

Abstract
The proteasome controls the turnover of most cellular proteins. Two structural features are typically required for proteins to be degraded: covalently attached ubiquitin polypeptides that allow binding to the proteasome, and an unstructured region in the targeted protein that initiates proteolysis. Here, we have tested the degradation of model proteins to further explore how the proteasome selects its substrates. Using purified yeast proteasome and mammalian proteasome in cell lysate, we have demonstrated that the two structural features can act in trans when separated onto different proteins in a multi-subunit complex. In such complexes, the location of the unstructured initiation site and its chemical properties determine which subunit is degraded. Thus, our findings reveal the molecular basis of subunit specificity in the degradation of protein complexes. In addition, our data provide a plausible explanation for how adaptor proteins can bind to otherwise stable proteins and target them for degradation.
The ubiquitin proteasome system (UPS) controls the cellular concentrations of regulatory proteins, degrades aberrant or misfolded polypeptides, and produces peptides as part of the adaptive immune response1. Most proteins are targeted to the proteasome by the covalent attachment of many copies of the small protein ubiquitin2. This modification is recognized directly by the proteasome3,4 which then degrades its substrates by running sequentially along the substrates' polypeptide chain5. Many UPS substrates, such as cyclins, cyclin-dependent kinase (CDK) inhibitors, transcription regulators (e.g., IκBα), and transcription factors, are parts of multimeric complexes before degradation1. In some of these cases, the proteasome degrades specifically only one of the subunits to create a remodeled complex with a new composition and function. This remodeling activity of the proteasome was first demonstrated through the observation of subunit specificity in the degradation of tetrameric βgal substrates in vitro6 and in vivo7. Two well-known physiological examples of subunit-specific degradation are the proteolysis of cyclins from their complex with CDK8 and the proteolysis of the CDK inhibitor Sic1 while bound to the cyclin / CDK complex9. Until now, the specificity of degradation by the proteasome has been assumed to result directly from the specificity of the ubiquitination reaction. In other words, the protease has been thought to begin to degrade its substrate at point of the ubiquitin modification and, therefore, to degrade only the ubiquitinated subunit.
However, protein ubiquitination, or protein binding to the proteasome, are not sufficient for effective degradation. For example, the ubiquitinated Cdc349-11 and proteasome-bound Rad2310,12,13 are stable proteins. The reason for this is probably that efficient proteasome-mediated proteolysis requires the presence of a second component of the degradation signal in the form of an unstructured initiation site in the substrate14. The ubiquitin tag allows the proteasome to bind target proteins3,4, and the unstructured region serves as the initiation site where the proteasome begins to degrade the substrate14. We now ask whether the two components of the targeting signal could work together when separated onto two different polypeptides chains in a complex. The existence of a trans-targeting mechanism of this type would require a new explanation for the observed subunit specificity of the proteasome.
We find that a substrate protein that lacks a ubiquitin modification can be targeted for rapid degradation when it associates with a ubiquitinated partner protein that serves as a proteasome adaptor. While the adaptor localizes the substrate onto the proteasome, the adaptor itself can remain stable and act catalytically. Insertion of an initiation site into the ubiquitinated adaptor leads to the degradation of the adaptor and the relative stabilization of the binding partner. Therefore, we propose that the subunit specificity of the proteasome results from the selection of the degradation initiation site and is not determined primarily by the location of the ubiquitin modification.
Results
Targeting and degrading subunits of a protein complex
To investigate the degradation of protein complexes by the proteasome, we constructed a series of UPS substrates from the ribonuclease barnase and its inhibitor barstar15 (Fig. 1a). The two proteins bind each other with a dissociation constant of ∼10−9 M (ref. 16) to form a well-characterized complex. First, we targeted proteins to degradation in reticulocyte lysate using the N-end rule degron developed and characterized previously (e.g., ref. 17-19). The N-degron contains both ubiquitination and initiation sites14 and induces the efficient degradation of proteins19. We fused the N-degron to the N terminus of barstar through a DHFR linker domain to make N-degron-DHFR-barstar (Fig. 1b). When we incubated the hybrid protein in reticulocyte lysate, the ubiquitin moiety at its N terminus was cleaved rapidly, and the remainder was ubiquitinated on Lys residues downstream in the degron (Supplementary Fig. 1 online). The ubiquitinated protein was then degraded by the proteasome (Fig. 1c). As expected for protein degradation by the UPS, proteolysis was inhibited by both the depletion of ATP and the addition of a lysine-less ubiquitin mutant, which prevents polyubiquitination (Fig. 1c). The proteasome inhibitor MG132 (1) also prevented degradation and led to the accumulation of the ubiquitinated substrate (Fig. 1c). Ubiquitination occurred in the linker region between the N-terminal ubiquitin and the DHFR-barstar fusion because deletion of a large part of the linker (indicated by the square bracket in Fig. 1b) prevented ubiquitination and degradation of the protein (Fig. 1c).
Figure 1.
