

Abstract
Background
Peanut allergy is the most common food-related cause of lethal anaphylaxis and, unlike other food allergies, typically persists into adulthood. Resistance to digestion and dendritic cell activation by the major peanut allergen, Ara h1, are reported to contribute to its allergenicity.
Objective
Evaluate whether peanut molecules may also promote anaphylaxis through an innate immune mechanism.
Methods
Naïve mice were treated with a β-adrenergic receptor antagonist and long-acting IL-4 to increase sensitivity to vasoactive mediators and injected with peanut extract (PE). Shock was detected and quantified by rectal thermometry. Gene-deficient mice and specific antagonists were used to determine the roles of specific cell types, complement, Fc receptors, and vasoactive mediators in shock pathogenesis.
Results
1) PE induces dose-dependent shock; 2) PE activates complement in vivo in mice and in vitro in mice and humans; 3) C3a, and, to a lesser extent, stimulatory immunoglobulin (Ig) receptors contribute to PE-induced shock; 4) PE-induced shock depends more on macrophages and basophils than on mast cells; 5) platelet activating factor and, to a lesser extent, histamine contribute to PE-induced shock; 6) PE induces shock in the absence of the adaptive immune system; 7) LPS contamination is not responsible for PE-induced shock; 8) PE and IgE-mediated mast cell degranulation synergistically induce shock; and 9) Tree nuts have similar effects to PE; skim milk and egg white do not.
Conclusion
Peanuts can contribute to shock by causing production of C3a, which stimulates macrophages, basophils and mast cells to produce PAF and histamine.
Keywords: peanut, C3a, complement, anaphylaxis, shock, macrophages, mast cells, basophils, PAF, histamine
Introduction
Peanut allergy affects ~1% of Americans1 and is the leading cause of food allergy-related death in the United States2, 3. Unlike most food allergies, which appear in children but resolve with age, childhood peanut allergy usually persists into adulthood4 and can reappear in individuals who have become peanut tolerant2, 4, 5. These observations suggest that peanuts may have special characteristics that increase their allergenicity.
Previous studies suggest that more than one peanut characteristic contributes to allergenicity. Roasting, which is used to process most peanuts consumed in the United States, increases peanut allergenicity6. Peanuts contain relatively large quantities of at least 8 proteins that express strong B and T cell epitopes and elicit IgE antibody (Ab) responses7, 8. Resistance of peanut allergens to digestion7 increases the likelihood that sufficient allergen will be absorbed systemically to induce an IgE antibody response and trigger IgE-mediated anaphylaxis. Ara h1, a strong peanut allergen, activates dendritic cell signaling pathways that induce these cells to present antigens in a way that promotes a pro-allergic Th2 response by binding to ICAM-1-grabbing non-integrin molecule (CD209)9.
The ability of peanuts to induce an IgE antibody response does not depend entirely on the 8 major peanut allergens, however, because purified major peanut allergens are considerably less immunogenic than a crude peanut extract (PE). This suggests that other, “matrix” components of peanuts may have adjuvant properties that promote a Th2 immune response 10. These considerations suggest that PE has proinflammatory effects that mediate its adjuvant activity and raise the possibility that such effects might even contribute directly to peanut-induced shock. Studies performed to investigate this possibility demonstrate that PE indeed contributes to shock induction by Ig-independent activation of the complement system with production of the anaphylatoxin C3a.
Materials and Methods
Allergen extracts
Commercial roasted peanuts were ground in a blender (Scovill) in 0.1M pH 9.0 ammonium bicarbonate. Insoluble material was removed by centrifugation after 4 hr incubation at room temperature. The supernatant was dialyzed overnight at 4°C against 0.15M NaCl, then fractionated by ammonium sulfate precipitation with retention of the fraction that was soluble in 25% saturated ammonium sulfate and insoluble in 80% saturated ammonium sulfate. This fraction was resuspended in water and dialyzed 4 times against 0.15 M NaCl. Refrigerated PE solutions were brought to room temperature to dissolve cryoprecipitates prior to injection into mice.
Extracts were prepared through a similar process from commercial roasted almonds, cashews and walnuts. Egg white was aspirated directly from a fresh chicken egg. Fresh commercial skim milk was centrifuged at 10,000 RPM to remove residual fat and particulate matter and filtered through a 0.45 μ2 filter prior to used.
LPS removal
LPS was removed from PE with an Endotrap red purification system (Profos AG) according to manufacturer’s protocol. More than 99 % of the LPS in PE preparations was eliminated by this treatment, as determined by an LAL assay with the LAL QCL-1000 kit from Cambrex Bio Sciences. Purified LPS was a gift of Stephanie Vogel.
Mice
BALB/c wild-type and FcRγ-deficient mice11 were purchased from Taconic; C57BL/6 μMT12, mixed background C3-13, BALB/c C5aR-14, BALB/c C3aR-15 and BALB/c Rag1-deficient16 and BALB/c SCID17 mice were purchased from the Jackson Laboratory. C57BL/6 toll-like receptor (TLR)4-18, TLR2-19 and MyD88-deficient mice20 were a gift of Christopher Karp (Cincinnati Children’s Hospital Medical Center, CCHMC). BALB/c FABPi-IL-9 transgenic mice21 were bred in the animal facility at CCHMC. Mixed background FcRγ/C3 double-deficient mice were generated by breeding C3-deficient mice with FcRγ-deficient mice. The PCR primers 5′-ACC CTA CTC TAC TGT CGA CTC AAG; 5′-CTC GTG CTT TAC GGT ATC GCC and 5′-CTC ACG GCT GGC TAT AGC TGC CTT were used to detect homozygous FcRγ-deficient mice. A genotyping protocol recommended by Jackson Laboratory’s on-line site was used to detect homozygous C3-deficient mice. Four 8–12 week old mice were used per group, except where noted otherwise. All experiments were performed with prior approval by the CCHMC Institutional Animal Care and Use Committee.
ELISAs
Mouse mast cell protease 1 (MMCP-1) levels were measured in serum obtained from blood drawn 2 hours after challenge (unless other specified) with an ELISA kit purchased from Moredun Scientific. Plasma histamine levels were determined with an ELISA kit purchased from IBL Hamburg. The amount of C3a protein in mouse and human plasma was determined by ELISA. For the mouse C3a assay, microtiter plate wells were coated with purified rat anti-mouse C3a monoclonal antibody (mAb) (BD-Pharmingen, catalog number 558250), followed sequentially by biotin-labeled rat anti-mouse C3a antibody (BD-Pharmingen, catalog number 558251), HRP-streptavidin and SuperSignal ELISA substrate (Pierce Biotechnology). Purified native mouse C3a (BD-Pharmingen, catalog number 558618) was utilized as a standard. For the human C3a assay, microtiter plate wells were coated with purified mouse anti-human C3a monoclonal antibody (Affinity BioReagents, catalog number GAU 013-16-02), followed by biotin-labeled mouse anti-human C3a monoclonal antibody (Affinity BioReagents, catalog number GAU 017-01-02B), HRP-streptavidin and SuperSignal ELISA substrate (Pierce Biotechnology). Purified native human C3a (Binding Site) was used as a standard.
Cell and mediator antagonists
Histamine, PAF, and macrophage function were inhibited as described22–24. In some experiments, mice were injected with 0.5 mg/kg of the C5a receptor (R) antagonist A8Δ71–7325 20 min before challenge or with 30 mg/kg of the C3aR antagonist SB29015726 (EMD Chemicals, Inc) 3 hrs before challenge. Mice were depleted of mast cells by injecting 1 mg of anti–c-kit Ab (ACK2)27, i.v., then i.p., every other day for 14 days. Control mice received equal doses of an isotype-matched control Ab (J1.2).
