

Abstract
HIV-associated dementia (HAD) is the most common AIDS-associated neurological disorder and is characterized by the development of synaptodendritic injury to neurons. To advance HAD therapy, it is crucial to identify the mechanisms and factors involved. The viral protein HIV-1 Tat is among those factors and is released by HIV-1-infected cells and can be taken up by adjacent neuronal cells leading to neurotoxic effects. Multiple cellular host proteins have been identified as Tat cofactors in causing neuronal injury. Interestingly, most of these factors function through activation of the p53 pathway. We have now examined the ability of Tat to activate the p53 pathway leading to the induction of endogenous p53 and p73 in neuronal cells. We found that Tat induced p53 and p73 levels in SH-SY5Y cells and that this induction caused retraction of neurites. In the absence of either p53 or p73, Tat failed to induce dendritic retraction or to activate the proapoptotic proteins, such as Bax. Further, we found that p53-accumulation in Tat-treated cells depends on the presence of p73. Therefore, we conclude that Tat contributes to neuronal degeneration through activation of a pathway involving p53 and p73. This information will be valuable for the development of therapeutic agents that affect these pathways to protect CNS neurons and prevent HAD.
Keywords: HIV-1 Tat, neuronal degeneration, p53, apoptosis, p73, phosphorylation, HIV-associated dementia (HAD)
Introduction
HIV infection is pandemic with more than 30 million people infected today worldwide. In the USA following the onset of AIDS in patients, 10-15% per year will develop HIV-associated dementia (HAD), a severe neurocognitive and motor abnormality during the latter stage of infection.1 In recent years, in vitro and ex vivo studies have attempted to characterize the mechanisms that underlie the relationship between HIV infection and HAD. Accumulating evidence indicates that the major reservoir of HIV infection in the CNS is localized in macrophages and microglia, producing deleterious effects on neuronal tissue via indirect mechanisms.2 The neuropathogenesis of HIV-infection still remains to be fully understood and effective therapeutic approaches for neuroAIDS are sorely needed. Available data suggest that the mechanism(s) that lead to damage in the brain of AIDS patients might involve combined effects of more than one neurotoxic factor. In particular, evidence suggests that viral proteins, such as Tat, secreted from HIV-1 infected cells are among these factors.3,4
Tat is a viral transactivator that binds to an RNA stem-loop structure called TAR at the 5' end of all viral transcripts.5,6 Tat binds the cyclin T1 component of the positive transcription elongation factor b (pTEFb) and recruits cyclin-dependent kinase 9 to elongating HIV transcripts.7,8 Tat is mainly active in the nucleus and is secreted at high-level in vitro.9 Secreted Tat can cause direct or indirect injury to neurons and thus Tat may contribute to HAD neuropathogenesis. The approximate amount of Tat released from HIV-infected cells and taken up by non-infected cells has been measured by capillary electrophoresis and is at a concentration that has been shown to be biologically active (Deshmane et al., submitted). However, the pathways that are activated by Tat leading to neuronal degeneration remain unclear. In this regard, other studies have suggested that Tat causes neuronal degeneration through activation of several pathways. These include increase of intracellular calcium release, induction of reactive oxygen species production2 and induction of apoptotic pathways leading to caspase activation.10 In addition to these pathways, Tat has also been shown to alter the expression and distribution of tight junction proteins and contributes to the disruption of the blood brain barrier.11,12 However, despite the identification of these pathways and the cellular factors involved, all attempts to inhibit Tat-mediated neurodegeneration have thus far failed. Interestingly, most of the cellular cofactors for Tat have been shown to cooperate with the tumor suppressor protein p53 and this cooperation is necessary for the induction of neuronal degeneration.13-18 Support for this hypothesis comes from studies demonstrating p53 accumulation in the brains of HAD patients, in cell culture models and in animal models. Taken together, these data suggest that co-operation of Tat with p53 is integral for the pathogenesis of HAD.
p53 is a central transcription factor that plays key roles in cell cycle regulation, differentiation and apoptosis.17 Under normal conditions, wild-type p53 protein is expressed at low levels, and these levels are mainly regulated by Mdm2, which represses p53 transcriptional activity,19 mediates ubiquitination of p53,20 and targets p53 for degradation.21 In contrast, p53 levels are augmented in response to a variety of cellular stress signals including DNA damage, hypoxia or oncogene activation leading to cell cycle arrest and/or apoptosis to prevent cells from multiplying.22 Further, activated p53 binds to specific DNA sequences, regulates the expression of many genes and downstream targets including the cyclin-dependent protein kinase inhibitor, p21WAF1.23 In addition to the above-mentioned functions, p53 has also been shown to be involved in the neurodegeneration phenomena often observed in AIDS patients. Evidence linking p53 to the neuropathogenesis of HAD include: (i) activation of p53 in HIV-1-infected cells (in culture and in the brain of infected patients,24 (ii) accumulation of p53 in neurons from HAD patients (44),25 (iii) interaction of p53 with viral factors such as Nef, (iv) effect of p53 on HIV-1 reverse transcriptase (RT) function, and (v) requirement for p53 expression in the induction of caspase 3 enzymatic activity by gp120 in neuronal cells. Although, p53 accumulates in microglia and astrocyte nuclei in a subset of AIDS patients without dementia, increased neuronal p53 was only observed in HAD cases. The mechanisms leading to p53 accumulation and activation remain unclear. Studies have reported the involvement of HIV-1 gp120 and Tat proteins. In support of this hypothesis, co-culture of neurons and Tat-stably transfected monocytes promoted secretion of toxins leading to p53 accumulation in neurons as well as neuronal degeneration.26 In addition, extracellular Tat has been shown to be rapidly internalized by neurons leading to p53 accumulation.27 Studies on cell types that express Tat have shown that Tat promoted cell cycle arrest by interacting with p53.28 Further, phosphorylation of p53 on serines 15 and 46 is necessary for its apoptotic functions.29 Finally, p53 was unable to cause cell death in the absence of p73.30 This illustrates that p73 is vital for p53-induced apoptosis and that p73 itself is an important component of the tumor suppressor activity of p53.