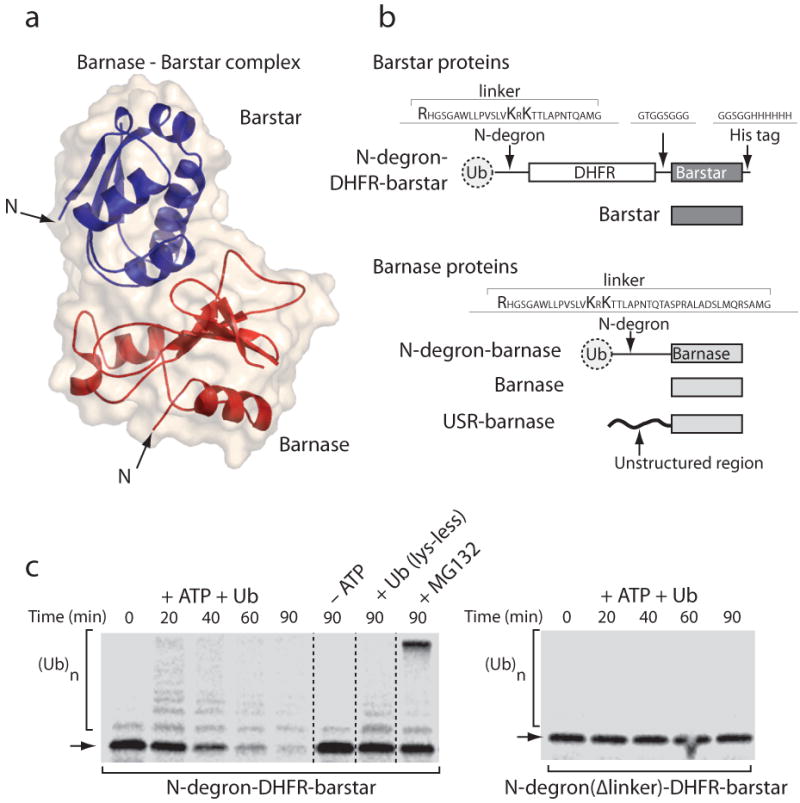
Proteasome substrates and N end rule degron-mediated degradation. (a) Structure of barnase bound to its inhibitor barstar [PDB ID 1BRS] and the molecular surface of this complex shown in cartoon representation using PyMol (DeLano Scientific LLC, Palo Alto, CA; www.pymol.org). (b) Barstar and barnase hybrid proteins. Barstar was targeted to the proteasome in reticulocyte lysate by an N end rule tag, which was composed of an N-terminal ubiquitin followed by an unstructured extension containing two lysine residues (N-degron). The N-degron was attached to the N terminus of barstar through an intervening DHFR domain. Barnase was targeted to the proteasome through an N end-rule degron in which the linker contained an additional 15 amino acids. Other barnase constructs contained N-terminal unstructured initiation sites derived from cytochrome b2 or lac repressor. (c) Proteolysis of barstar by the ubiquitin proteasome system in rabbit reticulocyte lysate. Incubation of N-degron-DHFR-barstar in reticulocyte lysate leads to its rapid degradation, which was inhibited in the absence of ATP, by addition of an excess of a ubiquitin mutant that inhibits poly-ubiquitination because it lacks all Lys residues, and by the proteasome inhibitor MG132 (1). Vertical lines separate radiogram images from different gels that were acquired simultaneously and set to the same exposure. Ubiquitination of substrates occurs at the lysine residues in the linker region of the N-degron, and deletion of the linker region in N-degron eliminates substrate ubiquitination as well as its degradation.
Attaching the N-end rule degron next to the N terminus of barnase (Fig. 1b) led to rapid ubiquitination and degradation of barnase in rabbit reticulocyte lysate, even when barnase was bound to barstar5 (Fig. 2a). Thus, the proteasome was able to extract its substrate from the complex as expected5-9. Next, we removed the N-degron from barnase and transferred it back to the N terminus of barstar through the DHFR linker (to make N-degron-DHFR-barstar; Fig. 1b). As a result, the bound barnase was no longer degraded because it lacked both a ubiquitin modification and a degradation initiation site (Fig. 2b). However, surprisingly, barnase was degraded again when we appended unstructured regions to its N terminus (Fig. 2c). Degradation was due to the UPS because degradation depended on the presence of the N-degron on barstar (Fig. 2d), and degradation was inhibited by any one of three events: ATP depletion, the addition of an excess of ubiquitin lacking Lys residues, or the proteasome inhibitor MG132 (1) (Fig. 2e).
Figure 2.
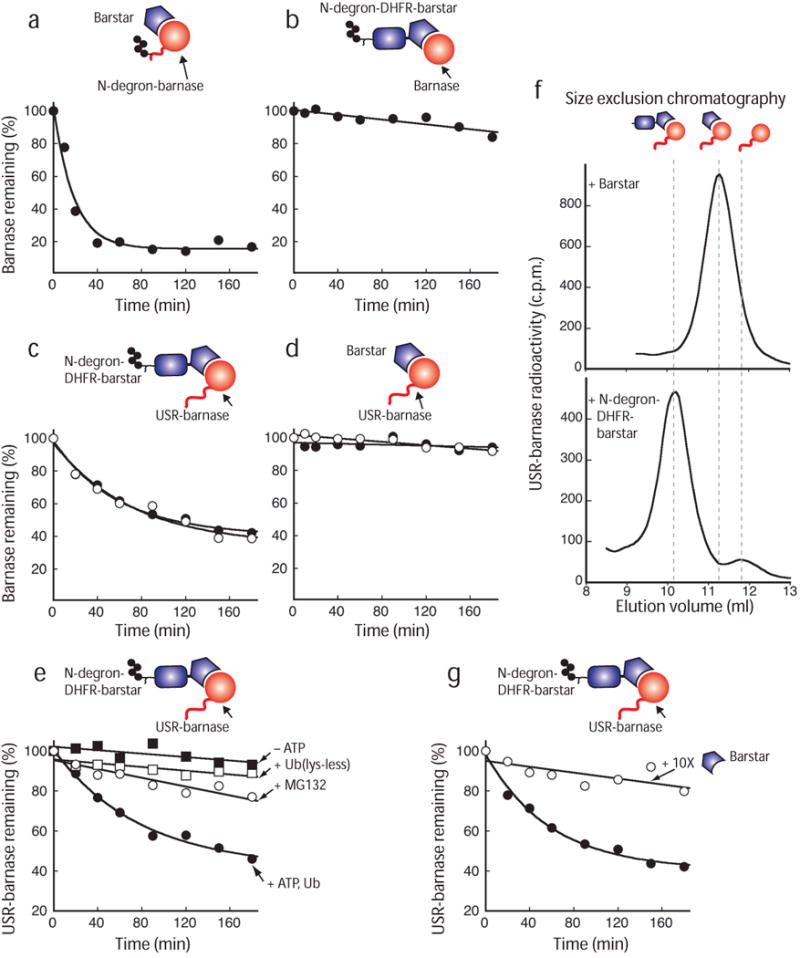
The proteasome degrades an unubiquitinated subunit in a heterodimeric complex. Radiolabeled barnase protein bound to its ligand barstar or to barstar containing the N-degron was degraded by the proteasome at 30 °C. (a) Barnase containing an N-degron and an initiation site was efficiently degraded by the proteasome when in complex with its ligand barstar. The N-degron is modified with many ubiquitin moieties, but, for simplicity, the polyubiquitin modification is indicated by four ubiquitin moieties in the schematic representation. (b) Barnase bound to N-degron-DHFR-barstar was not degraded by the proteasome unless (c) initiation sites derived from lac repressor (open circles) or cytochrome b2 (filled circles) were attached to barnase's N terminus. (d) Barnase proteins containing N-terminal initiation sites were not degraded when bound to barstar was not ubiquitinated because it lacked the N-degron. (e) Proteolysis of barnase bound to N-degron-DHFR-barstar adaptor was dependent on an active ubiquitin-proteasome system and was inhibited in the absence of ATP (filled squares) by the proteasome inhibitor MG132 (1) (open circles) and by the addition of excess of a ubiquitin mutant that inhibits poly-ubiquitination because it lacks all Lys residues (open squares). (f) Size exclusion chromatography elution profiles of [35S]-labeled barnase containing an unstructured initiation site bound to barstar or a barstar hybrid containing an N-degron. (g) Barnase containing an initiation site was assembled with N-degron-DHFR-barstar in the absence (filled circles) or presence (open squares) of 10-fold excess of untagged barstar and was degraded by the proteasome in rabbit reticulocyte lysate at 30 °C. Graphs show one representative dataset of at least three independent experiments.