Complement activation
Mouse or human plasma was prepared by centrifuging blood that had been collected in EDTA-coated tubes and was immediately used or frozen until use. To test for activation of complement, plasma (50 μl) was mixed with 2 μl of 1M CaCl2, 2 μl of 1M MgCl2 and 5 μl of peanut extract, then incubated for 10 min at 37 °C. The reaction was stopped by adding 11 μl of 0.7M EDTA, pH 8.0 and cooling to 0°C.
Antibody treatment
IgE-mediated anaphylaxis was induced by injecting mice i.v. with 80 μg of EM-95 (rat IgG2a anti–mouse IgE mAb)28.
Characterization of shock
Severity of shock was assessed by rectal thermometry 22, 29. IL-4C. IL-4/anti-IL-4 mAb complexes (IL-4C) were prepared by mixing IL-4 and BVD4-1D11 anti-IL-4 mAb at a 2:1 molar ratio. IL-4C slowly dissociate in vivo to release free IL-430. These complexes are unable to activate complement, bind more avidly than free IgG to FcγRs, or interact simultaneously with FcγRs and cytokine receptors because they contain a single IgG antibody molecule and the anti-IL-4 mAb used, BVD4-1D11, blocks IL-4 binding to IL-4Rα30.
Induction of shock with PE
Mice were pre-treated for 24 hrs with IL-4C (1 μg IL-4 plus 5 μg BVD4-1D11 rat IgG2b anti-mouse IL-4 mAb/mouse) and for 20 min with propranolol (35 μg/mouse). Both propranolol and IL-4C were injected i.v. Mice were challenged i.v. with PE (250 μg/mouse unless other specified).
Oral inoculation
This was performed with intragastric (i.g.) feeding needles (Thermo Fisher Scientific; cat. 01–290-2B). Mice were deprived of food for 3–4 hrs before each i.g. challenge.
Basophil depletion
Mice were injected how i.v. with 35 μg of Ba103, a non-activating, depleting mAb to the basophil-specific antigen, CD200R331,32. Preliminary experiments demonstrated ~80 % depletion of splenic and bone marrow basophils, which were identified as FcεRI+c-kit− cells33.
Worm inoculation
BALB/c mice were inoculated subcutaneously with 500 Nippostrongylus brasiliensis third stage infectious larvae (L3)34.
Statistics
Data were analyzed for statistical significance with the ANOVA and Fisher’s protected least significant difference tests, using Statview. p values < 0.05 were considered statistically significant.
Results
PE induces shock in IL-4C/propranalol-pre-treated mice
Because peanuts are responsible for such a large percentage of severe anaphylaxis in most developed countries, we hypothesized that they may induce shock through an innate immune mechanism in addition to the classical IgE/mast cell/vasoactive mediator pathway. To investigate this possibility, we evaluated whether injecting non-immune mice with water soluble PE would cause shock (detected as hypothermia22). When PE-treated mice failed to develop hypothermia (not shown), we increased the sensitivity of our model by pre-treating mice with a long-acting form of IL-4 (IL-4C), which decreases the amounts of vasoactive mediators required to induce vascular leak that causes hypovolemic hypotension29, 35, and with the β-adrenergic antagonist propranalol, which can exacerbate anaphylactic shock36. Mice pre-treated in this way developed severe, exquisitely dose-dependent shock in response to i.v. PE injection (Figure 1A). Although different batches of PE varied in their potency, the results shown in Figure 1A are typical, with considerable hypothermia induced by 200, but not 100 μg of PE and lethal shock induced by 250 μg. In contrast to i.v. injection of PE, ingestion of PE failed to induce shock in otherwise healthy mice, even after sensitization with IL-4C and propranalol. Because this suggested that PE must be absorbed systemically to induce shock, we evaluated whether shock is induced by PE ingestion in mice in which increased intestinal permeability has been induced by infection with the intestinal worm parasite Nippostrongylus brasiliensis37. Significant hypothermia was induced in these mice by oral inoculation of PE (Figure 1B), but not by oral saline inoculation (not shown).
Figure 1. PE induces shock in IL-4/propranalol-pre-treated mice.
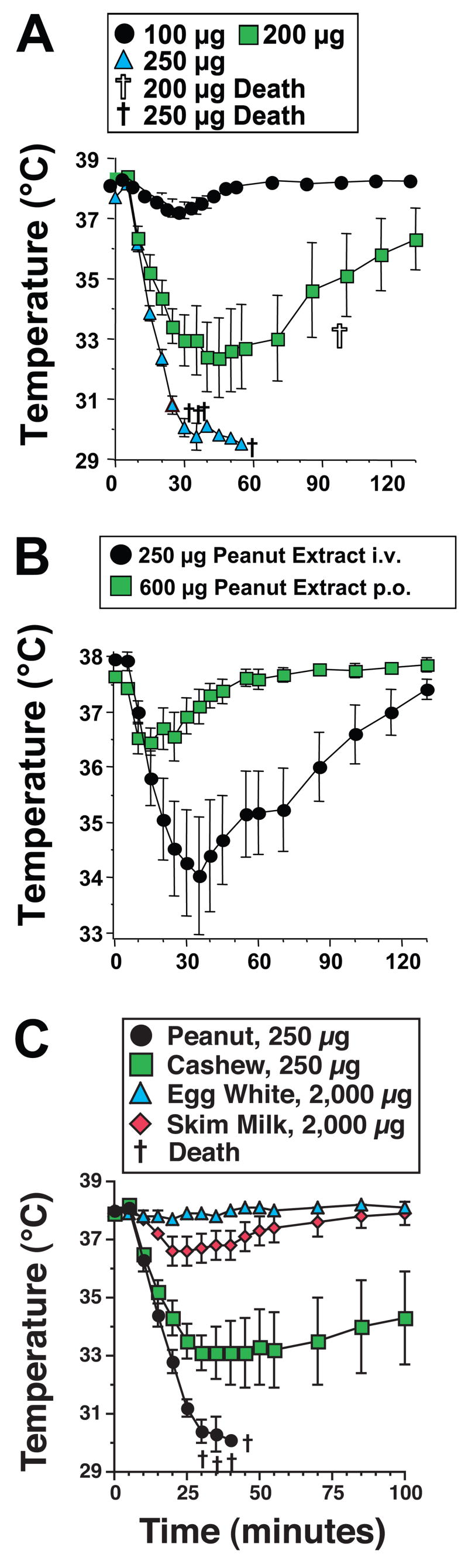
A. BALB/c mice were pre-treated with IL-4C and propranolol, then challenged i.v. with the doses of PE shown and followed for 2 hr by rectal thermometry. B. BALB/c mice were inoculated subcutaneously on day 0 with 500 Nippostrongylus brasiliensis third stage infectious larvae. These mice were injected with 35 μg of propranalol on day 9 and 23 min later were injected i.v. with 250 μg of PE or were administered 600 μg of PE by oral gavage. C. BALB/c mice were pre-treated with IL-4C and propranolol, then challenged i.v. with the doses of PE, cashew extract, egg white, or skim milk shown and followed for 100 min. by rectal thermometry.