p73 belongs to the family of p53-related transcription factors including p53, p73 and p63. The overall structure and sequence homology indicates that a p63/p73-like protogene is the ancestral gene, whereas p53 evolved later in higher organisms.31 p73 functions in a manner analogous to p53 by inducing tumor cell apoptosis and participating in the cell cycle checkpoint control through transactivating an overlapping set of p53/p73-target genes.32 Genetic studies of p73 protein revealed three functional domains including transactivation, DNA binding and oligomerization domains, which are common to the α and β variants of p73. p73 is stabilized in response to a subset of DNA-damaging agents in a way that is distinct from that of p53, and exerts pro-apoptotic activity. In sharp contrast to p53, however, p73 is expressed as two NH2-terminally distinct isoforms including transcriptionally active (TA) and inactive (ΔN) forms. Alternative splicing of the human p73 gene generates at least seven variants, α, β, δ, γ, ε, ζ and η (TA proteins) and at least four inactive, alternatively spliced N-terminal isoforms initiated at different ATG (ΔNp73 proteins).33 ΔNp73 isoforms have oncogenic potential and act in a dominant negative manner against TAp73 as well as p53.33 Although p73 expression is low, the p73 gene is expressed in all normal tissues. However, the TA and the ΔN proteins are the predominant forms of p73 in human and mouse brain, respectively.34 The importance of ΔNp73 in this system is demonstrated by the observation that increasing the levels of ΔNp73 alone is able to rescue sympathetic neurons from cell death. It should be noted that in neuroblastoma cells, ΔNp73 failed to inhibit p73, however, it did physically interact with and inhibited p53. Similar to p53, overexpression of p73 induces transcription of p21WAF1 and other genes involved in apoptosis.35 Finally, p73 mimics p53 by being phosphorylated, acetylated and interacting with mdm2. However, in contrast to p53, mdm2 does not induce the degradation of p73.36,37
In the present study, we examined the ability of Tat to cause neurodegeneration through a pathway that involved activation of p53 and p73. This information will be valuable for the development of therapeutic agents that affect these pathways to protect CNS neurons and prevent HAD.
Results
Induction of p53 protein levels in the brain of HIV-1-infected patients
To test our hypothesis that HIV-1 Tat protein might affect neuronal degeneration through the p53 pathway, we first sought to examine the levels of p73 and p53 in the brain of AIDS patients. Previously, we have reported increase in the levels of p73 (mRNA and protein) in the brain of AIDS patients.38,41 This gave us a rationale to examine the level and subcellular localization of p53 protein in brain tissue of HIV encephalitis (HIVE) patients. As shown in Figure 1, immunohistochemistry showed induction and the presence of p53 in the nucleus and the cytoplasm of perivascular macrophages and astrocytes, respectively, and in the nucleus of neurons of HIVE. Although the morphology of specific cell types in the brain, especially neurons, are very characteristic, we chose to confirm the neuronal nature of the cells using cell type-specific markers. Therefore we performed double labeling studies of neurons or astrocytes with p53 (red) and neurofilaments (neurons, green) or GFAP (astrocytes, green) to confirm their phenotype.
Figure 1.
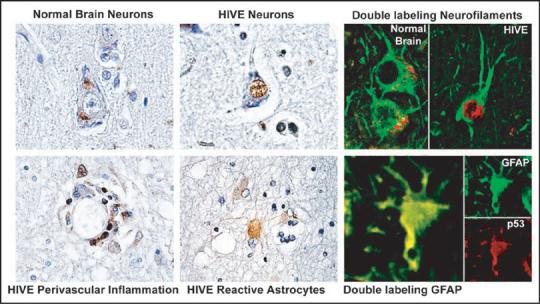
Subcellular localization of p53 in HIV-1-infected patients. Immunohistochemistry for p53 in gray and white matter from HIV-1 uninfected brain (control) or HIV-1-infected HAD brain showing p53 localization (arrows). While p53 is weakly expressed in the cytoplasm of neurons in the normal control brain, it is robustly expressed in the nuclei of neurons in HIVE. Also in HIVE, reactive astrocytes and perivascular cuffs of inflammatory cells show nuclear and cytoplasmic p53. Double labeling of neurons for neurofilaments (green) and p53 (red) in normal brain and HIVE and astrocytes with GFAP (green) and p53 (red) in HIVE. Original magnification × 400 or × 1000.
Tat induces endogenous levels of p53 and p73 proteins
Next, we examined the levels of p53 and p73 proteins in Tat-treated SH-SY5Y cells. The rationale for choosing SH-SY5Y cells is that they mirror pathways involved in neurodegenerative process associated with HIVE.42 Cells were plated at 1 × 105 cells/ml and cultured in DMEM. Confluent cells were replated for different experiments and differentiated by treatment with 10 μM retinoic acid for seven days with medium changes every two days. For all of the experiments, cells were serum starved for 6 h in the presence of 10 μM RA prior to treatment with Tat. In these experiments, we also used a highly pure toxin-free recombinant Tat (rTat, 101 aa). Further, we have previously demonstrated that the approximate amount of Tat released from HIV-infected cells and taken up by neuronal cells is about 1 pg (Deshmane et al., manuscript submitted). SH-SY5Y cells were treated with 1 μg, 1 ng or 1 pg/ml rTat for 24 h, and cell extracts prepared. Cell extracts were subject to Western blot analysis using anti-p73α, -p53, -Tat or -Grb-2 antibodies (Fig. 2A). Endogenous levels of p73α and p53 proteins were induced in the cell extracts prepared from Tat-treated but not from the control cells (p73 and p53 panels, compare lanes 2-4 to lane 1). Tat was only detected in extracts prepared from cells treated with 1 μg of rTat (Tat panel, lane 2). Note that the limit of detection for rTat by Western blot is 10 ng (data not shown). For equal protein loading, anti-Grb2 antibody was used.
Figure 2.
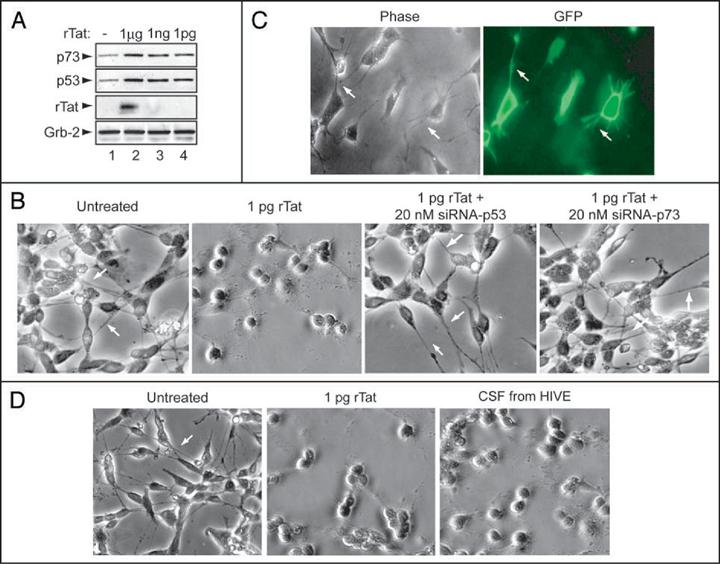
rTat upregulates expression of p53 and p73 in human neurons. (A) Cell lysates were prepared from untreated and rTat-treated SH-SY5Y cells as indicated. Fifty micrograms of total cell lysate were used in Western blot with α-p73, -p53, -Tat or anti-Grb-2 antibodies. (B-D) Phase contrast and fluorescence microscopy of SH-SY5Y cells treated with rTat (B), transfected with GFP (C) or HN (D) treated with rTat alone or with CSF deriving from HIVE patient showed rounding with shortened neurite. In contrast, untreated neurons or Tat-treated/transfected with siRNA-p53 or p73 resulted in maintenance of cellular architecture with extended neuronal processes (indicated by arrows).