Barnase had to be bound to barstar containing the ubiquitination tag to be degraded. Gel filtration of the complex showed that barnase and barstar associated (Fig. 2f), and displacement of the ubiquitinated barstar from barnase through the addition of an excess of unmodified barstar prevented barnase degradation (Fig. 2g). Barnase containing the appended unstructured region remained stable when not bound to barstar (Supplementary Fig. 2 online).
It is unlikely that barnase itself became ubiquitinated in these experiments because it contained only one Lys residue, which was buried at the center of the barstar-binding site. Some proteins become ubiquitinated at their N-terminal α-amino group20, but this mechanism is unlikely to explain the degradation of the barnase subunit here. N-terminal ubiquitination usually depends on specific amino acid sequences near the N terminus20, yet we found that two unrelated sequences at the N terminus of barnase (from yeast cytochrome b2 and E. coli lac repressor) led to its degradation (Fig. 2c). Furthermore, barnase was not degraded when bound to barstar lacking the N-degron (Fig. 2d) or to barstar in which the N-degron was inactivated by a deletion of the 24 amino acid flexible linker following the Arg residue18 (data not shown). Finally, and most importantly, degradation can be reconstituted with purified components in the absence of ubiquitination machinery (see below).
Altogether, the observations described above suggest that the proteasome degrades the unmodified barnase subunit by engaging the initiation site on barnase in trans to the ubiquitin modification on a binding partner.
The adaptor acts catalytically and in trans
Next, we reconstituted the degradation of purified barstar and barnase protein complex by purified yeast 26S proteasome (Fig. 3a–e). In these experiments, barstar was targeted to the proteasome (Fig. 3a) by a tag consisting of four ubiquitin moieties fused in the same reading frame to barstar's N terminus21,22 (Ub4-barstar; Fig. 3b,c). Barnase contained a 35 amino-acid long unstructured region at its N terminus to serve as an initiation site (USR1-barnase, where USR stands for unstructured region; Fig. 3b,c). When mixed together, USR1-barnase bound to Ub4-barstar and was degraded by the purified proteasome (Fig. 3d).
Figure 3.
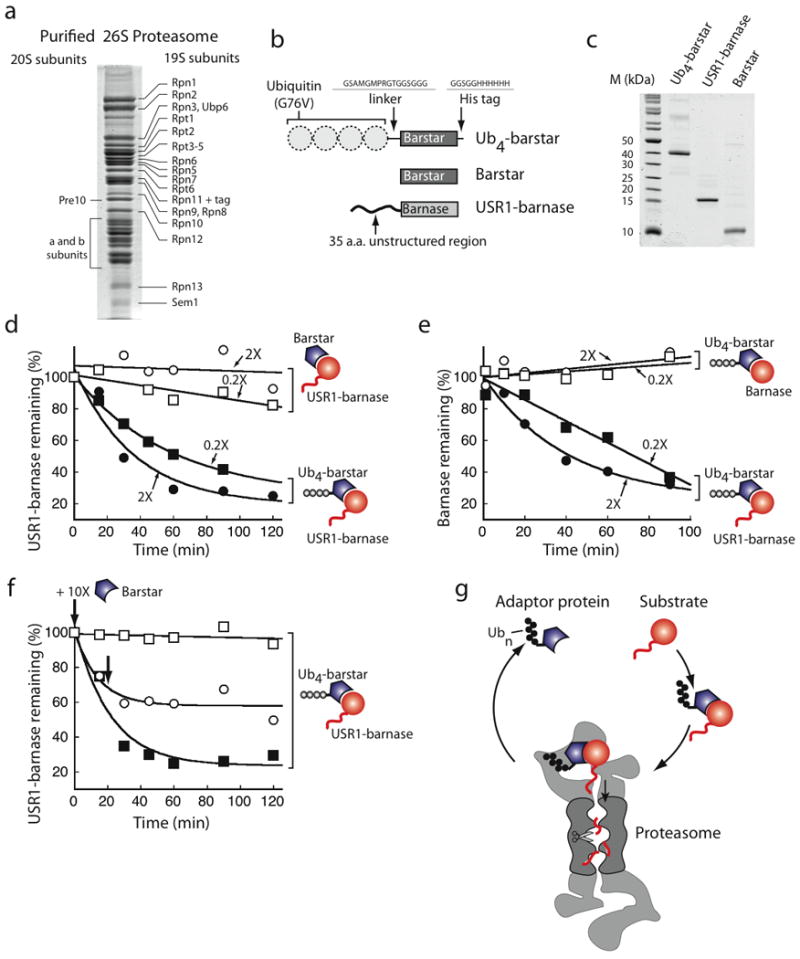
trans initiation by the proteasome. (a) Coomassie blue stained SDS-PAGE gels separating 15 μg of purified yeast 26S proteasome. Proteasome subunits are labeled following published reports49. (b) Barstar was targeted to the proteasome by an N-terminal Ub4 degron composed of four ubiquitin moieties fused in frame and then through a 16-residue linker to barstar. Barnase contained a 35 amino acid N-terminal unstructured region derived from cytochrome b2 (USR1-barnase). (c) Coomassie blue stained SDS-PAGE gels separating 1 μg of indicated proteins. (d) 0.5 μM USR1-barnase was assembled with 1 μM (2X) or 0.1 μM (0.2X) barstar (open symbols) or Ub4-barstar (filled symbols), and the complex was degraded by purified yeast proteasome. The graph shows the amount of barnase remaining (quantified by Western blotting) as a function of reaction time. (e) 0.5 μM USR1-barnase was assembled with Ub4-barstar (0.2 μM; filled squares) and degraded. A 10-fold excess of untagged barstar (open symbols) was added at the beginning of (squares) or 20 min after starting the reaction (circles), as indicated by bold arrows. (f) Degradation required an unstructured initiation site on the barnase substrate. The proteasome did not degrade barnase that lacked an unstructured region even when bound to 1 μM (2X) or 0.1 μM (0.2X) Ub4-barstar. (g) Schematic representation of trans initiation. A ubiquitinated protein may act as a recycling adaptor to shuttle its binding partner to the proteasome. The sketch shows a proteasome particle capped by two PA700 regulatory particles but other forms also exist. Graphs show one representative dataset of at two (e) or three independent experiments.