To determine whether other allergens might share the effects of PE, mice pre-treated with IL-4C and propranalol were challenged with fresh egg white or skim milk or water soluble extracts of cashews, walnuts, and almonds. Shock was induced by 250 μg of PE and, to a lesser extent by 250 μg of cashew, walnut and almond extracts (Figure 1C and data not shown). In contrast, 2,000 μg of egg white protein failed to induce shock and 2,000 μg of skim milk protein had only a trivial effect (Figure 1C).
PE-induced shock is TLR-independent
Because our preparations of PE contain LPS, we were concerned that contamination with LPS or other TLR ligands might be responsible for PE-induced shock. Three different approaches were used to investigate this possibility; all make it unlikely. First, PE injection induced severe hypothermia in MyD88-, TLR2-, and TLR4-deficient mice, despite the failure of LPS and/or bacterial lipoprotein to induce shock in these mice18, 19, 38 (Figure 2A). Secondly, removal of >99% of LPS from PE did not alter its ability to induce hypothermia (Figure 2B). Thirdly, shock induced by injecting mice with large quantities of LPS developed much more slowly than PE-induced shock (Figure 2C). Thus, shock induction by PE is not a result of LPS contamination.
Figure 2. PE-induced shock is MyD88-, TLR2-, TLR4- and LPS-independent.

A. C57BL/6 wild-type and MyD88-deficient mice (left panel) or wild-type, TLR2- and TLR4-deficient mice (right panel) were pre-treated with IL-4C and propranalol and challenged with 250 μg of PE, then followed by rectal thermometry for development of shock. B. BALB/c mice pre-treated with IL-4C and propranolol were injected i.v. with 300 μg of PE that contained 500 ng of LPS or with 300 μg of LPS-depleted PE, that contained 1.5 ng of LPS and followed for development of shock. C. BALB/c mice pre-treated with IL-4C and propranolol were injected i.v. with 10 μg or 50 μg of S. typhimurium LPS or 300 μg of PE and followed for development of shock.
PE induces shock in the absence of Ab
PE might induce shock by reacting with natural IgG or IgE Abs. PE was injected into Rag1-deficient mice, which lack B and T cells, or μMT mice, which lack B cells, to investigate this possibility. Results demonstrate that PE induces shock in both Ab-deficient mouse strains (Figure 3). SCID mice also develop shock in a similar manner (data not shown). Thus, PE induces shock in the absence of the adaptive immune system.
Figure 3. PE induces shock in the absence of B and T cells.
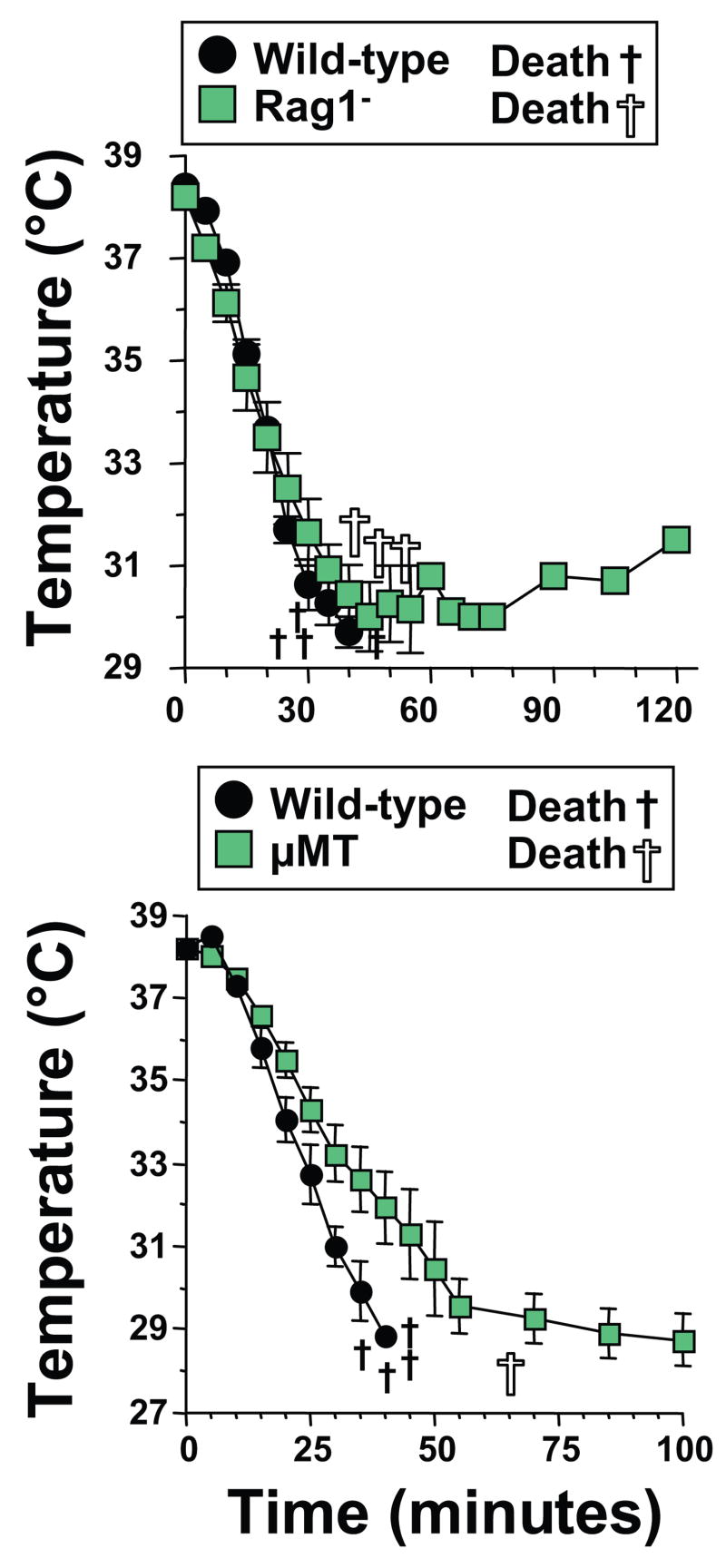
Age-, sex-, and genetic background-matched BALB/c wild-type and Rag1-deficient mice (left panel) or C57BL/6 wild-type and μMT mice (right panel) were pre-treated with IL-4C and propranolol and injected i.v. with 250 μg of PE and followed by rectal thermometry for development of hypothermia.
PE-induced shock is more macrophage- and basophil than mast cell-dependent
Because shock can be mediated by macrophages, basophils and mast cells in Ab-dependent anaphylaxis22, 32, we evaluated whether these cell types contribute to PE-induced shock. Elimination or inactivation of most macrophages with either gadolinium23 or clodronate-containing liposomes24 considerably reduced the severity of PE-induced shock (Figure 4A). Elimination of most basophils with a specific, non-activating antibody to CD200R3 (Ba103), also partially suppressed PE-induced shock and the combination of Ba103 and gadolinium had a slightly greater suppressive effect than gadolinium alone (Figure 4B). In contrast, although PE induces some mast cell degranulation, as shown by slightly increased serum levels of mouse mast cell protease 1 (MMCP1, Figure 4A), mast cell depletion with anti-c-kit mAb did not detectably inhibit shock severity (Figure 4C). Mast cell depletion in this experiment was sufficient to decrease the MMCP1 and histamine responses to anti-IgE mAb by ~95% and 80%, respectively (Figure 4C) and almost totally blocked the histamine response to PE (Figure 4D). Inhibition of mast cell degranulation by treating mice with 30 mg/kg of cromolyn every 12 hours for 60 hours21 also had no effect on PE-induced shock (not shown). Thus, macrophages and basophils appear to be more important than mast cells in PE-induced shock.