Tat requires both p73 and p53 to cause neuronal degeneration
Induction of the endogenous levels of p53 and p73 in Tat-treated cells led us to examine their involvement in neuronal degeneration. Treatment of SH-SY5Y cells with rTat (1 pg/ml) for 24 h caused neurite retraction (Fig. 2B, compare Tat-treated and untreated panels). Interestingly, in cells transfected with 20 nM siRNA directed against p53 or p73 then treated with rTat (1 pg/ml), no neuronal degeneration was observed, leading to the conclusion that Tat failed to cause neurite retraction in the absence of either p53 or p73. Similar results have been obtained with HIV-1 gp120 protein, which is unable to cause cell death in p53-/- cells.27 In order to demonstrate that the transfection protocol itself did not affect neurite retraction, we also performed the transfection procedure using a control plasmid that expressed GFP. As shown in Figure 2C, the transfection procedure did not affect neurite length as observed in the GFP panel.
Next, we examined whether Tat derived from cerebrospinal fluid (CSF) or added as recombinant protein had the capability to cause neuronal damage. In addition, to further extend our data, we used primary human neurons (HN) in this experiment. HN (purchased from ScienCell Research Laboratories, Carlsbad, CA) are isolated from human brain and characterized by immunofluorescent method with antibodies to neurofilament, MAP2, and β-tubulin III. They are negative for HIV-1, HBV, HCV, mycoplasma, bacteria, yeast and fungi. Cells were grown in a complete medium designed for optimal growth of normal human neurons in vitro (ScienCell Research Lab). HN cells were treated with 1 pg/ml of rTat or with 5 μl of CSF from an HIVE patient (Fig. 2D). Addition of CSF from HIVE or addition of rTat resulted in neurite retraction after 24 h relative to untreated cells. These results confirm our observation regarding the ability of Tat to cause neuronal damage observed in panel B.
In the next series of experiments, we examined whether Tat can cause neuronal injury in the presence of p73 in the SK-N-MC cell line. Importantly, the neuroblastoma cell line, SK-N-MC is one of the few neuronal cell lines that shows neither 1p deletion nor p73 loss of heterozygosity (LOH).43 The cells were transfected with Tat and/or p73 expression plasmids as shown in Figure 3A and B. As a control, the cells were also transfected with small interfering RNA directed against p53 or p73 as indicated. As shown in panel A, TUNEL assay indicates that Tat caused massive cell death in the presence of p73 (~59%) confirming previously published data.38 When Tat was co-transfected with p73α-siRNA, the pro-apoptotic effect of Tat was reduced to 13% (p ≤ 0.001). Approximately 1-3% of SK-N-MC cells undergo apoptosis in normal culture or in cells transfected with pcDNA3. Tat or p73α caused cell death in the presence of p53 (~55 and 76%, respectively) but not in its absence (~10 and 12%, respectively) (Panel B). The number of apoptotic cells was calculated from positively transfected cells (pDsRed1-mito). Note that >500 positively transfected cells in 10 randomly selected microscopic fields were counted. These results represent the average of three experiments. Panels C and D show the efficacy of siRNA to p53 or 73 to silence p53 and p73 genes, respectively, with (C, D, lanes 1, 2) and without Tat (lanes 2, 4).
Figure 3.
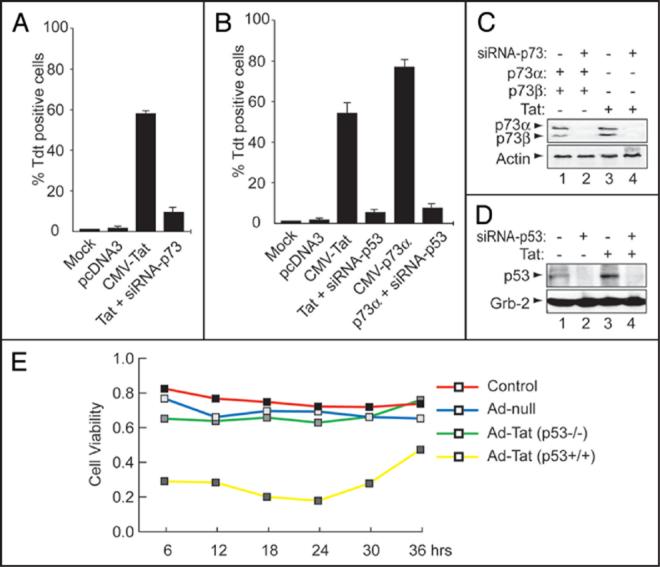
Tat requires the presence of p53 and p73 to cause apoptosis. (A and B) Analysis of apoptosis in SK-N-MC cells was performed by TUNEL assay. The TdT reaction was performed at 48 h after transfection and cells positive for DNA strand breaks (green nuclear fluorescence) were counted. Only pDsRed-mito positive cells (red fluorescence) were included in the calculation of the percentage of apoptotic cells. The pDsRed1-mito plasmid labels the mitochondria of transfected cells. (C and D) Effect of transfection of SH-SY5Y cells on protein expression. Cells were transfected with 1.0 μg of CMV-p73α, CMV-p73β (E, lanes 1, 2) or CMV-Tat (C and D, lanes 3) expression plasmids +/- 20 nM of p53 or p73 siRNA3 (lanes 2-4). Fifty micrograms of extract were assayed by Western blot with anti-p53, -p73, -Grb2 or -actin antibodies. Arrows indicate the positions of each protein. (E) Analysis of cell viability in HCT116 cells. MTT assay was used as a measure for viable cells. Data are expressed as the mean ± S.D *p < 0.05, **p < 0.01 vs. respective control.