To be degraded, USR1-barnase had to be bound to Ub4-tagged barstar and remained stable when bound to untagged barstar (Fig. 3d). USR1-barnase was degraded even when the Ub4-barstar adaptor was present at five times lower concentration than the USR1-barnase substrate (Fig. 3d). This suggests that the adaptor was recycled and could act catalytically. Degradation of USR1-barnase was prevented when excess untagged barstar competed with Ub4-barstar for barnase binding. Addition of excess untagged barstar inhibited degradation rapidly when added at the beginning of the reaction or at a delay after the reaction was started (Fig. 3e).
USR1-barnase also had to contain an unstructured region to be degraded (Fig. 3f). Barnase lacking an unstructured region bound to Ub4-barstar (not shown) but remained stable (Fig. 3f). These observations show that barnase is degraded after it forms a complex with ubiquitinated barstar only when it contains an initiation site for the proteasome. They suggest that a ubiquitin modification can target a protein to degradation by acting in trans. The ubiquitinated barstar acts as an adaptor that is recycled and used multiple times (Fig. 3g).
Substrate selection through the initiation site
Next we analyzed how the proteasome selects its substrate when more than one subunit contains an initiation site. In experiments in reticulocyte lysate (Fig. 2), the N-degron-DHFR-barstar adaptor protein was degraded together with the barnase substrate (Fig. 4) because both the ubiquitinated barstar adaptor and the barnase substrate contain degradation initiation sites. The adaptor and proteasome were present in molar excess over that of the barnase substrate: barstar was added to 0.25 μM, barnase was added to a concentration of less than 20 nM, and the concentration of the proteasome in reticulocyte lysate has been estimated to be above 1 μM (ref. 23). Stabilizing the DHFR domain against unfolding with the ligand methotrexate (2) protected the N-degron-DHFR barstar adaptor from degradation24 but did not affect degradation of the barnase substrate (Fig. 4). Thus, the stability of the adaptor did not affect its ability to target barnase for degradation when both the adaptor and the proteasome were present in molar excess over the barnase substrate (Fig. 4).
Figure 4.
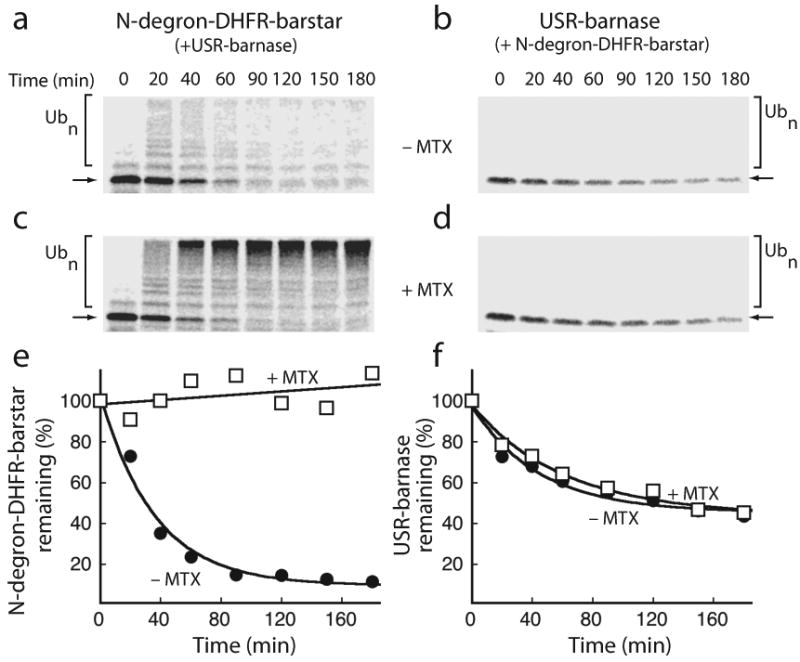
Substrate selection in reticulocyte lysate. (a-d) [35S]-methionine labeled barnase containing an initiation site was bound to an excess of N-degron-DHFR-barstar (0.25 μM) and degraded by the proteasome in reticulocyte lysate at 30 °C in the absence (a, b) or presence (c, d) of 100 μM methotrexate (2). Lanes in the autoradiograms contain equal samples of the reaction at the indicated times. (a, c) Trace amounts of radiolabeled N-degron-DHFR-barstar were added to reactions to visualize its stability in the reaction by SDS-PAGE and autoradiography. (b, d) Degradation of the barnase substrate was followed in parallel by SDS-PAGE and autoradiography. (e, f) Quantifications of the full-length protein (indicated by arrows) and its ubiquitinated forms (indicated by square brackets) in the autoradiograms are plotted as the amounts of protein remaining as a percentage of the total protein at the beginning of the reaction. Both rapidly degrading and methotrexate (2)-stabilized barstar subunit (e) functioned similarly in targeting the associated barnase for degradation (f). Graphs show one representative dataset of five independent experiments.
We then investigated how the proteasome selects substrates within a complex using the purified degradation system. In this series of experiments, the barstar adaptor contained different spacers between the ubiquitin tag and the barstar domain, and the barnase substrate contained a 35 amino acid long unstructured initiation site derived from cytochrome b2 (Fig. 3). We first used barstar adaptor at a concentration ten-times lower than that of the barnase substrate. When the barstar adaptor contained no extended unstructured regions, it remained stable and only the barnase substrate was degraded, presumably because the proteasome was unable to initiate degradation of the adaptor (Fig. 5a). Introducing a long unstructured linker into the adaptor led to the adaptor's degradation and thus turned the adaptor it into a substrate (Fig. 5b). At the same time, proteolysis of the barnase substrate was inhibited (Fig. 5b). The linker in barstar did not prevent complex formation (data not shown), and the introduction of a folded domain instead of the unstructured region into the adaptor did not destabilize the barstar adaptor and did not inhibit the degradation of barnase substrate (Fig. 5c). Thus, the explanation for the inverted stability of the adaptor and substrate is probably that the unstructured region in the adaptor is a better proteasome initiation site than the tail on the substrate. The adaptor is present at much lower concentrations than the substrate, therefore, the substrate is stabilized as the adaptor is depleted rapidly.
Figure 5.