Figure 4. PE-induced shock depends more on macrophages and basophils than on mast cells but is accompanied by mast cell activation.
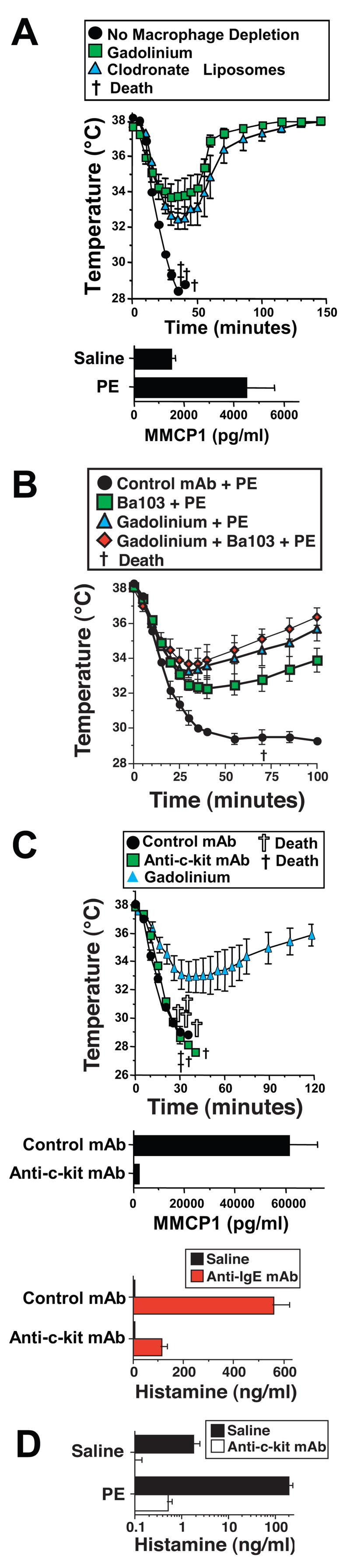
A. BALB/c mice were treated with saline (no macrophage depletion), or with gadolinium, or clodronate-containing liposomes to deplete macrophages; then pre-treated with IL-4C and propranolol, injected i.v. with 250 μg of PE and followed by rectal thermometry. Serum from mice injected with saline or PE was analyzed for MMCP1 content by ELISA. B. BALB/c mice were treated with gadolinium to deplete macrophages, 35 μg of Ba103 anti-basophil mAb to deplete basophils, both gadolinium and Ba103, or a control mAb isotype-matched to Ba103. Mice were then pre-treated with IL-4C and propranolol, injected i.v. with 250 μg of PE and followed by rectal thermometry. C. BALB/c mice were treated for 15 days with 0.5mg/mouse of anti-c-kit mAb by intraperitoneal injection every other day to deplete mast cells or an isotype-matched control mAb, then pre-treated with IL-4C and propranolol and challenged i.v. with 250 μg of PE. Sera obtained from mice after injection of an activating anti-IgE mAb were assayed for MMCP1 content or histamine content by ELISA. D. BALB/c mice, treated with saline or anti-c-kit mAb as in “B,” were challenged with saline or PE and bled 5 min later. Sera were assayed for histamine content by ELISA. * signifies p <0.05 as compared to saline in “A” and “C” and control mAb in “B.” † signifies p <0.05 for anti-c-kit mAb as compared to saline in “C.”
PE-induced shock is predominantly PAF-dependent but partially histamine-dependent
Mast cells, basophils and macrophages predominantly contribute to the pathogenesis of anaphylaxis through their secretion of histamine and PAF22, 32. The involvement of basophils and macrophages, in PE-induced shock led us to evaluate whether the same mediators are also involved in this process. An experiment performed with a specific PAF receptor antagonist demonstrated that PE-induced shock is substantially ameliorated by a PAF receptor antagonist (Figure 5). Surprisingly, in view of our studies with anti-c-kit mAb (Figure 4), anti-histamine treatment also inhibited PE-induced shock and the combination of PAF and histamine antagonists suppressed shock more completely than the PAF antagonist alone (Figure 5). Thus, PAF is more important than histamine in PAF-induced shock pathogenesis, consistent with the greater role of macrophages and basophils than mast cells, but both mediators appear to be involved.
Figure 5. PE-induced shock is predominantly PAF-dependent but partially histamine-dependent.

BALB/c mice were pre-treated with IL-4C and propranolol, then treated with saline, triprolidine (an antihistamine) or CV6209 (a PAF antagonist) or both triprolidine and CV6209. Mice were then challenged i.v. with 250 μg of PE and followed by rectal thermometry.
Peanut extract induced shock is predominantly C3-dependent
Taken together, our observations indicate that PE can induce shock by stimulating macrophage, basophil and mast cell production of PAF and histamine through an Ig-independent mechanism. Because macrophages and mast cells can be activated to secrete vasoactive mediators by C3a and C5a39–41, which are produced by complement activation42, this suggested that PE might activate complement through the alternative or lectin pathway43. To test the possibility that PE induces shock through a complement-dependent mechanism, we first compared the ability of PE to induce shock in wild-type mice and C3-deficient mice, which lack the complement component that is central to all 3 complement activation pathways13. To evaluate an alternative possibility, that macrophages, basophils and mast cells are activated by PE through an FcR-dependent pathway, we compared shock induction by PE in wild-type vs. FcRγ-deficient mice, which lack the polypeptide essential for all known mouse activating Fc receptors11. Results (Figure 6) demonstrate that PE induces shock normally in FcRγ-deficient mice, to only a slight extent in C3-deficient mice, and not at all in mice deficient in both C3 and FcRγ. C3aR- and C5aR-deficient mice and antagonists to these two receptors were used to evaluate the roles of each C-generated anaphylatoxin in PE-induced shock. Results of these experiments (Figure 6B and C) demonstrate an important role for C3a, but not for C5a. Analysis of serum from mice injected with saline or PE confirmed that PE rapidly activates C with production of C3a (Figure 6D).
Figure 6. PE-induced shock is mediated predominantly by C3a.