The number of viable cells was also measured using mitochondrial reductase activity (MTT) assay, which is also an indicator of mitochondrial function.44 To examine the role of p53 without the use of transfection or siRNA, in the next experiment, we made use of the human carcinoma HCT116 (p53+/+) and HCT116 (p53-/-) cell lines. HCT116 (p53-/-) and the parental cell line HCT116 (p53+/+) cells were infected with 1 MOI of Ad-null or Ad-Tat. As shown in Figure 3E, no cell death was observed with the control (red), Ad-null (blue), or Ad-Tat HCT116 (p53-/-) cells (green). Infection of HCT116 (p53+/+) with Tat led to considerable cell death (yellow). The following important conclusions emerge from these results (i) Tat cannot cause neuronal degeneration when either p53 or p73 is absent, (ii) p53 and p73 require each other to induce apoptosis. These observations correlate with other reports that p53 fails to cause cell death in the absence of p73.30
p73 is essential for Tat-induction of p53 and pro-apoptotic proteins
The results in Figures 2 and 3 show that Tat induces p53 and p73 at the protein level and that Tat requires the presence of both p53 and p73 to cause degeneration. Thus, we further examined the dependency of Tat on these proteins and the status of pro-apoptotic proteins such as Bax and caspase 3 in Tat-treated cells. SH-SY5Y cells were transfected with 20 nM of siRNA-p53 or siRNA-p73 for 24 h, after which 1 pg/ml of rTat protein was added to the cells. After 24 h, cell extracts were prepared and analyzed by Western blot. rTat induced endogenous levels of both p53 and p73 (Fig. 4A, lane 2). However, this induction was abolished by siRNA-p73 (lane 4). siRNA-p53 had no effect (lane 3). These results indicate that p73 is necessary for Tat-induction of p53, that p53 is a downstream target of p73 and that Tat induces p53 via a pathway that requires p73.
Figure 4.

Activation of pro-apoptotic proteins by Tat in p53-null cells. Fifty micrograms of cell lysate prepared from H1299 (B and C) and SH-SY5Y (A and B) cells were analyzed by Western blot. (D) H1299 cells were transfected with Bax-luc ± p73α, 20 nM of siRNA-p73 or infected with Ad-p53 or treated with rTat, as indicated. Luciferase and b-gal activities were determined after 48 h. All values were normalized for β-gal. Data represent the mean of 3 experiments.
Next, we examined a human lung carcinoma cell line (H1299), which lacks endogenous p53.45 Cells were treated with rTat and cell lysates were prepared and analyzed by Western blot with anti-p73 (Fig. 4B), -Bax, -caspase 3 (total and cleaved) (Fig. 4C) and anti-Grb2 antibodies. As shown in Figure 4B, rTat induced endogenous p73 in extracts prepared from Tat-treated H1299 cells (compare lanes 1 and 2). H1299 cells were then infected with adeno-p53 24 h prior to rTat treatment. In addition, Figure 4C shows that rTat induced Bax, caspase 3 and promoted cleavage of caspase 3 in the presence of adeno-p53 but not in its absence (compare lane 4 to lane 5 and lane 5 to lane 6). rTat also induced Bax, caspase 3 and promoted cleavage of caspase 3 (panel C, compare lanes 4 and 5). Transfection of 20 nM siRNA-p73 24 h prior to the addition of rTat did not affect the status of Bax or caspase 3 proteins (compare lanes 5 and 7). Similarly, SH-SY5Y cells were treated with rTat or transfected with 20 nM of siRNA-p73 24 h prior to rTat addition. Western blot shows induction of Bax, caspase 3 or cleavage of caspase 3 proteins in the presence of Tat (compare lanes 1 and 2). However, Tat was inactive in the absence of p73 (compare lane 2 to lane 3).
Next, we examined whether Tat can activate Bax directly. Thus, we examined the effect of Tat on Bax transcription in p53-/- cells. H1299 cells were transfected with 0.1 μg Bax-Luc alone or in combination with of 0.25 μg of p73α expression plasmid. In addition to p73α plasmid, some cells were also transduced with 1 MOI of Ad-p53 or Ad-null (data not shown), transfected with p73-siRNA or treated with 1 pg/ml of rTat as indicated. As a control for the transfection efficiency, 1 μg of pCMV-β-gal plasmid was also included. As shown in Panel D, in the absence of p53, Tat (lane 2) failed to activate the Bax promoter even when co-expressed with p73α (lane 4). Similar results were obtained with p73α in the absence of p53 (lane 3). However, cells infected with Ad-p53 either alone or with rTat (lanes 5, 6) activated the Bax promoter (lanes 5 and 6). Interestingly, Tat failed to activate the Bax promoter in the absence of p73 even when p53 is expressed (lane 8). Similarly, p53 also failed to activate the Bax promoter in the absence of p73 (lane 7). Thus activation of the Bax promoter by Tat is dependent on both p53 and p73.
Phosphorylation of p53 on serines 15 and 46 in the presence of Tat
Induction of cell death requires (i) phosphorylation of p53 by ATM or DNA-PK (on serines 15 and 46, respectively), (ii) accumulation of p53 in the nucleus, (iii) subsequent activation of p53-target genes (e.g., Bax, p21WAF), and (iv) dissociation from phosphorylated Mdm2 on serine 395.34,46 Further, phosphorylation of the p38MAPK pathway was shown to promote p53 accumulation.13 Therefore, we examined the phosphorylation status of p53 on serines 15 and 46 in SH-SY5Y cells. Cells were mock-treated or treated with 1 pg/ml of rTat and/or SB-202190 (an inhibitor of p38MAPK). The cells were also transfected with 20 nM of siRNA-p73 for 24 h prior to rTat addition. After 24 h, cell extracts were prepared and analyzed by Western blot for phospho-p53 (Ser15 and 46), cleaved caspase 3, p38MAPK (total and phosphorylated) or Grb2. As shown in Figure 5, Tat increased p53 phosphorylation on both serines, induced phosphorylation of p38MAPK and promoted cleavage of caspase 3 (lane 2) when compared to untreated cells (lane 1). Interestingly, Tat failed to promote these modifications in the presence of p38 inhibitor or siRNA-p73 (lanes 4 and 5). These results indicate that these modifications may be involved in Tat-induction of neuronal death.
Figure 5.
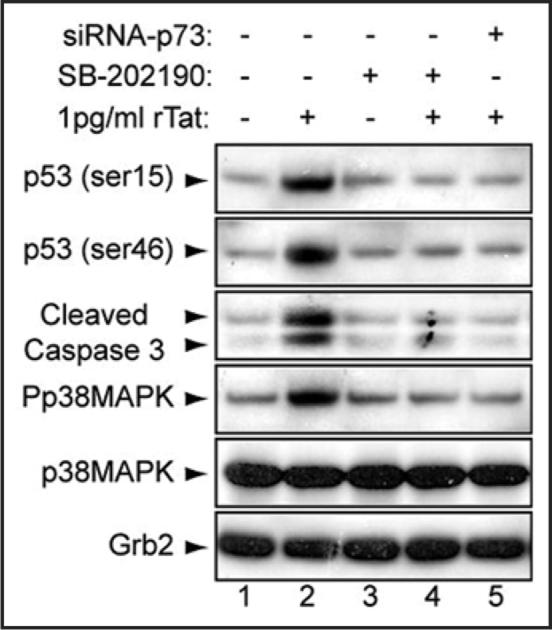
Tat phosphorylates p53 on ser15 & ser46 in neuronal cells. Cells were untreated, Tat- and/or p38-inhibitor-treated or transfected with siRNA-p73 as indicated, lysates were prepared and analyzed by Western blot with anti-p53, anti-phospho-p53 (serine 15 and 46), anti-cleaved caspase 3, anti-p73, anti-p38MAPK (total and phosphorylated), or anti-Grb-2 antibodies.