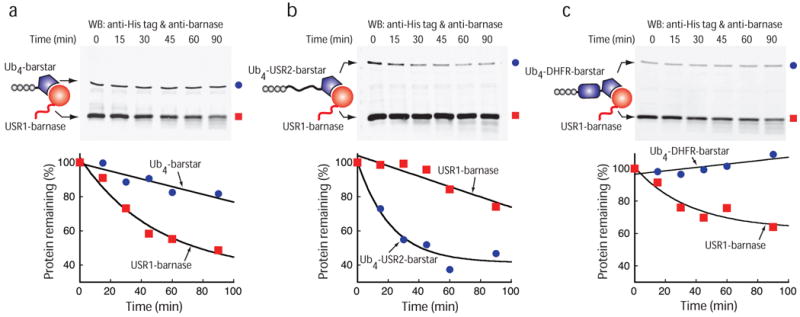
Substrate selection by the purified proteasome. (a-c) 0.5 μM barnase containing a N-terminal initiation site (red square) was bound to 0.05 μM Ub4-barstar (a, blue circles) or 0.05 μM Ub4-USR2-barstar (b, blue circles) or 0.05 μM Ub4-DHFR-barstar (c, blue circles) and the protein complex was degraded by purified proteasome at 30 °C and analyzed by Western blotting (top). Quantifications of the gels are shown as graphs plotting the amount of barnase (red) and barstar (blue) protein remaining at different reaction times as a percentage of protein amounts at the beginning of the reaction. Note that the barnase substrate is present in 10 fold molar excess over the barstar adaptor. Graphs show one representative dataset of two (b) or three independent experiments.
When both subunits are present at the same concentration, the proteasome also selects its substrate for degradation by the initiation site. If only the barnase substrate has an effective initiation site, either because the adaptor contains no unstructured initiation site (Fig. 6a,b), or because its unstructured region has an amino acid composition that does not support effective initiation25 (Fig. 6c), then only the substrate is degraded. If a better initiation site is introduced into the adaptor, then the adaptor is degraded more effectively than the substrate, although both subunits are accessible to the proteasome to some extent (Fig. 6d). The protease may be selecting the subunit to degrade stochastically, digesting either one or the other of the subunits in any one complex (i.e., for each complex choosing either pathway 1 or 2 in Fig. 6e). Alternatively, the proteasome might be able to degrade both subunits simultaneously in each complex (pathway 3 in Fig. 6e). Together, these results show that both subunits in the complex are accessible to the proteasome and that the protease selects the subunit with the preferred initiation site for degradation (Fig. 6e).
Figure 6.
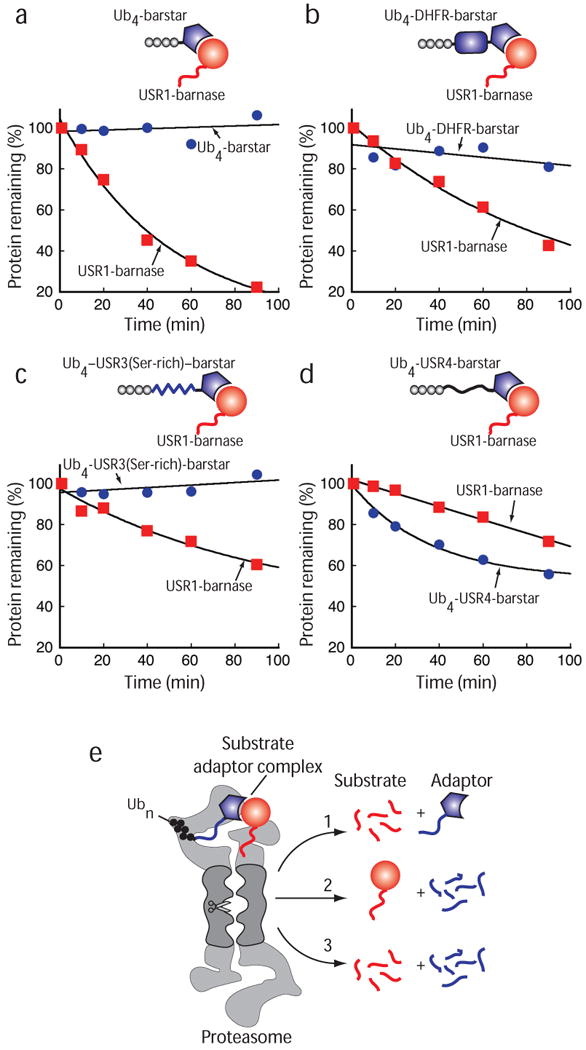
Subunit-specific degradation by the proteasome. (a-d) 0.5 μM Barnase containing a N-terminal initiation site (red squares) was bound to 0.5 μM Ub4-barstar (a, blue circles) or 0.5 μM Ub4-DHFR-barstar (b, blue circles) or 0.5 μM Ub4-USR3(Ser-rich)-barstar (c, blue circles) or 0.5 μM Ub4-USR4-barstar (d, blue circles) and degraded by purified proteasome at 30 °C. Reactions were separated by SDS PAGE and analyzed by Western blotting. The graphs plot the amount of barnase substrate (red) and barstar adaptor (blue) protein remaining at different reaction times as a percentage of protein amounts at the beginning of the reaction. Note that the barnase substrate is present in equimolar concentration to the barstar adaptor. (e) Schematic representation of the substrate selection by the proteasome. Here, both subunits in the complex are accessible to the proteasome and the protease selects the subunit with the preferred initiation site for degradation (path 1 and 2). If more than one subunit contains a proteasome initiation site, multiple subunits might be degraded simultaneously (path 3) or the proteasome might switch stochastically between pathways 1 and 2. The sketch shows a proteasome particle capped by two PA700 regulatory particles. Proteasome particles may contain only one PA700 regulator or they may be activated by complexes that do not bind ubiquitin. Graphs show averages of two (c) or three independent experiments.
Discussion
We find that the two components of the proteasome targeting signal, the ubiquitination and initiation sites, can function together after being separated onto two different polypeptide chains and thus act in trans. A protein that itself is not ubiquitinated can be degraded efficiently if it contains an initiation site and binds to a ubiquitinated partner. The ubiquitinated subunit functions as an adaptor that localizes the complex to the proteasome, which in turn engages and unravels the subunit that contains the initiation site.
Degradation by trans initiation may provide a mechanism by which some proteins convert stable cellular factors into proteasome substrates by binding to both proteasome and the factor. For example, the human papillomavirus protein E7 causes uncontrolled cell growth by binding to the retinoblastoma tumor suppressor protein (Rb) and targeting it for degradation through an unresolved mechanism26. E7 itself is known to be ubiquitinated and to bind to the proteasome regulatory cap directly through its C-terminal region27. Mutations in E7 that prevent it from binding to the proteasome or to Rb also prevent E7 from targeting Rb for degradation and from transforming cells26. Thus, it is likely that E7 functions as a proteasome adaptor protein that targets Rb for degradation through the trans-initiation mechanism described above. Other proteasome interacting proteins such as gankyrin and Mdm2 are found to accelerate the destruction of their binding partners by the proteasome. They, too, may act as adaptors and bring their substrates' initiation sites near the proteasome28. More recently, it was shown that the cytidine deaminase APOBEC3G can be targeted for proteasome degradation by ubiquitinated viral infectivity factor (VIF) even when APOBEC3G does not contain lysine residues, which again suggests that VIF might act as a proteasome adaptor29. Finally, some adaptor proteins such as Rad23 and Dsk2, as well as several E3 ligases, bind to both ubiquitinated proteins and the proteasome to target them for destruction, presumably by the adaptor mechanism described above30-33. Although substrates bound to this class of adaptor proteins are ubiquitinated, the ubiquitin modification is used to bind to the adaptor, which in turn binds to the proteasome34.
