sequential oligomers whereas FGF-1 forms only dimers. FGF-2, which possesses an interface capable of protein association, forms a translationally related oligomer, whereas FGF-1, which does not have this interface, forms only a symmetrically related dimer. Taken together, these data show that FGF-1 and FGF-2, despite their sequence homology, differ in their mechanism of oligomerization.
Fibroblast growth factors (FGFs) play important roles during development and morphogenesis (1, 2). Aberrant FGF expression is central to progression of pathogenesis in several disease states, including cancer and chronic inflammation (3). Acidic FGF (FGF-1) and basic FGF (FGF-2) are the two prototypical members of a family of at least 18 FGFs identified to date (4). FGFs signal through their cognate receptors, and this interaction is mediated by a low affinity receptor belonging to a family of molecules known as heparan sulfate proteoglycans (HSPGs) (2, 5, 6). Heparin-like glycosaminoglycans (HLGAGs), the polysaccharide component of HSPGs, mediate FGF binding to FGF receptors, leading to a ternary complex crucial for signaling.
The dependence of FGF activity on HLGAGs has garnered significant attention in the past few years (7, 8). Biochemically, it has been observed that FGF-1 and FGF-2 can be crosslinked as dimers or oligomers (henceforth referred to as oligomers) in the presence of HLGAGs (9, 10), suggesting that HLGAG-mediated FGF oligomerization may be central to HLGAGs’ role in FGF-FGF receptor complex formation. Further, biophysical and biochemical experiments performed in the presence of HLGAG oligosaccharides or HLGAG analogs demonstrate that FGF-1 and FGF-2 indeed oligomerize in the presence of HLGAGs (11–15). However, all apo-FGF-2 crystals (16–19) and four cocrystal structures of FGF-2 liganded with HLGAGs or analogs thereof (20, 21) contain only monomers in the asymmetric unit, whereas apo-FGF-1 (16, 22) and sucrose octasulfate (SOS)-liganded FGF-1 structures contained up to eight monomers in the asymmetric unit (23). Thus, none of these structures were able to address the issue of HLGAG-mediated FGF oligomerization (8). However, DiGabriele et al. (15) recently presented FGF-1-liganded HLGAG decasaccharide cocrystal structure and dynamic light scattering experiments of FGF-1 with HLGAG analog SOS and HLGAG hexasaccharide and octasaccharide. That report represented the first crystal structure where HLGAG-mediated FGF dimerization was observed; with that as a basis the authors proposed a general model for HLGAG-mediated FGF dimerization for the FGF family of growth factors.
FGF-1 and FGF-2 are prototypical members of the FGF family, and the proteins display more than 80% sequence similarity with an identical three-dimensional structural fold and HLGAG binding sites. However, in this study we provide convincing evidence that HLGAG-mediated FGF-1 and FGF-2 oligomerization are distinct (8, 15). Through a technique involving matrix-associated laser desorption ionization mass spectrometry (MALDI-MS), chemical crosslinking, and crystal structure analysis, we show that FGF-1 and FGF-2 oligomerize differently. Importantly, these studies demonstrate that despite having identical HLGAG binding features, the individual FGF-1 and FGF-2 molecules differ inherently in their ability to oligomerize in the presence of HLGAGs. We suggest that this inherent physical-chemical difference may be one mechanism by which these two growth factors differ in their biological activity. The data presented here are consistent with in vivo as well as in vitro observations that have shown that HLGAGs modulate FGF-1 and FGF-2 activity very differently.
MATERIALS AND METHODS
Proteins and Reagents.
Recombinant human FGF-1 (with a protein concentration of 0.68 mg/ml in 20 mM sodium citrate, pH 6.8) and the FGF-2 mutant with cysteines C69 and C87 mutated to serines, hereafter referred to as the cysteine FGF-2 mutant, was provided in PBS buffer by Amgen (Thousand Oaks, CA). Recombinant human FGF-2 was provided by Scios (Mountain View, CA) in 20 mM sodium citrate, 1 mM EDTA, 9% sucrose, pH 5.0 with a protein concentration of 8.6 mg/ml. All proteins were stored as single-use aliquots at −70°C before use. Bis[sulfosuccinimidyl]suberate (BS3) was obtained from Pierce. Monoclonal FGF-1 antibody (clone FA-88) was from Sigma. Monoclonal FGF-2 antibody (clone 11.1) was kindly provided by Scios. Goat anti-mouse IgG alkaline phosphatase (AP)-conjugated antibodies and AP substrate were from Bio-Rad. Goat anti-mouse horseradish peroxidase-conjugated antibodies and Super Signal ULTRA substrate were from Pierce. Heparin was obtained in powdered form from Celsus Laboratories, Cincinnati, OH. Sinapinic acid was purchased from Aldrich. Acetonitrile was from Burdich and Jackson (Mukegon, MI).
Decasaccharide (ΔU2S-HNS,6S-I2S-HNS,6S-I2S-HNS,6S-I2S-HNS,6S-I2S-HNS,6S¶, MW = 2887) was a gift from K. Biemann of the Massachusetts Institute of Technology. Decasaccharide was received as a lyophilized powder and dissolved in deionized water at a concentration of 1 mg/ml. Hexasaccharide (I2S-HNS,6S-I2S-HNS,6S-I2S-Man6S, MW = 1654) was a gift from D. Tyrrell of Glycomed (Almeda, CA) and also was dissolved in deionized water.