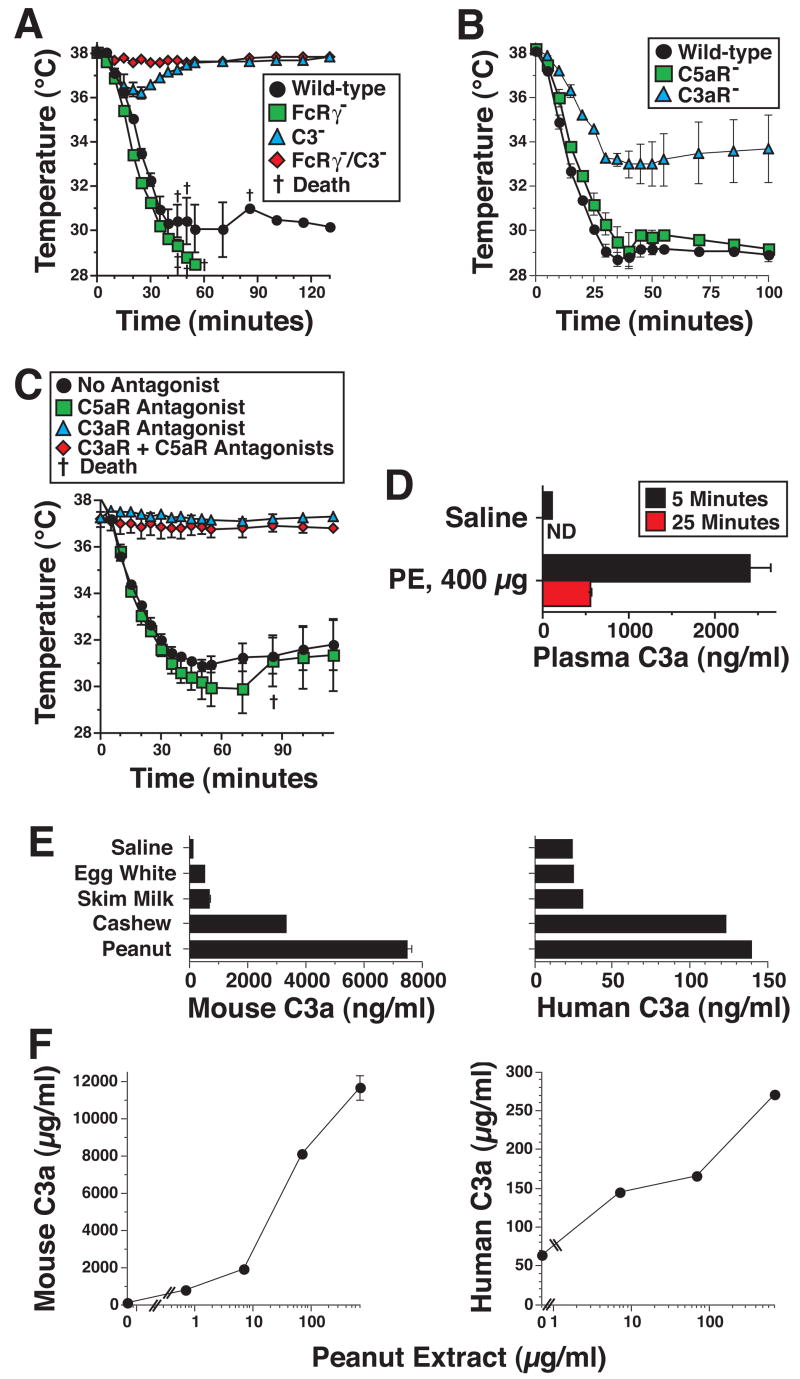
A. Wild-type, FcRγ-deficient, C3-deficient, and FcRγ/C3 double-deficient mice on the same genetic background were pre-treated with IL-4C and propranolol, injected i.v. with 250 μg of PE and followed for 2 hrs by rectal thermometry. B. C3aR-deficient, C5aR-deficient and wild-type mice on the same BALB/c genetic background were pre-treated with IL-4 and propranolol as described above, challenged i.v. with 250 μg of PE and followed for the next 2 hrs by rectal thermometry. C. BALB/c mice were pre-treated with propranalol and IL-4C. Mice were also injected i.p. with 0.6 mg of the C3aR antagonist SB290157, 3 hr prior to challenge with PE and/or i.v. with 20 μg of the C5aR antagonist, A8Δ71–73, 20 min prior to challenge with PE. Mice were followed by rectal thermometry for 115 min. after i.v. challenge with 400 μg of PE. D. Sera from mice injected i.v. with saline or with 300 μg of PE were analyzed for C3a concentration by ELISA. * signifies p <0.05 as compared to saline. † signifies p <0.05 as compared to 5 minutes. E. The abilities of saline, egg white, skim milk, cashew extract and PE to activate mouse and complement with production of C3a were determined. F. Mouse and human plasma were treated with the amounts of PE indicated and C3a levels were determined by ELISA.
To determine whether these observations with a mouse model might be relevant to humans, we also evaluated the ability of PE, cashew extract, egg white and skim milk to activate mouse and human complement in vitro, using C3a-specific ELISAs as a readout. PE and cashew extract increased C3a levels in both mouse and human plasma (Figure 6E) in a dose-dependent manner (Figure 6F). In contrast, skim milk and egg white, which fail to induce shock in IL-4C/propranalol-pretreated mice, had little effect. The mouse assay was considerably more sensitive than the human assay. This may reflect assay properties rather than a difference in the relative ability of PE to activate mouse vs. human complement, because a similar difference was observed when fixed gram negative bacteria were used to activate complement (data not shown).
Shock is synergistically induced by PE and IgE-stimulated mast cell degranulation
The requirement for presensitization with propranalol and IL-4C makes it unlikely that peanut ingestion can induce shock in normal individuals (or even those with increased intestinal permeability) solely by direct activation of complement. In contrast, it seemed possible that limited complement activation by PE might contribute to shock induction by acting synergistically with IgE-induced mast cell degranulation in individuals who have IgE antibodies to peanut Ags. To test this possibility, wild-type, C3-deficient, and FcRγ-deficient mice, pre-treated only with a low dose of propranalol, were challenged with anti-IgE mAb and/or PE and evaluated for development of shock. PE failed to induce detectable hypothermia in wild-type mice pre-treated with low dose propranalol (not shown), but considerably exacerbated shock when these mice were also challenged with anti-IgE mAb (Figure 7A, left panel). This synergism was C3-dependent, because PE did not exacerbate anti-IgE mAb-induced hypothermia in C3-deficient mice (Figure 7A, middle panel). Furthermore, PE had little effect on shock development in this system when anti-IgE mAb-induced mast cell degranulation was blocked by the absence of FcRγ(Figure 7A, right panel). Synergistic induction of shock by PE and anti-IgE mAb was also observed in wild-type mice in the absence of any sensitization with propranalol or IL-4C (Figure 7B) and when PE was administered orally rather than i.v. (Figure 7D). Oral administration of a large dose of PE caused a rapid increase in plasma C3a concentration that was small, but statistically significant (Figure 7C). This increase was considerably greater in mice that express an IL-9 transgene in their intestinal epithelium, which causes an increase in intestinal permeability21 than in normal mice (Figure 7C). Oral administration of PE also slightly but significantly enhanced the severity of shock induced by i.v. administration of anti-IgE mAb (Figure 7D).
Figure 7. PE acts synergistically with IgE-mediated mast cell degranulation to induce shock.

A. Wild-type, C3-deficient, and FcRγ-deficient mice on the same genetic background were pre-treated with propranolol, but not IL-4, and challenged i.v. with 80 μg of anti-IgE mAb ± 300 μg of PE and followed for the next 120 minutes by rectal thermometry. B. Naïve, non-pre-treated wild type BALB/c mice were challenged i.v. with 100 μg of anti-IgE ± 300 μg of PE and followed for the next 80 minutes by rectal thermometry. C. BALB/c wild-type mice and BALB/c mice that express an IL-9 transgene regulated by the small intestine-specific iFABP promoter were inoculated orally with saline or 6 mg of PE. Concentrations of C3a in plasma obtained 7 min later were determined by ELISA. D. BALB/c mice were pre-treated with a suboptimal dose of propranalol (15 μg) and inoculated orally with 400 μl of saline that did or did not contain PE (2.8 mg). Fifteen min later mice were challenged i.v. with 80 μg of EM-95 (anti-IgE mAb). Mice were followed for the next 120 min. by rectal thermometry. * signifies p <0.05 when compared to saline.