The observation of trans targeting also raises an important question. The proteasome remodels some protein complexes by specifically degrading individual subunits6,7. The finding that a ubiquitinated subunit can target a binding partner for degradation rules out that the location of the ubiquitination site itself determines degradation specificity. Instead, we propose that the proteasome primarily selects the subunit to degrade by choosing the degradation initiation site. In some cases, subunits may simply escape degradation because they lack any unstructured region that could serve as an initiation site. For example, ubiquitinated cyclins A and B are degraded when bound to CDKs while the CDKs remain stable8. The crystal structure of CDK2 shows it to have a compact tertiary structure devoid of disordered regions35; therefore, it lacks proteasome initiation sites. In contrast, the N-terminal region of cyclin is largely disordered and contains the ubiquitination site36,37. In human cyclin A, this region corresponds to amino acids 1 through 172 (ref. 35), so we predict that these amino acids serve as the proteasome initiation site in the degradation of the cyclin A in its complex.
Not all unstructured sequences can serve as proteasome initiation sites. For example, unstructured regions have to have a certain minimum length to function effectively and may have to be within a certain distance from the ubiquitin modification14. The sequence composition of the initiation site affects subunit selection too25. Some polypeptide sequences of low compositional complexity are poor proteasome initiation sites18,25 and can also attenuate progression of degradation by the proteasome25,38.
In some cases the situation is more complicated because several subunits may contain functional initiation sites. For example, the proteasome selectively degrades CDK inhibitor from its complex with CDK and cyclin to facilitate progression through the cell cycle9, even though cyclins are also proteasome substrates. CDK inhibitors such as p27 (Kip1) contain large natively unfolded segments next to the ubiquitin modification39. The situation here may be analogous to our observation that a ubiquitinated adaptor with a good initiation site is degraded rapidly so that a substrate with a less effective initiation site escapes degradation (Figs. 5 and 6). If both subunits contain similarly good initiation sites, both can be degraded (Fig. 6). The proteasome may do so by selecting its substrate stochastically in individual complexes, or it may be able to degrade two substrates at the same time. Previous experiments have shown that more than one polypeptide chain can be fed through the degradation channel simultaneously40 and that several substrate proteins can associate with the proteasome at the same time41.
Finally, our findings also suggest a way of designing artificial adaptor proteins that can target specific proteins for proteasome degradation in the cell42. Several strategies have been explored previously to silence protein activity by artificially forcing their degradation by the proteasome. Proteins were delivered to the proteasome by three methods: fusing domains of rapidly degraded polypeptides to the target protein43-46; introducing dimerization domains into target protein and proteasome47; and conjugating short peptides containing the recognition site for a ubiquitin ligase to small molecule ligands (Protacs) that bind to target proteins and induce their ubiquitination48. Our findings suggest a strategy in which a ubiquitin fusion tag would be attached to a protein ligand. Ligand binding, either spontaneous or induced, would then target the binding partner to degradation.
In summary, we find that the two elements of the proteasome targeting signal, ubiquitin modification and initiation site, can work together even when located in trans on two different molecules. We propose that the location of the degradation initiation site, rather than the ubiquitin modification, determines the specificity of subunit degradation.
Methods
Proteins
Proteasome substrates are derived from two proteins that form a tight complex in vitro: the ribonuclease barnase and its inhibitor barstar both from Bacillus amyloliquefaciens. Barnase contained the mutation H102A to inactivate the ribonuclease activity and the mutations K19,62,66,108R, K39A, and K49,98M to remove ubiquitin modification sites. Barstar contained the two mutations C40,82A that abolish dimerization. Proteins were targeted for degradation using two different degrons. In the experiments performed in reticulocyte lysate, ubiquitination and degradation were induced by the N end-rule degron (N-degron)17-19, which was composed of N-terminal wildtype ubiquitin followed by an arginine residue, a six amino acid linker (HGSGAW), an 18 amino acid region (residues 318-335) derived from an internal region of lac repressor protein, and a three amino acid linker (Ala-Met-Gly) (Fig. 1b). In the experiments performed with purified yeast proteasome, a Ub4-degron was used, which was composed of four ubiquitin moieties containing the mutation G76A that were connected by their N and C termini through a six-residue linker (GSGGGG)21. The Ub4 tag was attached in frame to the N terminus of barstar through a short, 16 amino acid linker (Ub4-barstar, Figs. 3, 5, 6), through E. coli DHFR, which is 159 amino acids long (Ub4-DHFR-barstar, Figs. 5c and 6b), or through a 210 amino acid unstructured region consisting of two copies of the 1-95 residues of the mitochondrial precursor protein cytochrome b2 (Ub4-USR2-barstar, Fig. 5b). USR stands for “unstructured region”. In the degradation experiments in which the barstar adaptor and barnase substrate are present in equimolar amounts (Fig. 6), the Ub4 tag is attached to barstar also through a 102 amino acid-long linker that was composed of 66% Ser residues and described previously25 (Ub4-USR3(Ser-rich)-barstar, Fig. 6c) or through a 129 amino acid unstructured region derived from NFκB subunit p105 (in frame fusion of p105 residues 352-403 and 367-425) (Ub4-USR4-barstar, Fig. 6d). The Ub4-USR4-barstar protein is more soluble than Ub4-USR2-barstar and therefore more suitable for the experiments in which adaptor and substrate are present in equimolar amounts because these have to be performed at higher protein concentrations. In addition, Ub4-USR4-barstar and Ub4-USR3(Ser-rich)-barstar are of similar length, making the comparison of these proteins more straightforward (Fig. 6). USR4 is rich in glycine, and a part of it has previously been described as a glycine-rich region, but glycine is not as overrepresented in USR4 as serine is overrepresented in USR3. In the barnase hybrid proteins, either the N end-rule degron, or the unstructured regions that served as initiation sites, were attached to the N terminus of the ribonuclease. In the experiments performed in reticulocyte lysate, the N-terminal initiation site in the barnase was derived from residues 1-95 of mitochondrial precursor cytochrome b2 or from two copies of an internal region of lac repressor (residues 318-348). In either initiation site, all lysine residues were mutated to arginine or glutamine. In the experiments performed with purified yeast proteasome, the N-terminal initiation site of barnase consisted of residues 1-35 of cytochrome b2 (USR1). The identities of the gene constructs were confirmed by DNA sequencing.