MALDI-MS.
Sinapinic acid (≈10 mg/ml) in 30% acetonitrile/water was used as a matrix solution. Seeded surfaces were prepared by a modification of the method of Xiang and Beavis (24). Previously, Juhasz and Biemann (25, 26) discovered that addition of a heparin-binding protein to a GAG chain results in the formation of a stable noncovalent complex that can be detected by MALDI-MS. Briefly, an equimolar concentration of either FGF-1 or FGF-2 was premixed with the basic peptide (arg-gly)15 in a large excess of matrix solution (although the basic peptide is not incorporated in the protein-saccharide complex, its presence significantly improved ionization of the complex). Then, 9 μl of FGF/peptide in matrix solution was added to 1 μl of 10 pmol/μl of aqueous decasaccharide. One microliter of the FGF/peptide/saccharide mixture was deposited on the seeded surface (stainless-steel plate). After drying, the sample was washed with water, dried under a stream of nitrogen gas, and placed into the mass spectrometer. MALDI mass spectra were acquired in the linear mode by using a Voyager Elite reflectron time-of-flight instrument (PerSeptive Biosystems, Framingham, MA) fitted with a 337-nm nitrogen laser. Delayed extraction was used to increase resolution (25 kV, grid at 91%, guide wire at 0.25%, pulse delay 350 ns, low mass gate at 2,000). Mass spectra were calibrated externally with myoglobin and BSA. For mass spectra in the absence of saccharide, 1 pmol of either FGF-2 or FGF-1 was deposited on the target, and the same collection parameters as outlined above were used to record spectra.
Crosslinking Reactions.
Before using chemical crosslinking methods to assess specific protein interactions, we ensured that either disulfide-bonded aggregates or crosslinking caused by chance protein interactions were not observed. To avoid disulfide-bonded aggregates, control crosslinking experiments were carried out with the cysteine FGF-2 mutant; in this case, the cysteine FGF-2 mutant had the same oligomerization profile as recombinant FGF-2 (data not shown). Additional control experiments were performed with recombinant FGF-2 in the presence as well as in the absence of 2-mercaptoethanol; no differences in the crosslinking profile were detected, again suggesting the absence of disulfide aggregates. Finally, a time-course experiment showed that after 5 min the crosslinking reaction was complete, ensuring that we measured equilibrium values.
To quantify the equilibrium association values for FGF-1 and FGF-2, crosslinking studies were performed with BS3. Varying concentrations of FGF-1 or FGF-2 were incubated with a 10:1 molar ratio of heparin for 15 min at room temperature in a 20-μl volume of 50 mM sodium phosphate, 100 mM NaCl, and 0.2% 2-mercaptoethanol, pH 7.0. One microliter of BS3 in 5 mM sodium citrate, pH 5.5, was added at a final concentration of 100 μM because a higher concentration resulted in the formation of nonspecific complexes. The reaction was allowed to proceed for 30 min at room temperature before quenching with 1 μl of 1 mol of glycine. The reaction products were run on 12% SDS-polyacrylamide gels. Proteins then were transferred to nitrocellulose membranes at 275 mA for 45 min. Membranes were blocked with 1% Tris-buffered saline/BSA, washed, and then incubated with the appropriate primary antibody for 90 min. Membranes then were washed and incubated with a goat anti-mouse alkaline phosphatase conjugate for 45 min and then developed with the Bio-Rad substrate. Alternatively, membranes were incubated with goat anti-mouse horseradish peroxidase conjugate as the secondary antibody and developed with Pierce chemiluminescent Super Signal ULTRA substrate. The blot then was viewed with a Stratagene Eagle Eye II video system, and band intensities were quantified. Protomer species were expressed as a fraction of total intensity per lane. It should be pointed out that the same amount of protein was loaded in all lanes to enable accurate quantification. These values then were used to determine equilibrium constants, by fitting to the following equation (assuming an isodesmic model) (27):
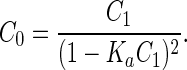 |
C0 is the total concentration of FGF-1 or FGF-2 and C1 is the concentration of monomer.
Analysis of FGF Crystal Structures.
The following FGF crystal structures were obtained from the Brookhaven data bank (http://www.pdb.bnl.gov): 1BFB, 1BFC (21), 1AXM (15), 2AXM (15), 1AFC (23), 1BAS (16), 1FGA (17), 2FGF (18), 4FGF (19), and 2AFG (22). The coordinates of the cocrystal of FGF-2 with α-l-iduronyl-α-d-glucosaminyl-β-d-glucuronic acid anomeric O-methyl ester and of the FGF-2 with β-d-glucuronyl-α-d-glucosamine anomeric O-methyl ester cocrystal were kindly provided by G. Waksman (20). By using one of these structures as the reference (arbitrarily chosen 1BAS.pdb), the Cα traces of all of the other structures were superimposed to the reference protein. Upon superposition, the position and the interaction of the HLGAGs in the cocrystal structures were compared. For the FGF-1 decasaccharide, three different modes of HLGAG binding have been reported (15) (see legend to Fig. 5B). All three of these FGF-1-decasaccharide complexes were generated for subsequent analysis. The orientation of the side chains of the amino acid residues involved in interaction with HLGAGs were compared after superposition of the Cα trace of the molecules. Similarly, the sulfate positions and orientation of the different HLGAGs were compared. For the analysis of FGF oligomerization, all of the molecules within the unit cell were generated by applying the space group transformations. FGF-FGF associations within the unit cell were examined as described (28).