Discussion
Our observations, when combined with those published by other investigators, suggest that the high incidence, persistence and severity of peanut allergy may result from a combination of properties that make it a “perfect” allergen: high content of several, poorly digestible proteins that have strong B and T cell epitopes7, 8, 44; the ability of at least one of these proteins to directly activate antigen presenting cells9; and the ability of soluble peanut molecules to rapidly activate complement with production of large amounts of the anaphylatoxin C3a. The rapid production of C3a through an antibody-independent pathway stimulates macrophages, basophils and, to a lesser extent, mast cells, to secrete PAF and histamine, which contribute to the induction of shock by increasing vascular permeability29. C5a, another complement-derived anaphylatoxin, does not appear to be important in this process, even though it strongly promotes leukocyte activation and migration in immune complex disease45 and both C3a and C5a can contribute to the pathogenesis of asthma, and sepsis46–51.
Although our studies consistently demonstrate that PAF contributes more than histamine to PE-induced shock, the results of our experiments that evaluate the importance of histamine appear inconsistent. A role for histamine is supported by evidence that PE induces a histamine response and the ability of the H1 antagonist, triprolidine, to decrease the severity of PE-induced shock. The suppressive effect of triprolidine was most obvious in experiments that compared the effect of a PAF antagonist with the combined effect of this antagonist plus triprolidine (Figure 5). In contrast, treatment of mice with anti-c-kit mAb, which kills mast cells by eliminating an essential growth factor27, had no effect on PE-induced shock, even though it almost completely blocked histamine production (Figure 4). This suggests that triprolidine inhibits PE-induced shock by blocking the effects of a mediator other than histamine (for example, bradykinin52 or that treatment of mice with anti-c-kit mAb enhances mast cell-independent pathways of inflammation (for example, c-kit can promote mast cell production of PAF acetylhydrolase, which catabolizes PAF53).
Although PE contains some LPS, which stimulates C3a production through the alternative complement activation pathway as well as the classical pathway54, PE-induced shock was not caused by LPS contamination. This was demonstrated by 3 sets of studies: 1) TLR2, TLR4 and MyD88 deficiency did not protect mice from PE-induced shock; 2) removal of >99% of LPS from PE also failed to diminish PE-induction of shock; and 3) LPS induction of hypothermia was shown to be less rapid and more prolonged than that induced by PE.
Detection of PE-induced shock required development of a system that allows a normally inapparent insult to become obvious. Systemic anaphylaxis in mice is mediated predominantly by vascular leak, which causes hypotension that is reflected by hypothermia22, 29. Development of hypotension is normally limited by the magnitude of vasoactive mediator effects on vascular endothelial cells that increase their permeability and by β-adrenergic-dependent increases in vascular tone, heart rate, and myocardial contractility that compensate for decreased intravascular volume55. With these observations in mind, we made mice more sensitive to PE-induced shock by pretreating them with IL-4, which increases sensitivity to vasoactive mediators29 and with propranolol, which blocks β-adrenergic compensatory mechanisms and can increase the severity of human anaphylaxis56. Pretreatment with IL-4C and propranalol does not induce shock by itself, but increases the ability of ingested allergens to induce IgE-dependent anaphylaxis (unpublished data).
The requirement for presensitization of mice with IL-4 and propranalol to allow PE to induce shock makes it unlikely that peanut-mediated complement activation induces shock by itself in the absence of other insults. This is consistent with the ability of normal rodents and most humans to eat large quantities of peanuts without untoward effects and the observation that a negative RAST test is highly predictive of peanut tolerance in humans57. In contrast, PE-induced C3a production probably acts synergistically with IgE/FcεRI-dependent mast cell degranulation to exacerbate anaphylaxis. This hypothesis is supported by our observation that the severity of IgE-mediated anaphylaxis is enhanced by PE, even in the absence of both IL-4C and propranolol and even when PE is administered orally. Although we do not know whether our mouse model results are directly relevant to peanut-induced anaphylaxis in humans, it is notable that peanut extract can activate complement in both mouse and human plasma in vitro (Figure 6). It is also notable that complement is activated by hymenoptera venom58, the most common cause of severe, non-food-related human anaphylaxis59, and by metabolites of penicillin60, the most common cause of severe drug-induced human anaphylaxis61. Thus, exacerbation of anaphylaxis by complement activation may not be restricted to mice or to our PE model; rather, complement activation products may have a general role in the pathogenesis of severe, IgE-mediated anaphylaxis.
In contrast to these allergens, peanuts and tree nuts, which activate complement and are associated with severe anaphylaxis, milk and egg white have little ability to activate complement in vivo or in vitro and typically cause relatively mild allergic reactions. It is tempting to speculate that this association between complement activation and induction of severe anaphylaxis is not coincidental, but reflects complement exacerbation of the effector phase of anaphylaxis.
Because activated complement is a potent adjuvant62, 63, peanut activation of complement may contribute to induction of the IgE response to peanut allergens in addition to the effector phase of peanut-induced shock. This possibility is consistent with the considerably poorer immunogenicity of purified major peanut allergens than unfractionated peanut extract10 and with our initial observations that two purified major peanut allergens, Ara h1 and Ara h2, have little or no ability to induce shock (data not shown). Instead, we find that a low molecular weight (<5 kDa) fraction of PE, which has a partial amino acid sequence that does not match that of any of the 8 major peanut allergens, is particularly effective at inducing shock when injected into mice i.v. (unpublished data). Studies are underway to identify this molecule or molecules and to determine whether it can act as an adjuvant that promotes Th2 responses to major peanut allergens. Identification of this putative adjuvant may provide a strategy for engineering peanuts that have reduced allergenicity.
Acknowledgments
We thank Laura Armstrong for her contributions to development of our C3a ELISA, Dr. Stephanie Vogel for her gift of purified LPS, Dr. Wesley Burks for generous gift of purified peanut allergens and Dr. Chistopher Karp for his gift of TLRs-deficient mice and help with endotoxin measurement.
Funding was provided by a merit award from the U.S. Department of Veterans Affairs and by NIH grants R21AI079947, R01GM083204, R01AI052099 (F.D.F.); AI059305 (J.K.), RO1AI073553 (S.H.) and P30DK078392 (R.S.).