Protein expression and purification
Barstar and barnase constructs were overexpressed in and purified from E. coli using standard methodology. Details of the purification protocols are described in the Supplementary Methods online.
Yeast proteasome was from the S. cerevisiae strain YYS40 (MATa rpn11∷RPN113FLAG-HIS3 leu2 his3 ura3 trp1 ade2 can1 ssd1) in which the lid subunit Rpn11 is tagged with three FLAG tags at its C terminus49. The 26S proteasome was purified using M2-agarose FLAG affinity beads (Sigma), according to the previously published protocol49.
Proteasome Degradation Assays
For degradation assays in rabbit reticulocyte lysate, the lysate (Green Hectares) was depleted of ATP as described19. In vitro translated [35S]-methionine labeled barnase protein was added to ATP-depleted reticulocyte lysate in buffer containing 5% [v/v] glycerol, 5 mM MgCl2, 50 mM Tris/Cl [pH 7.4] and supplemented with 1 mM DTT, 25 μM ubiquitin or 100 μM lysine-less ubiquitin (Boston Biochem). Purified barstar or barstar fused to the N-degron was used at 0.25 μM. Reactions were first incubated at 30 °C for 30 min to allow removal of the N-terminal ubiquitin. Ubiquitination and degradation were initiated by addition of 1 mM ATP and an ATP-regeneration system (10 mM creatine phosphate, 0.1 mg/ml creatine phosphokinase). Incubation was continued at 30 °C, and equal volume samples were withdrawn at the indicated times and added to SDS-PAGE sample buffer to stop the reaction. Samples were resolved by SDS-PAGE and analyzed and quantified by electronic autoradiography (Instant Imager, Packard).
Assays with purified proteasome contained 0.25 μM 26S yeast proteasome in 5% [v/v] glycerol, 5 mM MgCl2, 50 mM Tris/Cl [pH 7.4], 1 mM DTT, ATP-regeneration system (1 mM ATP, 10 mM creatine phosphate, 0.1 mg/ml creatine phosphokinase). Indicated amounts of purified barstar adaptor were bound to 0.5 μM barnase proteins at 30 °C for 15 min and the complex was added to the purified proteasome to start the degradation reaction. At the indicated times, samples of equal volumes were withdrawn and added to sample buffer and analyzed by SDS-PAGE followed by immunoblotting. Barnase and barstar proteins were visualized using rabbit antibodies specific to barnase and to barstar's His6 tag (Bethyl Labs), respectively, and Alexa-680 labeled secondary antibodies. Proteins were quantified using an Odyssey infrared imaging system (LI-COR Biosciences).
Size Exclusion Chromatography
Radioactive barnase protein was allowed to bind to 0.25 μM barstar or N-degron-DHFR-barstar at 30 °C for 30 min. The barnase-barstar complex was resolved by Sephacryl S-100 high-resolution matrix (Amersham) in a 70/50 column (VT = 20 ml, Vo = 7ml) (BioRad). 0.25 ml fractions were collected and precipitated with 10 % [w/v] trichloroacetic acid. The protein pellets were resolved by SDS-PAGE and quantified by electronic autoradiography (Instant Imager, Packard).
Supplementary Material
Acknowledgments
We thank Rich Carthew, Amy Rosenzweig, Jonathan Widom, and members of the Matouschek lab for advice and comments, as well as Ginger Leigh for editing the manuscript. The work was supported by grant R01GM64003 from the NIH, the Leukemia and Lymphoma Society, and the Robert H. Lurie Comprehensive Cancer Center at Northwestern University. T.I. gratefully acknowledges a Japanese Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad.
Footnotes
Author Contributions: S.P., T.I., and A.M. designed the experiments and wrote the manuscript. S.P. and T.I. performed most of the experiments. A.J.H. assisted S.P. in performing some of the degradation experiments, in purifying proteins, and in constructing many of the genes.
References
- 1.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 2.Hochstrasser M. Lingering mysteries of ubiquitin-chain assembly. Cell. 2006;124:27–34. doi: 10.1016/j.cell.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 3.Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem. 1994;269:7059–7061. [PubMed] [Google Scholar]
- 4.Lam YA, Lawson TG, Velayutham M, Zweier JL, Pickart CM. A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature. 2002;416:763–767. doi: 10.1038/416763a. [DOI] [PubMed] [Google Scholar]
- 5.Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell. 2001;7:627–637. doi: 10.1016/s1097-2765(01)00209-x. [DOI] [PubMed] [Google Scholar]
- 6.Johnson ES, Gonda DK, Varshavsky A. Cis-trans recognition and subunit-specific degradation of short-lived proteins. Nature. 1990;346:287–291. doi: 10.1038/346287a0. [DOI] [PubMed] [Google Scholar]
- 7.Hochstrasser M, Varshavsky A. In vivo degradation of a transcriptional regulator: The yeast alpha 2 repressor. Cell. 1990;61:697–708. doi: 10.1016/0092-8674(90)90481-s. [DOI] [PubMed] [Google Scholar]
- 8.Nishiyama A, et al. A nonproteolytic function of the proteasome is required for the dissociation of Cdc2 and cyclin B at the end of M phase. Genes Dev. 2000;14:2344–2357. doi: 10.1101/gad.823200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verma R, McDonald H, Yates JR, Deshaies RJ. Selective degradation of ubiquitinated Sic1 by purified 26S proteasome yields active S phase cyclin-Cdk. Mol Cell. 2001;8:439–448. doi: 10.1016/s1097-2765(01)00308-2. [DOI] [PubMed] [Google Scholar]
- 10.Elsasser S, et al. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat Cell Biol. 2002;4:725–730. doi: 10.1038/ncb845. [DOI] [PubMed] [Google Scholar]
- 11.Goebl MG, Goetsch L, Byers B. The Ubc3 (Cdc34) ubiquitin-conjugating enzyme is ubiquitinated and phosphorylated in vivo. Mol Cell Biol. 1994;14:3022–3029. doi: 10.1128/mcb.14.5.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heessen S, Masucci MG, Dantuma NP. The Uba2 domain functions as an intrinsic stabilization signal that protects Rad23 from proteasomal degradation. Mol Cell. 2005;18:225–235. doi: 10.1016/j.molcel.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Schauber C, et al. Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature. 1998;391:715–718. doi: 10.1038/35661. [DOI] [PubMed] [Google Scholar]
- 14.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol. 2004;11:830–837. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]
- 15.Hartley RW. Directed mutagenesis and barnase-barstar recognition. Biochemistry. 1993;32:5978–5984. doi: 10.1021/bi00074a008. [DOI] [PubMed] [Google Scholar]