Figure 5.

(A) Stereo diagram comparing the residues involved in saccharide binding for FGF-1 and FGF-2. The Cα trace of the following FGF cocrystal structures are superimposed: 1AFC, 1AXM, 1BFB, 1BFC, 1BAS, and the cocrystal of FGF-2 with disaccharides and trisaccharides. For sake of clarity the Cα trace of one FGF-2 molecule and one FGF-1 molecule are shown (green). The side-chain atom positions of the amino acid residues that interact with the HLGAG fragments in each of the structures are shown (violet). Note that more than eight of the side-chain conformations at the center of the heparin-binding region are almost identical and are found to interact with the HLGAG in all of the cocrystal structures (FGF-1 and FGF-2). Residues K27 and N102 of FGF-2 (on the left with only one side chain shown) are observed to interact only in the FGF-2 hexasaccharide cocrystal and have been identified as the secondary heparin binding site (21). Residues R116 and R119 of FGF-1 (on the right with side chain from only one structure shown) represent the additional FGF-1 residues identified to interact with SOS. Notably, the side chains of both FGF-1 and FGF-2 that interact with the cognate HLGAG fragments are topologically very similar. (B) Position of sulfates in bound to FGF-1 or FGF-2 in the cocrystal structures. The heparin-binding region of FGF-2 is represented as a Connolly surface (blue). Only the sulfate groups of the ligands are compared, and the HLGAG sugar atoms are not shown. The following ligands are shown: SOS (dark red), hexasaccharide (yellow), tetrasaccharide (green), decasaccharide 1 (light red, see below), decasaccharide 2 (orange, see below), and decasaccharide 3 (pink, see below). DiGabriele et al. (15) report three different modes of HLGAG decasaccharide binding to FGF-1, one in which the decasaccharide is bound with reversed polarity (decasaccharide 2) compared with the other (decasaccharide 1) and a third where the decasaccaharide is shifted by about two saccharide units (decasaccaharide 3). Notice that the position of two of the sulfates in all of these structures are identical (shown inside green boxes). For the box on the left, all of the sulfates (except SOS and decasaccaharides 2 and 3) are linked to the 2-O of the iduronate, whereas the decasaccaharide 2 sulfate belongs to the N position of the glucosamine and the decasaccaharide 3 sulfate belongs to the 6-O position of the glucosamine. Similarly for the box on the right, all of the sulfates except SOS, decasaccaharide 2, and decasaccaharide 3 belong to the N sulfate of the glucosamine whereas the decasaccaharide 2 and decasaccaharide 3 sulfates are from the 2-O position of the iduronate.
RESULTS
MALDI-MS Studies.
We analyzed the formation of FGF-HLGAG complexes with two saccharides by using MALDI-MS. Specifically, we chose to use the biologically active decasaccharide as it is known to potentiate the activity of both FGF-1 and FGF-2 in vitro (11, 15) and it recently was used for the FGF-1-decasaccharide cocrystal structure (15). Also used for comparison was the biologically inactive hexasaccharide. Like the decasaccharide, the disaccharide unit of the hexasaccharide is trisulfated, making the two saccharides chemically equivalent except for length. As a baseline for the complexation studies and to determine the sample preparation conditions that would maximize detection of both FGF-1 and FGF-2, these proteins were ionized in the absence of HLGAGs (Fig. 1). The m/z value for FGF-1 monomer was found to be 15,904 (theoretical m/z = 15,899). Similarly, the m/z value for FGF-2 monomer was found to be 17,083 (theoretical m/z = 17,103).
Figure 1.

(A) MALDI mass spectrum of 1 pmol of FGF-2. The theoretical m/z value of [M+H]+ for FGF-2 is 17,102. Present in the spectrum are dimers, trimers, tetramers, and pentamers. Also present is a peak at m/z 25,640 corresponding to a doubly charged species of the trimer. (B) MALDI mass spectrum of FGF-1. The theoretical m/z value of [M+H]+ for FGF-1 is 15,899. Present in the spectrum is only a weak dimer peak. The instrument was calibrated externally with myoglobin and BSA.
Importantly, for FGF-2 under mild ionization conditions, higher order oligomers—namely, dimers, trimers, tetramers, and pentamers—were observed (Fig. 1A). Several control experiments were run to ensure that the observed oligomers for FGF-2 are not simply a result of protein aggregation. First, higher-order oligomers are seen in the mass spectrum even if FGF-2 is run through a size exclusion column to remove any possible aggregates before placement on the target. Second, the cysteine-to-serine FGF-2 mutant (i.e., the mutant is unable to form disulfide-bonded aggregates) displays the same behavior under these conditions (data not shown). Finally, the spectrum of FGF-1, which shares high sequence homology, showed no oligomer higher than a dimer (Fig. 1B). Together, these results point to a specific protein association among FGF-2 protomers that leads to the formation of oligomers (see Discussion).