Abbreviations
- Ab
antibody
- FcγR
receptor for the Fc part of IgG
- FcRγ
Fc receptor γchain
- Ig
immunoglobulin
- i.g
intragastric
- IL-4C
complexes of interleukin-4 with anti-interleukin-4 monoclonal antibody
- IL-4Rα
interleukin-4 receptor α chain
- mAb
monoclonal antibody
- MMCP1
mouse mast cell protease 1
- PAF
platelet activating factor
- PE
peanut extract
- R
receptor
- TLR
toll-like receptor
Footnotes
Clinical Implications: Peanut-induced C3a may act synergistically with IgE-dependent mast cell activation to cause shock in peanut-allergic individuals and may contribute to peanut induction of an IgE response.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sicherer SH, Munoz-Furlong A, Sampson HA. Prevalence of peanut and tree nut allergy in the United States determined by means of a random digit dial telephone survey: a 5-year follow-up study. J Allergy Clin Immunol. 2003;112:1203–7. doi: 10.1016/s0091-6749(03)02026-8. [DOI] [PubMed] [Google Scholar]
- 2.Bock SA, Munoz-Furlong A, Sampson HA. Fatalities due to anaphylactic reactions to foods. J Allergy Clin Immunol. 2001;107:191–3. doi: 10.1067/mai.2001.112031. [DOI] [PubMed] [Google Scholar]
- 3.Bock SA, Munoz-Furlong A, Sampson HA. Further fatalities caused by anaphylactic reactions to food, 2001–2006. J Allergy Clin Immunol. 2007;119:1016–8. doi: 10.1016/j.jaci.2006.12.622. [DOI] [PubMed] [Google Scholar]
- 4.Skolnick HS, Conover-Walker MK, Koerner CB, Sampson HA, Burks W, Wood RA. The natural history of peanut allergy. J Allergy Clin Immunol. 2001;107:367–74. doi: 10.1067/mai.2001.112129. [DOI] [PubMed] [Google Scholar]
- 5.Palmer K, Burks W. Current developments in peanut allergy. Curr Opin Allergy Clin Immunol. 2006;6:202–6. doi: 10.1097/01.all.0000225161.60274.31. [DOI] [PubMed] [Google Scholar]
- 6.Beyer K, Morrow E, Li XM, Bardina L, Bannon GA, Burks AW, et al. Effects of cooking methods on peanut allergenicity. J Allergy Clin Immunol. 2001;107:1077–81. doi: 10.1067/mai.2001.115480. [DOI] [PubMed] [Google Scholar]
- 7.Maleki SJ, Kopper RA, Shin DS, Park CW, Compadre CM, Sampson H, et al. Structure of the major peanut allergen Ara h 1 may protect IgE-binding epitopes from degradation. J Immunol. 2000;164:5844–9. doi: 10.4049/jimmunol.164.11.5844. [DOI] [PubMed] [Google Scholar]
- 8.Sen M, Kopper R, Pons L, Abraham EC, Burks AW, Bannon GA. Protein structure plays a critical role in peanut allergen stability and may determine immunodominant IgE-binding epitopes. J Immunol. 2002;169:882–7. doi: 10.4049/jimmunol.169.2.882. [DOI] [PubMed] [Google Scholar]
- 9.Shreffler WG, Castro RR, Kucuk ZY, Charlop-Powers Z, Grishina G, Yoo S, et al. The major glycoprotein allergen from Arachis hypogaea, Ara h 1, is a ligand of dendritic cell-specific ICAM-grabbing nonintegrin and acts as a Th2 adjuvant in vitro. J Immunol. 2006;177:3677–85. doi: 10.4049/jimmunol.177.6.3677. [DOI] [PubMed] [Google Scholar]
- 10.van Wijk F, Nierkens S, Hassing I, Feijen M, Koppelman SJ, de Jong GA, et al. The effect of the food matrix on in vivo immune responses to purified peanut allergens. Toxicol Sci. 2005;86:333–41. doi: 10.1093/toxsci/kfi187. [DOI] [PubMed] [Google Scholar]
- 11.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γchain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–29. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 12.Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12:471–81. doi: 10.1016/s1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- 13.Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A. 1995;92:11490–4. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hopken UE, Lu B, Gerard NP, Gerard C. Impaired inflammatory responses in the reverse arthus reaction through genetic deletion of the C5a receptor. J Exp Med. 1997;186:749–56. doi: 10.1084/jem.186.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humbles AA, Lu B, Nilsson CA, Lilly C, Israel E, Fujiwara Y, et al. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature. 2000;406:998–1001. doi: 10.1038/35023175. [DOI] [PubMed] [Google Scholar]
- 16.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–77. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 17.Godfrey P, Rahal JO, Beamer WG, Copeland NG, Jenkins NA, Mayo KE. GHRH receptor of little mice contains a missense mutation in the extracellular domain that disrupts receptor function. Nat Genet. 1993;4:227–32. doi: 10.1038/ng0793-227. [DOI] [PubMed] [Google Scholar]
- 18.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–52. [PubMed] [Google Scholar]
- 19.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–51. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 20.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–50. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 21.Forbes EE, Groschwitz K, Abonia JP, Brandt EB, Cohen E, Blanchard C, et al. IL-9- and mast cell-mediated intestinal permeability predisposes to oral antigen hypersensitivity. J Exp Med. 2008;205:897–913. doi: 10.1084/jem.20071046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strait RT, Morris SC, Yang M, Qu XW, Finkelman FD. Pathways of anaphylaxis in the mouse. J Allergy Clin Immunol. 2002;109:658–68. doi: 10.1067/mai.2002.123302. [DOI] [PubMed] [Google Scholar]
- 23.Mizgerd JP, Molina RM, Stearns RC, Brain JD, Warner AE. Gadolinium induces macrophage apoptosis. J Leukoc Biol. 1996;59:189–95. [PubMed] [Google Scholar]
- 24.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 25.Otto M, Hawlisch H, Monk PN, Muller M, Klos A, Karp CL, et al. C5a mutants are potent antagonists of the C5a receptor (CD88) and of C5L2: position 69 is the locus that determines agonism or antagonism. J Biol Chem. 2004;279:142–51. doi: 10.1074/jbc.M310078200. [DOI] [PubMed] [Google Scholar]
- 26.Ames RS, Lee D, Foley JJ, Jurewicz AJ, Tornetta MA, Bautsch W, et al. Identification of a selective nonpeptide antagonist of the anaphylatoxin C3a receptor that demonstrates antiinflammatory activity in animal models. J Immunol. 2001;166:6341–8. doi: 10.4049/jimmunol.166.10.6341. [DOI] [PubMed] [Google Scholar]
- 27.Grencis RK, Else KJ, Huntley JF, Nishikawa SI. The in vivo role of stem cell factor (c-kit ligand) on mastocytosis and host protective immunity to the intestinal nematode Trichinella spiralis in mice. Parasite Immunol. 1993;15:55–9. doi: 10.1111/j.1365-3024.1993.tb00572.x. [DOI] [PubMed] [Google Scholar]
- 28.Baniyash M, Eshhar Z. Inhibition of IgE binding to mast cells and basophils by monoclonal antibodies to murine IgE. Eur J Immunol. 1984;14:799–807. doi: 10.1002/eji.1830140907. [DOI] [PubMed] [Google Scholar]
- 29.Strait RT, Morris SC, Smiley K, Urban JF, Jr, Finkelman FD. IL-4 exacerbates anaphylaxis. J Immunol. 2003;170:3835–42. doi: 10.4049/jimmunol.170.7.3835. [DOI] [PubMed] [Google Scholar]
- 30.Finkelman FD, Madden KB, Morris SC, Holmes JM, Boiani N, Katona IM, et al. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J Immunol. 1993;151:1235–44. [PubMed] [Google Scholar]
- 31.Kojima T, Obata K, Mukai K, Sato S, Takai T, Minegishi Y, et al. Mast cells and basophils are selectively activated in vitro and in vivo through CD200R3 in an IgE-independent manner. J Immunol. 2007;179:7093–100. doi: 10.4049/jimmunol.179.10.7093. [DOI] [PubMed] [Google Scholar]