- 16.Schreiber G, Fersht AR. Interaction of barnase with its polypeptide inhibitor barstar studied by protein engineering. Biochemistry. 1993;32:5145–5150. doi: 10.1021/bi00070a025. [DOI] [PubMed] [Google Scholar]
- 17.Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 18.Bachmair A, Varshavsky A. The degradation signal in a short-lived protein. Cell. 1989;56:1019–1032. doi: 10.1016/0092-8674(89)90635-1. [DOI] [PubMed] [Google Scholar]
- 19.Gonda DK, et al. Universality and structure of the N-end rule. J Biol Chem. 1989;264:16700–16712. [PubMed] [Google Scholar]
- 20.Ciechanover A, Ben-Saadon R. N-terminal ubiquitination: More protein substrates join in. Trends Cell Biol. 2004;14:103–106. doi: 10.1016/j.tcb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Stack JH, Whitney M, Rodems SM, Pollok BA. A ubiquitin-based tagging system for controlled modulation of protein stability. Nat Biotechnol. 2000;18:1298–1302. doi: 10.1038/82422. [DOI] [PubMed] [Google Scholar]
- 22.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beal RE, Toscano-Cantaffa D, Young P, Rechsteiner M, Pickart CM. The hydrophobic effect contributes to polyubiquitin chain recognition. Biochemistry. 1998;37:2925–2934. doi: 10.1021/bi972514p. [DOI] [PubMed] [Google Scholar]
- 24.Johnston JA, Johnson ES, Waller PR, Varshavsky A. Methotrexate inhibits proteolysis of dihydrofolate reductase by the N-end rule pathway. J Biol Chem. 1995;270:8172–8178. doi: 10.1074/jbc.270.14.8172. [DOI] [PubMed] [Google Scholar]
- 25.Tian L, Holmgren RA, Matouschek A. A conserved processing mechanism regulates the activity of transcription factors cubitus interruptus and NF-κB. Nat Struct Mol Biol. 2005;12:1045–1053. doi: 10.1038/nsmb1018. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez SL, Stremlau M, He X, Basile JR, Münger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berezutskaya E, Bagchi S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26S proteasome. J Biol Chem. 1997;272:30135–30140. doi: 10.1074/jbc.272.48.30135. [DOI] [PubMed] [Google Scholar]
- 28.Whitby FG, Hill CP. A versatile platform for inactivation and destruction. Structure. 2007;15:137–138. doi: 10.1016/j.str.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 29.Dang Y, Siew LM, Zheng YH. APOBEC3g is degraded by the proteasomal pathway in a Vif-dependent manner without being polyubiquitylated. J Biol Chem. 2008;283:13124–13131. doi: 10.1074/jbc.M708728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Madura K. Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol Cell Biol. 2002;22:4902–4913. doi: 10.1128/MCB.22.13.4902-4913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elsasser S, Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol. 2005;7:742–749. doi: 10.1038/ncb0805-742. [DOI] [PubMed] [Google Scholar]
- 32.McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control: Chaperones culling corrupt conformations. Nat Cell Biol. 2005;7:736–741. doi: 10.1038/ncb0805-736. [DOI] [PubMed] [Google Scholar]
- 33.Xie Y, Varshavsky A. Physical association of ubiquitin ligases and the 26S proteasome. Proc Natl Acad Sci USA. 2000;97:2497–2502. doi: 10.1073/pnas.060025497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verma R, Oania R, Graumann J, Deshaies RJ. Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome system. Cell. 2004;118:99–110. doi: 10.1016/j.cell.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 35.Jeffrey PD, et al. Mechanism of Cdk activation revealed by the structure of a Cyclin A-Cdk2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 36.King RW, Glotzer M, Kirschner MW. Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol Biol Cell. 1996;7:1343–1357. doi: 10.1091/mbc.7.9.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klotzbücher A, Stewart E, Harrison D, Hunt T. The ‘destruction box’ of cyclin A allows B-type cyclins to be ubiquitinated, but not efficiently destroyed. EMBO J. 1996;15:3053–3064. [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang M, Coffino P. Repeat sequence of Epstein-Barr virus-encoded nuclear antigen 1 protein interrupts proteasome substrate processing. J Biol Chem. 2004;279:8635–8641. doi: 10.1074/jbc.M310449200. [DOI] [PubMed] [Google Scholar]
- 39.Bienkiewicz EA, Adkins JN, Lumb KJ. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1) Biochemistry. 2002;41:752–759. doi: 10.1021/bi015763t. [DOI] [PubMed] [Google Scholar]
- 40.Lee C, Prakash S, Matouschek A. Concurrent translocation of multiple polypeptide chains through the proteasomal degradation channel. J Biol Chem. 2002;277:34760–34765. doi: 10.1074/jbc.M204750200. [DOI] [PubMed] [Google Scholar]
- 41.Sharon M, et al. 20S proteasomes have the potential to keep substrates in store for continual degradation. J Biol Chem. 2006;281:9569–9575. doi: 10.1074/jbc.M511951200. [DOI] [PubMed] [Google Scholar]
- 42.Stankunas K, Crabtree GR. Exploiting protein destruction for constructive use. Proc Natl Acad Sci USA. 2007;104:11511–11512. doi: 10.1073/pnas.0704762104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stankunas K, et al. Conditional protein alleles using knockin mice and a chemical inducer of dimerization. Mol Cell. 2003;12:1615–1624. doi: 10.1016/s1097-2765(03)00491-x. [DOI] [PubMed] [Google Scholar]
- 45.Pratt MR, Schwartz EC, Muir TW. Small-molecule-mediated rescue of protein function by an inducible proteolytic shunt. Proc Natl Acad Sci USA. 2007;104:11209–11214. doi: 10.1073/pnas.0700816104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsuzawa S, Cuddy M, Fukushima T, Reed JC. Method for targeting protein destruction by using a ubiquitin-independent, proteasome-mediated degradation pathway. Proc Natl Acad Sci USA. 2005;102:14982–14987. doi: 10.1073/pnas.0507512102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Janse DM, Crosas B, Finley D, Church GM. Localization to the proteasome is sufficient for degradation. J Biol Chem. 2004;279:21415–21420. doi: 10.1074/jbc.M402954200. [DOI] [PubMed] [Google Scholar]
- 48.Sakamoto KM, et al. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saeki Y, Isono E, Toh-E A. Preparation of ubiquitinated substrates by the PY motif-insertion method for monitoring 26S proteasome activity. Meth Enzymol. 2005;399:215–227. doi: 10.1016/S0076-6879(05)99014-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.