Fig. 2 shows the result of addition of decasaccharide to FGF-2. In this spectrum, clearly present are [M+H]+ species of m/z 37,094, 54,186, and 71,232 corresponding to 2:1, 3:1, and 4:1 complexes of FGF-2 and decasaccharide. Further experiments show that the ratio of FGF to decasaccharide can affect the observed complexed species (see crosslinking results). Studying the complexation efficiency between FGF-2 and decasaccharide indicates that the optimum ratio for observing complexed oligomers was at a FGF-2/saccharide ratio of 10:1.
Figure 2.

MALDI mass spectrum of 10 pmol of FGF-2 with 1 pmol of decasaccharide. As in Fig. 1, FGF-2 is found to oligomerize in the absence of HLGAGs. However, upon addition of decasaccharide, oligomer species corresponding to 2:1, 3:1, and 4:1 complexes of FGF-2 with decasaccharide also are formed.
To show that this observed complexation and oligomer formation by FGF-2 is not a nonspecific FGF-HLGAG interaction, similar experiments were completed with an HLGAG-derived hexasaccharide. Although a complex between FGF-2 and hexasaccharide is observed, significant complex is seen only at a protein/saccharide ratio of 20:1 (data not shown). At lower ratios, no complex is observed. Also, unlike with decasaccharide, even when a complex is observed between hexasaccharide and FGF-2, only a signal corresponding to a monomer or dimer plus hexasaccharide is observed.
In contrast to FGF-2, FGF-1 behaves very differently when complexed with decasaccharide. (Fig. 3). Similar to what was noted by DiGabriele et al. (15), at various protein/saccharide ratios a species corresponding to a 2:1 complex of FGF-1/decasaccharide ([M+H]+ calculated m/z 34,685) is observed. As compared with the signal for the FGF-2/decasaccharide complex, a weaker signal was seen for the dimer complex with FGF-1, suggesting that complexation occurs less readily. Similar to what was seen with FGF-2, the protein/saccharide ratio critically affected the amount of complex. Furthermore, again unlike FGF-2, FGF-1 does not form any complex with hexasaccharide (data not shown). Together the FGF-1 results presented here are consistent with what has been reported for FGF-1 in the presence of decasaccharide and hexasaccharide (15).
Figure 3.

MALDI mass spectrum of 20 pmol of FGF-1 with 1 pmol of decasaccharide. FGF-1 forms a 1:1 and a 2:1 complex with decasaccharide; a dimer peak also is seen in the absence of added decasaccharide.
These experiments clearly demonstrate that noncovalent associations between FGF and HLGAG can be observed by using MALDI-MS. Furthermore, the same HLGAG decasaccharide complexes distinctly with FGF-1 (where only 1:1 and 2:1 complexes are observed) compared with FGF-2 (where 1:1, 2:1, 3:1, and 4:1 complexes are readily detected) (Figs. 2 and 3).
To further investigate the differences in FGF-1 and FGF-2 oligomerization mediated by HLGAGs, and to extend the analysis to long chains of HLGAGs, we used classical chemical crosslinking approaches as described below.
FGF-1 and FGF-2 Chemical Crosslinking with BS3.
Chemical crosslinking has been a direct and well-established method to probe for associations between proteins (27, 29, 30). In addition, through accurate quantification of resultant oligomer bands, we can derive equilibrium values for oligomer transitions in both FGF-1 and FGF-2.
For the crosslinking studies, we chose the homobifunctional lysine crosslinking agent BS3 because of its high selectivity, moderate spacer length, aqueous solubility, and wide precedence in the literature (29, 30). In this case, it is important to ensure that analysis of covalent crosslinking is an accurate reflection of specific protein-protein associations and not an artifact of adventitious crosslinking. To this end, extensive control experiments were completed with and without the reducing agent and with a variety of protein/crosslinker ratios to demonstrate minimal nonspecific binding (see Materials and Methods).
Initial experiments were completed by varying the heparin/FGF ratio from 0.01 to 10 with both FGF-1 and FGF-2 (data not shown) to determine the oligomerization behavior of FGF in the presence of heparin. For both FGF-1 and FGF-2 there is a bell-shaped response to varying heparin ratios, with maximal oligomerization at a heparin/FGF ratio of 0.1. This finding is similar to what was reported by Mach et al. (12) for FGF-1-heparin interactions observed by dynamic light scattering.
Fig. 4 shows representative Western blots of FGF-1 and FGF-2 crosslinked with BS3 across a range of protein concentrations at an optimal protein/heparin ratio of 10:1. It can be seen that the amount of oligomer increases with increasing initial FGF concentration in both blots. However, it also can be seen that FGF-1 oligomers above dimers are not observed at any concentration, even with overstaining. This finding stands in contradiction to the prediction of FGF-1 aggregation above dimers by Mach et al. (12). In the FGF-2 blot, oligomers are observed up to tetramers at a concentration of 180 μM.
Figure 4.