Discussion
HIV-associated dementia (HAD) and the less severe disorder, minor cognitive motor disorder (MCMD) are the most common AIDS-associated neurological disorders and are characterized by the development of synaptodendritic injury. The introduction of highly active antiretroviral therapy (HAART) has improved the survival and reduced incidence of HAD.25 However, the incidence of MCMD in advanced HIV/AIDS exceeds 20% annually and may rise further with prolonged survival of patients receiving HAART. In addition, HAD and MCMD have a combined prevalence of ~30-50% in advanced HIV-1, suggesting HAART does not provide complete protection against neurological disorders.1 Hence, it is crucial to identify the mechanisms and the factors involved in causing neuronal injury. The viral protein Tat is among those factors, especially since it can be released by HIV-1-infected cells such as macrophages, microglia, and astrocytes and taken up by adjacent neuronal cells in quantities that are biologically active (Deshmane et al., manuscript submitted). The role of HIV-1 Tat protein in the pathogenesis of AIDS-associated disorders of the brain is now well established. Many studies from different groups, including our own, have demonstrated the ability of HIV-1-infected microglia, astrocytes and uninfected neurons to release and uptake Tat protein.2,3,9,11,14,41 However, until this time, the exact amount of Tat released and subject to uptake was unclear. We have now addressed this question by immunohistochemistry (IHC) and proteomics to show the presence and the amount of Tat involved. Tat was also found in cerebrospinal fluid from brains of HIV-1 infected patients with severe neurological symptoms.9 Many studies indicate that the degeneration of neurons involves the activation of the tumor suppressor protein p53. Based on these observations, we examined the ability of Tat to induce the p53 pathway and to cause neuronal degeneration through this pathway. We demonstrated that endogenous levels of p53 and p73 proteins are induced in the brain of AIDS patients and in neuronal cells treated with Tat (Figs. 1 and 2). These data have provided us with a strong rationale to examine p73 in the presence and absence of Tat in neuronal cells. Further, we show that even a very small amount of Tat can cause significant neurite retraction in human neurons (Fig. 2). Of note, we previously demonstrated that this same amount of Tat is functionally active (data not shown). We next assessed the role of Tat in neuronal degeneration with and without p53 and p73. Interestingly, we found that Tat induces p53 through a pathway that involved p73, and that induction of p53 requires the presence of p73 (Figs. 3 and 4). Therefore, one can conclude that induction of p53 by Tat is an indirect event. Finally, since accumulation of p53, stability and activity of p73 are mediated through the p38MAPK pathway,13,29 we found that Tat failed to induce p53 and/or p73 in the presence of inhibitor of p38MAPK (Fig. 5). Taken together, our data show that Tat causes neuronal degeneration through a pathway involving cofactors including p73 and p53 and that modifications of these factors at the transcriptional and post-translational levels leads to neuronal degeneration.
Our data correlate with the previously published data regarding the ability of p53 to cause neuronal degeneration in the brain of AIDS patients. However, the mechanisms of this event remain unclear. In this regard, it has been suggested that the HIV-1 gp120 protein might play a role in the induction of p53 leading to neuronal apoptosis.2,22,27 We examined this hypothesis, and found that gp120 did not affect endogenous levels of p73 in neuronal cells (data not shown) and thus suggest that Tat might be the HIV protein responsible for p73 induction. However, Tat and p73 failed to cause neuronal degeneration in the absence of p53 or in the presence of p38MAPK inhibitor. It is noteworthy to mention that in T cells, Tat was previously shown to inhibit p53 functions and to decrease its levels.28 Thus, it is likely that the regulation of p53 by Tat is cell type-specific. Further, it is interesting to mention that the presence of p53 is required for the transcriptional regulation and replication of HIV-1.47 Therefore, one can conclude that p53 plays a dual role, positive in HIV-infected cells and negative in uninfected neuronal cells exposed to Tat. In addition, our results regarding the impact of p53 on neuronal degeneration is not without a precedent. In this regard, it was shown that p53 cooperates with Moloney murine leukemia virus-ts1 to mediate neuronal degeneration.48 Further, p53 also causes neuronal apoptosis in the brain of mice infected with herpes simplex virus type 2.49 In both cases the mechanisms used by p53 to cause neuronal death are unknown. It should be noted that accumulation of p53 and p73 normally leads to the accumulation of Mdm2 leading to p53 degradation.19,20 However, it has been shown that Mdm2 can be recruited by p73 without promoting p73 degradation implying that Mdm2 contributes to the stability of p73 leading to post-translational modifications and stability of p53.50 This finding could explain the necessity of Tat and p53 to the p73 protein in order to cause cell injury (our data and ref. 30). In addition, we previously demonstrated that Mdm2 failed to interact with p53 in Tat-treated cells due to Mdm2 phosphorylation (Mukerjee et al., submitted).
Moreover, recent studies indicate that the transcriptional activities of p53 and p73 depend on their post-translation modifications and that these modifications contribute to their stabilization.45 Similar to p53, once activated and stable, p73 can cause cell cycle arrest and promote cell death. In accord with this observation, we previously demonstrated the ability of Tat to physically interact with p73 and to prevent its acetylation and hence render it incapable of causing cell death.41 Therefore, it is important to evaluate the stability and post-translation modifications of p73 in neurons exposed to Tat. Further, it should be noted that p73 is also frequently deleted in neuroblastoma and to be predominantly expressed as a truncated anti-apoptotic isoform (ΔNp73), which antagonizes the functions of both p53 and full-length p73 protein (TAp73).32,37 However,using a specific p73 antibody raised against the N-terminal domain of p73α, we found that full-length p73 was expressed in brain tissue and in SH-SY5Y cells, and that ΔNp73 failed to prevent Tat induction of p73 (data not shown).