- 32.Tsujimura Y, Obata K, Mukai K, Shindou H, Yoshida M, Nishikado H, Kawano Y, Minegishi Y, Shimizu T, Karasuyama H. Basophils play a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity. 2008;28:581–9. doi: 10.1016/j.immuni.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 33.Khodoun MV, Orekhova T, Potter C, Morris S, Finkelman FD. Basophils initiate IL-4 production during a memory T-dependent response. J Exp Med. 2004;200:857–70. doi: 10.1084/jem.20040598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Urban JF, Jr, Noben-Trauth N, Schopf L, Madden KB, Finkelman FD. Cutting edge: IL-4 receptor expression by non-bone marrow-derived cells is required to expel gastrointestinal nematode parasites. J Immunol. 2001;167:6078–81. doi: 10.4049/jimmunol.167.11.6078. [DOI] [PubMed] [Google Scholar]
- 35.Peitzman AB, Billiar TR, Harbrecht BG, Kelly E, Udekwu AO, Simmons RL. Hemorrhagic shock. Curr Probl Surg. 1995;32:925–1002. doi: 10.1016/s0011-3840(05)80008-5. [DOI] [PubMed] [Google Scholar]
- 36.Lang DM. Anaphylactoid and anaphylactic reactions. Hazards of β-blockers Drug Saf. 1995;12:299–304. doi: 10.2165/00002018-199512050-00002. [DOI] [PubMed] [Google Scholar]
- 37.Madden KB, Yeung KA, Zhao A, Gause WC, Finkelman FD, Katona IM, et al. Enteric nematodes induce stereotypic STAT6-dependent alterations in intestinal epithelial cell function. J Immunol. 2004;172:5616–21. doi: 10.4049/jimmunol.172.9.5616. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi O, Takeda K, Hoshino K, Adachi O, Ogawa T, Akira S. Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int Immunol. 2000;12:113–7. doi: 10.1093/intimm/12.1.113. [DOI] [PubMed] [Google Scholar]
- 39.Mencia-Huerta JM, Benveniste J. Platelet-activation factor and macrophages. I Evidence for the release from rate and mouse peritoneal macrophages and not from mastocytes. Eur J Immunol. 1979;9:409–15. doi: 10.1002/eji.1830090512. [DOI] [PubMed] [Google Scholar]
- 40.Cavaillon JM, Fitting C, Haeffner-Cavaillon N. Recombinant C5a enhances interleukin 1 and tumor necrosis factor release by lipopolysaccharide-stimulated monocytes and macrophages. Eur J Immunol. 1990;20:253–7. doi: 10.1002/eji.1830200204. [DOI] [PubMed] [Google Scholar]
- 41.Johnson AR, Hugli TE, Muller-Eberhard HJ. Release of histamine from rat mast cells by complement peptides C3a and C5a. Immunology. 1975;28:1067–80. [PMC free article] [PubMed] [Google Scholar]
- 42.Abbas Abul K, Lichtman Andrew H. Cellular and Molecular Immunology. 5. Philadelphia, PA, USA: Saunders, Elsevier Science; 2003. [Google Scholar]
- 43.Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305–10. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 44.Proust B, Astier C, Jacquenet S, Ogier V, Magueur E, Roitel O, et al. A single oral sensitization to peanut without adjuvant leads to anaphylaxis in mice. Int Arch Allergy Immunol. 2008;146:212–8. doi: 10.1159/000115889. [DOI] [PubMed] [Google Scholar]
- 45.Trendelenburg M, Fossati-Jimack L, Cortes-Hernandez J, Turnberg D, Lewis M, Izui S, et al. The role of complement in cryoglobulin-induced immune complex glomerulonephritis. J Immunol. 2005;175:6909–14. doi: 10.4049/jimmunol.175.10.6909. [DOI] [PubMed] [Google Scholar]
- 46.Hawlisch H, Wills-Karp M, Karp CL, Kohl J. The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol Immunol. 2004;41:123–31. doi: 10.1016/j.molimm.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 47.Nakano Y, Morita S, Kawamoto A, Suda T, Chida K, Nakamura H. Elevated complement C3a in plasma from patients with severe acute asthma. J Allergy Clin Immunol. 2003;112:525–30. doi: 10.1016/s0091-6749(03)01862-1. [DOI] [PubMed] [Google Scholar]
- 48.Ali H, Panettieri RA., Jr Anaphylatoxin C3a receptors in asthma. Respir Res. 2005;6:19. doi: 10.1186/1465-9921-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang M, Mackay CR, Zetoune FS, Gerard NP, Cianflone K, Köhl J, Berard C, Sarma JV, Ward PA. Functional roles for C5a receptors in sepsis. Nat Med. 2008;14:551–7. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baelder R, Fuchs B, Bautsch W, Zwirner J, Köhl J, Hoymann HG, Blaab T, Erpenbeck V, Krug N, Braun A. Pharmacological targeting of anaphylatoxin receptors during the effector phase of allergic asthma suppresses airway hyperresponsiveness and airway inflammation. J Immunol. 2005;174:783–9. doi: 10.4049/jimmunol.174.2.783. [DOI] [PubMed] [Google Scholar]
- 51.Kildsgaard J, Hollmann TJ, Matthews KW, Bian K, Murad F, Wetsel RA. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J Immunol. 2000;165:5406–9. doi: 10.4049/jimmunol.165.10.5406. [DOI] [PubMed] [Google Scholar]
- 52.Becker EL, Mota I, Wong D. Inhibition by antihistamines of the vascular permeability increase induced by bradykinin. Br J Pharmacol. 1968;34:330–6. doi: 10.1111/j.1476-5381.1968.tb07054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakajima K, Murakami M, Yanoshita R, Samejima Y, Karasawa K, Setaka M, et al. Activated mast cells release extracellular type platelet-activating factor acetylhydrolase that contributes to autocrine inactivation of platelet-activating factor. J Biol Chem. 1997;272:19708–13. doi: 10.1074/jbc.272.32.19708. [DOI] [PubMed] [Google Scholar]
- 54.Mondino BJ, Sumner HL. Generation of complement-derived anaphylatoxins in normal human donor corneas. Invest Ophthalmol Vis Sci. 1990;31:1945–9. [PubMed] [Google Scholar]
- 55.Cauwels A, Janssen B, Buys E, Sips P, Brouckaert P. Anaphylactic shock depends on PI3K and eNOS-derived NO. J Clin Invest. 2006;116:2244–51. doi: 10.1172/JCI25426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.TenBrook JA, Jr, Wolf MP, Hoffman SN, Rosenwasser LJ, Konstam MA, Salem DN, et al. Should β-blockers be given to patients with heart disease and peanut-induced anaphylaxis? A decision analysis. J Allergy Clin Immunol. 2004;113:977–82. doi: 10.1016/j.jaci.2004.02.043. [DOI] [PubMed] [Google Scholar]
- 57.Perry TT, Matsui EC, Kay Conover-Walker M, Wood RA. The relationship of allergen-specific IgE levels and oral food challenge outcome. J All Clin Immunol. 2004;114:144–9. doi: 10.1016/j.jaci.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 58.De Carolis C, Perricone R, De Sanctis G, Fontana L. Complement activation by Hymenoptera venom allergenic extracts. J Allergy Clin Immunol. 1982;70:219–20. doi: 10.1016/0091-6749(82)90045-8. [DOI] [PubMed] [Google Scholar]
- 59.Reisman RE. Stinging insect allergy. Med Clin North Am. 1992;76:883–94. doi: 10.1016/s0025-7125(16)30330-3. [DOI] [PubMed] [Google Scholar]
- 60.von Zabern I, Przyklenk H, Nolte R, Vogt W. Effect of different penicillin derivatives on complement components in human serum. Int Arch Allergy Appl Immunol. 1984;75:164–72. doi: 10.1159/000233608. [DOI] [PubMed] [Google Scholar]
- 61.Leone R, Conforti A, Venegoni M, Motola D, Moretti U, Meneghelli I, et al. Drug-induced anaphylaxis : case/non-case study based on an italian pharmacovigilance database. Drug Saf. 2005;28:547–56. doi: 10.2165/00002018-200528060-00006. [DOI] [PubMed] [Google Scholar]
- 62.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348–50. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 63.Stager S, Alexander J, Kirby AC, Botto M, Rooijen NV, Smith DF, et al. Natural antibodies and complement are endogenous adjuvants for vaccine-induced CD8+ T-cell responses. Nat Med. 2003;9:1287–92. doi: 10.1038/nm933. [DOI] [PubMed] [Google Scholar]