Representative blots of (a) FGF-1 or (c) FGF-2 crosslinking reactions. (b) Gel showing the purity of the uncrosslinked monomer of FGF-1 and FGF-2 (left and right, respectively). (a and c) The protein/heparin ratio was held constant at 10:1. Concentrations of FGF-1 in a are 240, 180, 120, 60, and 30 μM, left to right. For FGF-2 in c, the concentrations crosslinked are 30, 60, 120, and 180 μM, left to right. The blot of FGF-1 is overstained to demonstrate the clear lack of higher-order oligomers in the crosslinked products.
FGF-1 and FGF-2 were crosslinked at various protein concentrations (data not shown), and an association constant (Ka) for this data was calculated. Assuming an isodesmic model, the data can be used to determine C0, C1, and the Ka for oligomerization reaction of FGF-1 and FGF-2. In the case of FGF-1, the fitted Ka is 0.63 ± 0.12 mM−1; for FGF-2 the fitted Ka is 10.6 ± 1.3 mM−1. Thus, FGF-2 forms oligomers with an affinity of roughly 20 times that of FGF-1 in the presence of heparin under the conditions measured here. If an isodesmic model is not assumed for FGF-2 but instead the equilibrium values are calculated individually, they do not shift significantly (data not shown). Furthermore, a plot of the logarithm of mole fraction of polymer vs. the polymer size yields a straight line at each concentration of FGF-2 crosslinked, indicative of the formation of a linear species via interaction at a single, repeating interface (27). This result is consistent with orientation of FGF-2 oligomers along an association interface as found in the MALDI-MS experiments (see also below).
Thus, MALDI-MS and chemical crosslinking data of FGF-2 and FGF-1 under identical conditions taken together clearly demonstrate very distinct modes and degrees of oligomerization for the two proteins. To determine a possible mechanism by which HLGAGs mediate different effects on FGF-1 and FGF-2, we sought to investigate HLGAG binding to FGF-1 and FGF-2 by a rigorous analysis of the available crystal structures.
FGF-1 and FGF-2 Crystal Structures and FGF Oligomerization.
As stated earlier, several independent crystal structures of FGFs (FGF-1 and FGF-2) have been solved, both unliganded and liganded to HLGAG oligosaccharides (7, 15). Even though chemically and biologically different HLGAG oligosaccharides were used for the cocrystallization of FGF, the FGF amino acids involved in HLGAG binding in these cases are almost identical (Fig. 5A). In FGF-2, the surface (amino acid residues and their side-chain topological orientation) formed at the heparin binding region in the FGF-2-tetra, FGF-2-hexa, and FGF-2-tri (and FGF-2-di) structures are very similar (rms deviation of 1.5 Å) (Fig. 5A). Also, the amino acids corresponding to the heparin binding surface of FGF-2 are conserved in FGF-1. It is interesting to note that both the charge properties and the topological orientation of these side chains are virtually identical for FGF-1 and FGF-2 (Fig. 5A). This finding suggests that both FGF-1 and FGF-2 adapt a similar surface at the heparin binding site and that saccharides of different chemical composition and sequence are able to fit into this pocket (data not shown).
In addition, despite conformational flexibility, all of the saccharides in the cocrystal structures have hydrogen bond donor atoms that occupy almost identical spatial positions. As a validation of this observation, unliganded FGF-2 and FGF-1 crystal structures crystallized with sulfates or selenium (18, 19, 22) show a sulfate ion at exactly the same position as occupied by the sulfates of HLGAG fragments in the cocrystals (Fig. 5B). Thus, despite differences in the saccharide sequence, the protein surface at the heparin binding site in FGF is able to recognize a unique pattern of hydrogen bond donor groups, specifically sulfates regardless of whether they are N sulfates, 2-O sulfates, or 6-O sulfates.
The above observations clearly suggest that individual FGF-1 and FGF-2 molecules do not seem to discriminate between different backbone sequences provided the sulfate groups are presented in the correct spatial orientation. However, this finding raises the puzzling question of how HLGAGs seem to bind essentially in the same manner to individual FGF-1 and FGF-2 molecules yet play very different roles in modulating FGF-1 or FGF-2. We therefore reexamined the crystal structures in light of the above observation.
In all of the HLGAG-FGF-2 cocrystal structures, FGF-2 has been reported to be a monomer in the asymmetric unit, thereby raising the question of how FGF-2 molecules are assembled on a HLGAG chain leading to FGF-2 oligomerization (7). Crystal structure analysis leading to inferences of possible modes of FGF-2-HLGAG interactions leading to FGF-2 dimerization appears to be the only way to reconcile the x-ray crystal data with the significant biochemical and biophysical observations of FGF dimerization/oligomerization in the presence of HLGAGs and HLGAG analogs (28).
In an earlier study (28), we investigated all possible modes of FGF-2 oligomerization that could be mediated by HLGAGs, through an exhaustive analysis of HLGAG liganded as well as unliganded FGF-2 crystal structures. Our analysis revealed a characteristic FGF-2 molecular association involving translationally related (side-by-side) FGF-2 molecules in all of the crystal structures. This conserved association is such that the internal 3-fold axis of the molecule is approximately parallel, and HLGAG chain can be easily accommodated to bridge neighboring FGF-2 molecules, thereby facilitating FGF-2 oligomerization.