In summary, our data (schematized in Fig. 6) and previous studies51 indicate that HIV-1 Tat causes synaptodendritic injury leading to neuronal degeneration and the development of HIV-1 associated dementia (Fig. 2). We have found strong evidence for involvement of p53 and p73 in this process. Experiments with brain tissue from HIV-1-infected patients (with or without dementia) revealed increase in the endogenous levels of p53 (Fig. 1). This observation was further confirmed with the use of Tat protein (Figs. 2 and 4). Interestingly, Tat caused neuronal degeneration in the presence of both proteins but not in their absence (Fig. 3). Surprisingly, Tat required the presence of p73 in order to induce endogenous levels of p53 (Fig. 4). In addition, p73 induction by Tat is p38-dependent and Tat cannot induce p53 or Bax in the absence of either p73 or p38 (Figs. 4 and 5). These results are in agreement with other published reports and indicate that p53, p73 and Tat play a role in the induction of synaptodendritic injury, neuronal degeneration and the development of HAD. Thus, we strongly believe that we identified the pathway used by Tat to cause synaptodendritic injury contributing to the development of AIDS neuropathogenesis. These results may provide possible therapeutic options for the neurodegeneration often observed in AIDS patients.
Figure 6.
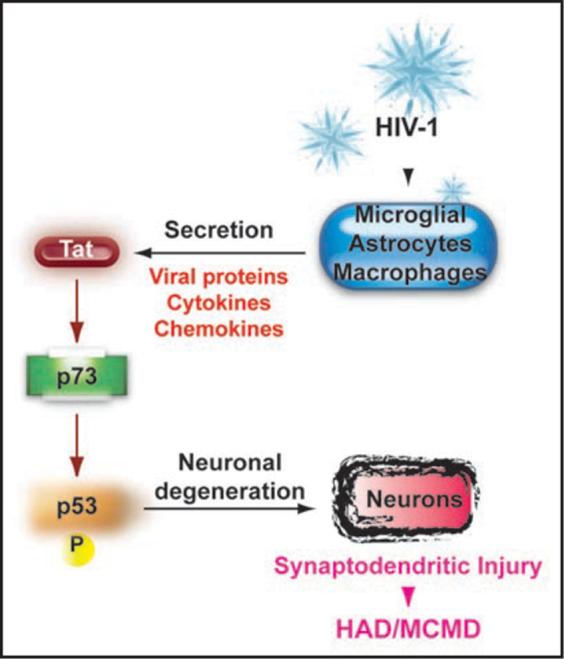
Potential pathway used by HIV-Tat to cause neurodegeneration. Schematic representation of the pathway used by Tat to cause synaptodendritic injury. Briefly, HIV-1 infects microglia, astrocytes and macrophages. Infected cells release viral proteins including Tat as well as several other toxins such as cytokines and chemokines. Once released from infected cells, proteins/toxins taken up by neuronal cells promote activation of the p53 family members leading to synpatodendritic injury and the development of HAD and/or MCMD.
Materials and Methods
Plasmids
The pcDNA3-Tat, CMV-p73α and p73β, and pDsRed1-mito plasmids have been previously described.38 The Bax-luciferase promoter was kindly provided by J. Manfredi (Columbia University, NY).
Cell culture, and transfection assays
Human neuroblastoma (SH-SY5Y and SK-N-MC) and H1299 lung carcinoma cell lines were maintained in DMEM + 10% FBS (Gibco), 100 units/ml penicillin, 50 μg/ml streptomycin. Human carcinoma HCT116 and HCT116 (p53-/-) cell lines were kindly provided by B. Vogelstein (Johns Hopkins University, MD) and maintained in McCoy's medium containing 10% FBS (Life Technologies, Grand Island, NY). Confluent SH-SY5Y were replated at 1-5 × 105 cells/ml for different experiments and induced to differentiate by treatment with 10 μM retinoic acid (Sigma, St. Louis, MO) for 7 d with medium changes every two days. For all of the experiments cells were serum starved for 6 h in the presence of 10 mM RA prior to treatment with rTat or transfection. Cells were transfected with 0.1 μg of Bax-luc reporter plasmid or cotransfected with 0.25 μg of various expression cDNAs as previously described.39 The cells were also transfected with 1.0 μg of CMV-β-galactosidase plasmid. The amount of DNA used for each transfection was normalized with pcDNA3 vector plasmid. Each transfection was repeated multiple times with different plasmid preparations. Cell extracts were prepared 48 h after transfection and luciferase and β-gal determined as previously described.40 All values were normalized for β-gal.
Recombinant adenoviruses
p53 or Tat cDNA (1179 and 300 bp) were excised from pcDNA3-p53 or Tat and cloned into the EcoRI and NheI sites of the adenovirus-shuttle plasmid pDC515 under the control of the murine cytomegalovirus promoter (purchased from Microbix Inc., Ontario, Canada). Recombinant shuttle plasmids containing p53 or Tat sequences (pDC515-p53 or Tat) were transfected into HEK-293 cells with pBHGfrt (del) E1, 3FLP, and a plasmid that provides adenovirus type-5 genome deleted in E1 and E3 genes. Plaques of recombinant adenovirus arising as a result of frt/FLP recombination were isolated, grown and purified by cesium chloride density equilibrium banding. Empty shuttle plasmid, pDC515, was used to construct control adenoviral vector (Adeno-null, a virus without a transgene). Adeno-p53, adeno-Tat or adeno-null were used at an MOI of 1.0 plaque forming units per cell. [MOI = multiplicity of infection].
Western blotting
SH-SY5Y or H1299 cells were treated with recombinant highly purified toxin-free Tat protein as indicated (rTat; 101 aa) (highly pure toxin-free Tat purchased from Immuno Diagnostics, Inc., Woburn, MA). Twenty-four hours post-treatment, 50 μg of cell extracts were used for Western blot analysis as described previously.41 Anti-Grb2 antibody was used as a control for equal protein loading.
RNA interference
SmartPool small interfering RNA against p53 (siRNA-p53) or p73 (siRNA-p73) were transfected at a concentration of 20 nM into approximately 1 × 105 SH-SY5Y in serum free media RNAiFect transfection reagents (Qiagen). To evaluate siRNA-p53 or siRNA-p73 efficiency, SH-SY5Y cells were co-transfected with siRNA-p53 or siRNA-p73 and cell lysates were analyzed by Western blot assay using anti-p53 or p73 antibodies.