On the other hand, analysis of FGF-1 crystal structures indicates that the translationally related conserved molecular association observed for FGF-2 is not observed in any of the FGF-1 crystal structures. Interestingly, a comparison between the FGF-1 and FGF-2 amino acid corresponding to the conserved interface leading to the characteristic molecular association shows that in the first site all of the amino acids except residue R60 (of FGF-2) are conserved. However, residues from the second site of the interface are not conserved for FGF-1 when compared with FGF-2. Mutations of FGF-2 at this site with the amino acids corresponding to the FGF-1 site results in loss of activity with saccharides but not with full-length heparin, suggesting the biological relevance of this site and presumably the characteristic interface in FGF-2 activity (8). It is important to point out that a rotationally related molecular association about the conserved first site is observed in the FGF-1 crystal structure, and this symmetrical FGF-1 dimer would require an inversion of polarity of HLGAG binding site between the two side-by-side FGF-1 dimers. However, unlike FGF-2, the recently solved cocrystal structure of FGF-1 with the decasaccharide shows a HLGAG-mediated FGF-1 dimer with no protein-protein interface or contacts (15). In this dimer, two molecules of FGF-1 bind on opposite sides of the heparin, with a quasi-dyad perpendicular to that heparin helix axis relating the two FGF-1 molecules. In addition, the crystal structure reveals flexibility in relative position of the two FGF-1 molecules about the heparin chain. It is important to point out that such FGF-HLGAG interactions, namely with no protein contacts, thus far are not observed for FGF-2.
The above taken together lead us to suggest that, for individual FGF-1 and FGF-2 molecules, although the molecular interactions between protein and the saccharide are essentially identical, the mode of molecular association leading to FGF oligomerization is distinct for the two proteins. Moreover, we find the protein molecular associations to be distinct for FGF-1 when compared with FGF-2, in both the HLGAG-liganded as well as the unliganded structures. For proteins with such high sequence similarities, it is less likely that crystal packing forces may be responsible for the observed distinct similarities and differences in packing or molecular associations of FGFs both in the presence and in the absence of HLGAG (8).
DISCUSSION
Through a combination of MALDI-MS, chemical crosslinking and crystal structure analysis, we have completed a direct comparison of the oligomerization characteristics of FGF-1 and FGF-2. We have shown that differences in the oligomerization profiles of the two proteins cannot be simply attributed to differences in extent of oligomerization or to differential HLGAG binding, but rather must be attributed to FGF-1 and FGF-2 differing fundamentally in their modes of oligomerization.
MALDI-MS, Chemical Crosslinking, and Crystal Structure Analysis.
To probe FGF-HLGAG interactions at picomole levels of protein, we have developed a MALDI-MS technique. A similar technique was used to probe the interaction of decasaccharide with the basic protein angiogenin and was found to lead to efficient detection of protein/saccharide complexes, including oligomer species (26). By using a modification of this initial MALDI-MS procedure, we have shown that FGF-2, up to tetramers, can complex with biologically active decasaccharide. Importantly, these interactions were found to be highly specific and not an artifact of MALDI-MS sample preparation. It should be kept in mind that even after bringing the sample solution to dryness the protein molecules are still highly diluted by the large excess of matrix molecules (matrix isolation).
As opposed to FGF-2, FGF-1, in the presence of this same decasaccharide, formed only dimers, consistent with previous observations (12, 15). Also consistent with previous observations is that no FGF-1 dimers are seen when hexasaccharide is added to FGF-1, whereas FGF-2 forms dimers (15). Furthermore, these observations were supported by crosslinking experiments of FGF-1 and FGF-2 in the presence of full-length heparin. Crosslinking studies were completed not only to corroborate the MALDI-MS studies but also to quantify the oligomerization processes in these cases. Together these results demonstrate that FGF-1 and FGF-2 possess a different propensity to oligomerize in the presence of HLGAG fragments.
One possible explanation for these differences is that FGF-1 and FGF-2 interact with HLGAG fragments in a very different fashion. However, upon extensive analysis of the crystal structure information for both FGF-1 and FGF-2, we find that essentially the same molecular interactions occur when individual FGF-1 molecules bind to HLGAGs as when individual FGF-2 molecules bind to them. An extensive analysis of all available crystal structures indicates that FGF-1 and FGF-2, upon binding to HLGAGs, oligomerize by a very different process, explaining the observed differences in the MALDI-MS profiles and the chemical crosslinking data.
Fig. 1, which depicts the oligomerization of FGF-1 and FGF-2 in the absence of HLGAGs, supports the above model for FGF-1 and FGF-2 oligomerization processes. Consistent with the model that FGF-2 possesses a protein association involving a translationally related side-by-side interface, the FGF-2 mass spectrum shows the formation of sequential oligomers up to pentamers. Similar oligomer formation in the absence of HLGAGs is seen for FGF-2 in crosslinking experiments (data not shown). As would be expected for a translationally related sequential oligomer, the amount of oligomer species falls off at a regular interval. Unlike FGF-2, no higher-order oligomers were seen for FGF-1 consistent with the observation that FGF-1 lacks the conserved protein association interface.
The pattern of FGF oligomerization in the presence and absence of HLGAGs appear to be the same. Only dimers are formed for FGF-1, while dimer and multimers are formed for FGF-2. Analysis of the crystal data of FGF-1 and FGF-2 in the HLGAG complexed and uncomplexed form provide an explanation for the difference in the FGF-1 and FGF-2 oligomerization. It remains to be seen if there are other possible explanations for the data.
FGF-2 and FGF-1 Oligomerization.
The data presented in this study are consistent with our earlier investigations into the very diverse crystal forms of apo-FGF-2 structures and the FGF-2-trisaccahride/disaccharide cocrystal structures, which led to an important observation that in all of these structures a characteristic protein-protein interacting interface for FGF-2 was observed (28). These observations, along with others, led us to rationalize that FGF-2 preferentially self-associates in a head-to-tail side-by-side or cis fashion, such that the self-associated sequential FGF-2 oligomers assemble along a HLGAG chain.
Thus, in the case of FGF-2, it can be reasoned that protein-protein interactions facilitate the oligomerization of the protein with HLGAGs that are long enough (octa or longer) to facilitate a cis dimer. However, with fragments shorter than octasaccharides (i.e., hexasaccharides), where the proposed protein association cannot occur, a different dimer type must form. At a very high protein/hexasaccharide ratio, FGF-2 forms a presumably less stable trans-dimer with no protein-protein contact, similar to the trans dimer proposed by Moy et al. (11) for FGF-2/tetrasaccharide. On the other hand, the presence of sequential FGF-2 oligomers in the absence of HLGAGs, coupled with the formation of trimers, as well as dimers and tetramers in the presence of HLGAGs, supports the proposition of a translationally related cis oligomer as proposed by Venkataraman et al. (28). However, the data presented in this study do not support the model for decasaccharide-mediated FGF-2 oligomerization proposed by Moy (11). In Moy et al., model decasaccharide is hypothesized to induce two head-to-head cis dimers in a trans configuration. Thus, four symmetry-related FGF-2 protomers form a tetramer, precluding the formation of a trimer species and dismissing the presence of the characteristic FGF-2 association interface. It is plausible that because we see FGF-2 self-association at very low protein concentrations (Fig. 1A), perhaps FGF-2 already exists self-associated at high protein concentrations necessary for NMR experiments, thereby complicating the interpretation of the NMR data (11).
Unlike for FGF-2, FGF-1 decasaccharide interaction does not lead to the formation of the expected higher-order oligomers, but only the formation of a trans dimer with no protein-protein contact. FGF-1 does not form dimers with tetrasaccharides or hexasaccharides, whereas, tetrasaccharides and hexasaccharides induce FGF-2 dimerization. Essentially, it appears that FGF-1 and FGF-2 differ from each other in terms of the topological position of the FGF molecule along the HLGAG chain and that of protein contact.
HLGAG modulation of the inherent physical-chemical difference between FGF-1 and FGF-2, leading to distinct protein oligomerization, may be responsible for their unique biological activities (31–34).
Acknowledgments
We are grateful to Dr. Judy Abraham (Scios) and Dr. T. Arakawa (Amgen) for the generous supply of recombinant FGF-2 and mutant FGF-2 and recombinant FGF-1, respectively. This work was supported by National Institutes of Health Training Grant ES 07020 (to J.C.D.), Whitaker Foundation (Z.S.), and Sloan-Cabot Foundation, Massachusetts Institute of Technology.
ABBREVIATIONS
- FGF
fibroblast growth factor
- HLGAG
heparin-like glycosaminoglycan
- SOS
sucrose octasulfate
- BS3
bis[sulfosuccinimidyl]suberate
- MALDI-MS
matrix-assisted laser desorption ionization mass spectrometry
Footnotes
The abbreviations used to describe the decasaccharide and hexasaccharide are: I, α-l-iduronic acid; H, d-glucosamine; ΔU, Δ4,5-uronic acid; 2S, 2-O sulfation; 6S, 6-O sulfation; NS, N sulfation.