Immunohistochemistry
The tissue, which had been previously formalin fixed and paraffin embedded, was sectioned at 5 μm thickness and placed on electromagnetically charged glass slides. Sections were deparaffinized in xylene and re-hydrated through descending grades of alcohol up to water. Immunohistochemistry was performed utilizing an Avidin-Biotin-Peroxidase kit, according to the manufacturer's instructions (Vectastain Elite ABC Peroxidase kit; Vector Laboratories Inc., Burlingame, California). Antigen retrieval was performed in citrate buffer for 30 min at 95°C in a vacuum oven. After a 20 min cooling period, sections were rinsed with PBS and endogenous peroxidase was quenched with 3% H2O2 in methanol for 30 min. Sections were then rinsed with PBS and a blocking step was performed with normal goat or normal rabbit serum at room temperature in a humidified chamber for 2 h. Primary antibodies were incubated overnight at room temperature. After rinsing with PBS, sections were incubated for one hour at room temperature with biotinylated anti-rabbit or anti-goat secondary antibodies. The tissue was subsequently incubated with Avidin-Biotin-Peroxidase complexes for 1 h at room temperature according to the manufacturer's instructions (Vector Laboratories, Burlingame, CA) and finally, the sections were developed with a diaminobenzidine substrate (Sigma Laboratories, Saint Louis, MO), counterstained with hematoxylin and coverslipped with Permount (Fisher Scientific, Pittsburg, PA).
Apoptosis assays
SK-N-MC cells were co-transfected with pDsRed1-mito to evaluate the efficiency of transfection and with 1.0 μg of CMV-Tat or CMV-p73 expression plasmids separately or in combination. pcDNA3 empty vector was used as a negative control. Forty-eight hours after transfection, cells were collected and evaluated for the presence of apoptosis and efficiency of transfection. For detection of terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL), cells were immunolabeled and counted according to the manufacturer's descriptions by the in situ Cell Death Detection Kit (Boehringer Mannheim).
Acknowledgements
This work was supported by grants awarded by NIH to B.E.S., S.A. and L.D.V.
References
- 1.Kaul M, Lipton SA. Mechanisms of neuroimmunity and neurodegeneration associated with HIV-1 infection and AIDS. J Neuroimmune Pharmacol. 2006;1:138–51. doi: 10.1007/s11481-006-9011-9. [DOI] [PubMed] [Google Scholar]
- 2.Kaul M. HIV's double strike at the brain: neuronal toxicity and compromised neurogenesis. Front Biosci. 2008;13:2484–94. doi: 10.2741/2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gendelman HE, Persidsky Y, Ghorpade A, Limoges J, Stins M, Fiala M, Morrisett R. The neuropathogenesis of the AIDS dementia complex. AIDS. 1997;11:35–45. [PubMed] [Google Scholar]
- 4.Pulliam L, Herndier BG, Tang NM, McGrath MS. Human immunodeficiency virus-infected macrophages produce soluble factors that cause histological and neurochemical alterations in cultured human brains. J Clin Invest. 1991;87:503–12. doi: 10.1172/JCI115024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baba M. Inhibitors of HIV-1 gene expression and transcription. Curr Top Med Chem. 2004;4:871–82. doi: 10.2174/1568026043388466. [DOI] [PubMed] [Google Scholar]
- 6.Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998;17:7056–65. doi: 10.1093/emboj/17.23.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garber ME, Wei P, Kewal Ramani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, Jones KA. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998;12:3512–27. doi: 10.1101/gad.12.22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaehlcke K, Dorr A, Hetzer-Egger C, Kiermer V, Henklein P, Schnoelzer M, Loret E, Cole PA, Verdin E, Ott M. Acetylation of Tat defines a cyclinT1-independent step in HIV transactivation. Mol Cell. 2003;12:167–76. doi: 10.1016/s1097-2765(03)00245-4. [DOI] [PubMed] [Google Scholar]
- 9.Ma M, Nath A. Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol. 1997;71:2495–9. doi: 10.1128/jvi.71.3.2495-2499.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallace DR. HIV Neurotoxicity: Potential Therapeutic Interventions. J Biomed Biotechnol. 2006;3:65741. doi: 10.1155/JBB/2006/65741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andras IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res. 2003;74:255–65. doi: 10.1002/jnr.10762. [DOI] [PubMed] [Google Scholar]
- 12.Pu H, Tian J, Flora G, Lee YW, Nath A, Hennig B, Toborek M. HIV-1 Tat protein upregulates inflammatory mediators and induces monocyte invasion into the brain. Mol Cell Neurosci. 2003;24:224–37. doi: 10.1016/s1044-7431(03)00171-4. [DOI] [PubMed] [Google Scholar]
- 13.Brown L, Benchimol S. The involvement of MAPK signaling pathways in determining the cellular response to p53 activation: cell cycle arrest or apoptosis. J Biol Chem. 2006;281:3832–40. doi: 10.1074/jbc.M507951200. [DOI] [PubMed] [Google Scholar]
- 14.Buscemi L, Ramonet D, Geiger JD. Human immunodeficiency virus type-1 protein Tat induces tumor necrosis factor-alpha-mediated neurotoxicity. Neurobiol Dis. 2007;26:661–70. doi: 10.1016/j.nbd.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Djebaïli M, Rondouin G, Baille V, Bockaert J. p53 and Bax implication in NMDA induced-apoptosis in mouse hippocampus. Neuroreport. 2000;11:2973–6. doi: 10.1097/00001756-200009110-00029. [DOI] [PubMed] [Google Scholar]
- 16.Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem. 1999;73:578–86. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- 17.Okorokov AL. p53 in a crosstalk between DNA repair and cell cycle checkpoints. Cell Cycle. 2003;2:233–5. [PubMed] [Google Scholar]
- 18.Sui Z, Sniderhan LF, Schifitto G, Phipps RP, Gelbard HA, Dewhurst S, Maggirwar SB. Functional synergy between CD40 ligand and HIV-1 Tat contributes to inflammation: implications in HIV type 1 dementia. J Immunol. 2007;178:3226–36. doi: 10.4049/jimmunol.178.5.3226. [DOI] [PubMed] [Google Scholar]
- 19.Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 20.Lim MS, Elenitoba-Johnson KS. Ubiquitin ligases in malignant lymphoma. Leuk Lymphoma. 2004;45:1329–39. doi: 10.1080/10428190410001663635. [DOI] [PubMed] [Google Scholar]
- 21.Hay TJ, Meek DW. Multiple sites of in vivo phosphorylation in the MDM2 oncoprotein cluster within two important functional domains. FEBS Lett. 2000;478:183–6. doi: 10.1016/s0014-5793(00)01850-0. [DOI] [PubMed] [Google Scholar]
- 22.Garden GA, Morrison RS. The multiple roles of p53 in the pathogenesis of HIV associated dementia. Bioch Biophy Res Com. 2005;331:799–809. doi: 10.1016/j.bbrc.2005.03.185. [DOI] [PubMed] [Google Scholar]