References
- 1.Baird A, Bohlen P. Peptide Growth Factors and Their Receptors I. New York: Springer; 1990. [Google Scholar]
- 2.Conrad H E. Heparin Binding Proteins. San Diego: Academic; 1998. [Google Scholar]
- 3.Basilico C, Moscatelli G. Adv Cancer Res. 1992;59:115–165. doi: 10.1016/s0065-230x(08)60305-x. [DOI] [PubMed] [Google Scholar]
- 4.Naski M C, Ornitz D M. Front Biosci. 1998;3:D781–D794. doi: 10.2741/a321. [DOI] [PubMed] [Google Scholar]
- 5.Schlessinger J, Lax I, Lemon M. Cell. 1995;83:357–360. doi: 10.1016/0092-8674(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 6.Nugent M A, Edelman E R. Biochemistry. 1992;31:8876–8883. doi: 10.1021/bi00152a026. [DOI] [PubMed] [Google Scholar]
- 7.Faham S, Linhardt R J, Rees D C. Curr Opin Struct Biol. 1998;8:578–586. doi: 10.1016/s0959-440x(98)80147-4. [DOI] [PubMed] [Google Scholar]
- 8.Waksman G, Herr A B. Nat Struct Biol. 1998;5:527–530. doi: 10.1038/778. [DOI] [PubMed] [Google Scholar]
- 9.Ornitz D M, Yayon A, Falnagan J G, Svahn C M, Levi E, Leder P. Mol Cell Biol. 1992;12:240–247. doi: 10.1128/mcb.12.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spivak-Kroizman T, Lemmon M S, Dikic I, Ladbury J E, Pinchasi D, Huang J, Jaye M, Crumley G, Schlessinger J, Lax I. Cell. 1994;79:1015–1024. doi: 10.1016/0092-8674(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 11.Moy F J, Safran M, Seddon A P, Kitchen D, Bohlen P, Aviezer D, Yayon A, Powers R. Biochemistry. 1997;36:4782–4791. doi: 10.1021/bi9625455. [DOI] [PubMed] [Google Scholar]
- 12.Mach H, Volkin D B, Burke C J, Middaugh C R, Linhardt R J, Fromm J R, Loganathan D, Mattson L. Biochemistry. 1993;32:5480–5489. doi: 10.1021/bi00071a026. [DOI] [PubMed] [Google Scholar]
- 13.Herr A B, Ornitz D M, Sasisekharan R, Venkataraman G, Waksman G. J Biol Chem. 1997;272:16382–16389. doi: 10.1074/jbc.272.26.16382. [DOI] [PubMed] [Google Scholar]
- 14.Pantoliano M W, Horlick R A, Springer B A, Van Dyk D E, Tobery T, Wetmore D R, Lear J D, Nahapetian A T, Bradley J D, Sisk W P. Biochemistry. 1994;33:10229–10248. doi: 10.1021/bi00200a003. [DOI] [PubMed] [Google Scholar]
- 15.DiGabriele A D, Lax I, Chen D I, Svahn C M, Jaye M, Schlessinger J, Hendrickson W A. Nature (London) 1998;393:812–817. doi: 10.1038/31741. [DOI] [PubMed] [Google Scholar]
- 16.Zhu X, Komiya H, Chirino A, Faham S, Fox G M, Arakawa T, Hsu B T, Rees D C. Science. 1991;251:90–93. doi: 10.1126/science.1702556. [DOI] [PubMed] [Google Scholar]
- 17.Eriksson A E, Cousens L S, Matthews B W. Proc Natl Acad Sci USA. 1991;88:3441–3445. doi: 10.1073/pnas.88.8.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Cousens L S, Barr P J, Sprang S R. Proc Natl Acad Sci USA. 1991;88:3446–3450. doi: 10.1073/pnas.88.8.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eriksson A E, Cousens L S, Matthews B W. Protein Sci. 1993;2:1274–1284. doi: 10.1002/pro.5560020810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ornitz D M, Herr A B, Nilsson M, Westman J, Svahn C-M, Waksman G. Science. 1995;268:432–436. doi: 10.1126/science.7536345. [DOI] [PubMed] [Google Scholar]
- 21.Faham S, Hileman R E, Fromm J R, Lindhardt R J, Rees D C. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- 22.Blaber M, DiSalvo J, Thomas K A. Biochemistry. 1996;35:2086–2094. doi: 10.1021/bi9521755. [DOI] [PubMed] [Google Scholar]
- 23.Zhu X, Hsu B T, Rees D C. Structure. 1993;1:27–34. doi: 10.1016/0969-2126(93)90006-3. [DOI] [PubMed] [Google Scholar]
- 24.Xiang F, Beavis R C. Rapid Commun Mass Spectrom. 1994;8:199–204. [Google Scholar]
- 25.Juhasz P, Biemann K. Proc Natl Acad Sci USA. 1994;91:4333–4337. doi: 10.1073/pnas.91.10.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Juhasz P, Biemann K. Carbohydr Res. 1994;270:131–147. doi: 10.1016/0008-6215(94)00012-5. [DOI] [PubMed] [Google Scholar]
- 27.Carter G J, van Holde K. Biochemistry. 1998;37:12477–12488. doi: 10.1021/bi980716v. [DOI] [PubMed] [Google Scholar]
- 28.Venkataraman G, Sasisekharan V, Herr A B, Ornitz D M, Waksman G, Cooney C L, Langer R, Sasisekharan R. Proc Natl Acad Sci USA. 1996;93:845–850. doi: 10.1073/pnas.93.2.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Handler C G, Eisenberg R J, Cohen H. J Virol. 1996;70:6067–6075. doi: 10.1128/jvi.70.9.6067-6070.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf B B, Gonias L. Biochemistry. 1994;33:11270–11277. doi: 10.1021/bi00203a024. [DOI] [PubMed] [Google Scholar]
- 31.Ornitz D M, Xu J, Colvin J S, McEwen D G, MacArthur C A, Coulier F, Gao G, Goldfarb M. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 32.Nurcombe V, Ford M D, Wildschut J A, Barlett P F. Science. 1993;260:103–106. doi: 10.1126/science.7682010. [DOI] [PubMed] [Google Scholar]
- 33.Brickman Y, Ford M D, Gallagher J T, Nurcombe V, Barlett P F, Turnbull J E. J Biol Chem. 1998;273:4350–4359. doi: 10.1074/jbc.273.8.4350. [DOI] [PubMed] [Google Scholar]
- 34.Sasisekharan R, Ernst S, Venkataraman G. Angiogenesis. 1997;1:45–54. doi: 10.1023/A:1018318914258. [DOI] [PubMed] [Google Scholar]