- 23.Liang SH, Clarke MF. Regulation of p53 localization. Eur J Biochem. 2001;268:2779–83. doi: 10.1046/j.1432-1327.2001.02227.x. [DOI] [PubMed] [Google Scholar]
- 24.Thompson KA, McArthur JC, Wesselingh SL. Correlation between neurological progression and astrocyte apoptosis in HIV-associated dementia. Ann Neurol. 2001;49:745–52. doi: 10.1002/ana.1011. [DOI] [PubMed] [Google Scholar]
- 25.Silva C, Zhang K, Tsutsui S, Holden JK, Gill MJ, Power C. Growth hormone prevents human immunodeficiency virus-induced neuronal p53 expression. Ann Neurol. 2003;54:605–14. doi: 10.1002/ana.10729. [DOI] [PubMed] [Google Scholar]
- 26.New DR, Ma M, Epstein LG, Nath A, Gelbard HA. Human immunodeficiency virus type 1 Tat protein induces death by apoptosis in primary human neuron cultures. J Neurovirol. 1997;3:168–73. doi: 10.3109/13550289709015806. [DOI] [PubMed] [Google Scholar]
- 27.Garden GA, Guo W, Jayadev S, Tun C, Balcaitis S, Choi J, Montine TJ, Moller T, Morrison RS. HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 2004;18:1141–3. doi: 10.1096/fj.04-1676fje. [DOI] [PubMed] [Google Scholar]
- 28.Li CJ, Wang C, Friedman DJ, Pardee AB. Reciprocal modulations between p53 and Tat of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1995;92:5461–4. doi: 10.1073/pnas.92.12.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perfettini JL, Castedo M, Nardacci R, Ciccosanti F, Boya P, Roumier T, Larochette N, Piacentini M, Kroemer G. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J Exp Med. 2005;201:279–89. doi: 10.1084/jem.20041502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu J, Baron V, Mercola D, Mustelin T, Adamson ED. A network of p73, p53 and Egr1 is required for efficient apoptosis in tumor cells. Cell Death Differ. 2007;14:436–46. doi: 10.1038/sj.cdd.4402029. [DOI] [PubMed] [Google Scholar]
- 31.Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–4. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 32.Melino G, Lu X, Gasco M, Crook T, Knight RA. Functional regulation of p73 and p63: development and cancer. Trends Biochem Sci. 2003;28:663–70. doi: 10.1016/j.tibs.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 33.Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, Cesareni G, Melino G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836–48. doi: 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–6. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 35.Jost CA, Marin CM, Kaelin WG., Jr p73 is a human p53-related protein that can induce apoptosis. Nature. 1997;389:191–4. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 36.Fulco M, Costanzo A, Merlo P, Mangiacasale R, Strano S, Blandino G, Balsano C, Lavia P, Levrero M. p73 is regulated by phosphorylation at the G2/M transition. J Biol Chem. 2003;278:49196–202. doi: 10.1074/jbc.M304921200. [DOI] [PubMed] [Google Scholar]
- 37.Minty A, Dumont X, Kaghad M, Caput D. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J Biol Chem. 2000;275:36316–23. doi: 10.1074/jbc.M004293200. [DOI] [PubMed] [Google Scholar]
- 38.Saunders M, Eldeen MB, Del Valle L, Reiss K, Peruzzi F, Mameli G, Gelman BB, Khalili K, Amini S, Sawaya BE. p73 modulates HIV-1 Tat transcriptional and apoptotic activities in human astrocytes. Apoptosis. 2005;10:1419–31. doi: 10.1007/s10495-005-2467-x. [DOI] [PubMed] [Google Scholar]
- 39.Mukerjee R, Sawaya BE, Khalili K, Amini S. Association of p65 and C/EBPbeta with HIV-1 LTR modulates transcription of the viral promoter. J Cell Biochem. 2007;100:1210–6. doi: 10.1002/jcb.21109. [DOI] [PubMed] [Google Scholar]
- 40.Mameli G, Deshmane SL, Ghafouri M, Cui J, Simbiri K, Khalili K, Mukerjee R, Dolei A, Amini S, Sawaya BE. C/EBPbeta regulates human immunodeficiency virus 1 gene expression through its association with cdk9. J Gen Virol. 2007;88:631–40. doi: 10.1099/vir.0.82487-0. [DOI] [PubMed] [Google Scholar]
- 41.Amini S, Mameli G, Del Valle L, Skowronska A, Reiss K, Gelman BB, White MK, Khalili K, Sawaya BE. p73 interacts with HIV-1 Tat in astrocytic cells and prevents its acetylation on Lys28. Mol Cell Biol. 2005;25:8126–38. doi: 10.1128/MCB.25.18.8126-8138.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanders VJ, Everall IP, Johnson RW, Masliah E. Fibroblast growth factor modulates HIV coreceptor CXCR4 expression by neural cells. J Neurosci Res. 2000;59:671–9. doi: 10.1002/(SICI)1097-4547(20000301)59:5<671::AID-JNR10>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 43.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, Ferrara P, McKeon F, Caput D. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–19. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 44.Chen LH, Hsu CY, Weng CF. Involvement of p53 and Bax/Bad triggering apoptosis in thioacetamide-induced hepatic epithelial cells. World J Gastroenterol. 2006;12:5175–81. doi: 10.3748/wjg.v12.i32.5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koida N, Ozaki T, Yamamoto H, Ono S, Koda T, Ando K, Okoshi R, Kamijo T, Omura K, Nakagawara A. Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. J Biol Chem. 2008 doi: 10.1074/jbc.M710608200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park JS, Kim EJ, Lee JY, Sin HS, Namkoong SE, Um SJ. Functional inactivation of p73, a homolog of p53 tumor suppressor protein, by human papillomavirus E6 proteins. Int J Cancer. 2001;91:822–7. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1130>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 47.Pauls E, Senserrich J, Clotet B, Esté JA. Inhibition of HIV-1 replication by RNA interference of p53 expression. J Leukoc Biol. 2006;80:659–67. doi: 10.1189/jlb.0306189. [DOI] [PubMed] [Google Scholar]
- 48.Kim HT, Tasca S, Qiang W, Wong PK, Stoica G. Induction of p53 accumulation by Moloney murine leukemia virus-ts1 infection in astrocytes via activation of extracellular signal-regulated kinases ½. Lab Invest. 2002;82:693–702. doi: 10.1097/01.lab.0000017373.82871.45. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe D, Honda T, Nishio K, Tomita Y, Sugiura Y, Nishiyama Y. Corneal infection of herpes simplex virus type 2-induced neuronal apoptosis in the brain stem of mice with expression of tumor suppressor gene (p53) and transcription factors. Acta Neuropathol. 2000;100:647–53. doi: 10.1007/s004010000240. [DOI] [PubMed] [Google Scholar]
- 50.Ozaki T, Hosoda M, Miyazaki K, Hayashi S, Watanabe K, Nakagawa T, Nakagawara A. Functional implication of p73 protein stability in neuronal cell survival and death. Cancer Lett. 2005;228:29–35. doi: 10.1016/j.canlet.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 51.Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]