

Abstract
Using human immune globulins made from antihepatitis C virus (HCV)-positive plasma, we recently identified two antibody epitopes in the E2 protein at residues 412–426 (epitope I) and 434–446 (epitope II). Whereas epitope I is highly conserved among genotypes, epitope II varies. We discovered that epitope I was implicated in HCV neutralization whereas the binding of non-neutralizing antibody to epitope II disrupted virus neutralization mediated by antibody binding at epitope I. These findings suggested that, if this interfering mechanism operates in vivo during HCV infection, a neutralizing antibody against epitope I can be restrained by an interfering antibody, which may account for the persistence of HCV even in the presence of an abundance of neutralizing antibodies. We tested this hypothesis by affinity depletion and peptide-blocking of epitope-II-specific antibodies in plasma of a chronically HCV-infected patient and recombinant E1E2 vaccinated chimpanzees. We demonstrate that, by removing the restraints imposed by the interfering antibodies to epitope-II, neutralizing activity can be revealed in plasma that previously failed to neutralize viral stock in cell culture. Further, cross-genotype neutralization could be generated from monospecific plasma. Our studies contribute to understanding the mechanisms of antibody-mediated neutralization and interference and provide a practical approach to the development of more potent and broadly reactive hepatitis C immune globulins.
Most hepatitis C virus (HCV)-infected patients fail to clear the virus and, despite the presence of neutralizing antibodies (NAbs), develop chronic infections. These chronically HCV-infected patients are at risk of developing cirrhosis and liver cancer (1, 2). Although current standard treatment with pegylated IFN and ribavirin results in cures in as many as 50% of patients, neither antibody-based prophylaxis nor an effective vaccine is available.
The mechanism by which HCV persists in the presence of NAbs is unknown. Heterogeneity, a prominent feature of HCV, has been considered important in immune escape. Previously we identified an antigenic region in the E2 envelope glycoprotein of hepatitis C virus that contains two important epitopes, i.e., epitope I and epitope II. Epitope I has been recognized by us and others as an important neutralization site (3, 4). We showed that antibody to epitope II interfered with antibody to epitope I, inhibiting neutralization of the virus (4). In this study, we have further characterized these epitopes and identified the amino acid residues in epitope I important for antibody binding. By absorbing out antibody to epitope II in plasma from a chronically infected HCV patient, we show that neutralizing activity is not only enhanced but also broadened to include additional genotypes of the virus. Furthermore, by using plasma from 2 chimpanzees that had been vaccinated with recombinant E1 and E2 envelope glycoproteins of a genotype 1a HCV, we demonstrate that a monotypic immune response contained cross-neutralizing capability that could be revealed only following depletion of the antibodies to epitope II.
Results
Amino Acid Specificities of Antibody Directed Against Epitope I.
Epitope I is now recognized as a major antibody neutralization target (3–9). However, little is known about the antibody specificities that mediate neutralization. We mapped the key amino acid residues responsible for antibody binding to epitope I by screening a random peptide phage display library with eluate I, derived by affinity purification of experimental immune globulin IV made from anti-HCV-positive plasma (HCIGIV) with epitope I peptide (Fig. 1A; see Materials and Methods). Eluate I reacted with peptides, containing residues Q, L, S, and W, which mimicked epitope I (Fig. 1A), suggesting that these residues are sufficient for eluate I recognition regardless of their spatial arrangement. ELISA analysis revealed that mutants bearing QL413>AA or SW420>AA exhibited reduced antibody binding, whereas HIN423>AAA did not affect antibody binding (Fig. 1 B and C). When alanine was substituted at Q412, L413, S419, or W420 (Fig. 1 B and C), we discovered that L413 and W420 were the 2 discontinuous residues within epitope I indispensable for the binding of NAb. This finding was corroborated by the fact that both L413 and W420 are among the most conserved residues in all HCV genotypes, suggesting that cross-genotype neutralization might be achieved if antibody can effectively bind to epitope I.
Fig. 1.

Key residues for the binding of epitope-I-specific neutralizing antibody. (A) Eluate I, an antibody solution, was obtained by affinity purification of HCIGIV with the chemically synthesized peptide containing epitope I (4). With eluate I, peptidyl mimics of epitope-I were selected by screening a random peptide phage display library using a panning method (4). The key residues within both epitope I and peptidyl mimics are indicated (bold and underline). The numbers indicate the position of these peptides in HCV polyprotein. (B) Chemically synthesized peptides containing epitope I and its mutants at key residues are indicated. (C) ELISA analysis revealed critical residues in epitope I. Biotin-conjugated peptides listed in B were added to streptavidin-coated 96-well plates (200 ng/well), individually. Eluate I at 1:50 dilution or HCIGIV at 1:2,000 dilution was used as the primary antibody in the ELISA. The x axis indicates individual peptides tested, and the y axis shows corresponding absorbance at 450 nm (A450 nm) of each mutant relative to that of epitope I (in percent of control).
Presence of Epitope-I- and Epitope-II-Specific Antibodies in Plasma from Chronically HCV-Infected Patients and in HCIGIV Preparations.
We assayed the levels of epitope-I- and epitope-II-specific antibodies in plasma from 9 chronically HCV-infected patients by ELISA (Fig. 2A). Of these, 2 (H77 and no.1) had detectable antibodies against epitope I, whereas 4 (H77, nos. 1, 4, and 5) had antibodies against epitope II (Fig. 2A). The appearance of epitope-I-specific antibodies in these patients coincided with the presence of high levels of epitope-II-specific antibodies. None of these samples reacted with the epitope I mutant, SW420>AA, indicative of the specificity of the antibody.
Fig. 2.

Presence of HCV epitope-specific antibodies in plasma of patients with chronic HCV infection. (A) Biotin-conjugated peptides encompassing epitope II, epitope I, or the mutant of epitope I, SW>AA (Fig. 1 B and C) were added to streptavidin-coated 96-well plates (200 ng/well), individually. SW>AA was used as a NC for the assay. Human patient plasma samples, H77 and numbers 1 to 8, were diluted 1:800 and 1:100, respectively, and used as the primary antibody. Diluted HCIGIV lot A (1:2,000), eluate I, and eluate II, an HCIGIV fraction affinity-purified by using epitope II peptide (4), were used at 1:50 dilution as controls. The y axis indicates A450 nm obtained in the ELISA, representing specific binding of a given plasma to each individual peptide. (B) HCIGIV lots (A–F) were diluted 1:2,000 and used as the primary antibodies in the ELISA, in which biotin-conjugated peptides encompassing epitope II, epitope I, or SW>AA were added to streptavidin-coated 96-well plates (200 ng/well). Peptide SW>AA was used as a NC. The ratios of antibody against epitope II versus that against epitope I are presented. (C) Plasma samples collected from patient H before and after HCV infection were diluted 1:100 except for the sample of d 5266, which was diluted at 1:400, and used as the primary antibodies. Plasma levels of antibodies directed against epitope II, epitope I, or SW>AA, as indicated by values of A450 nm, were determined in the ELISA. SW>AA was used as a negative peptide control. Diluted HCIGIV lot A at 1:2,000 and eluates I and II at 1:50 were used as controls for primary antibodies.
We then measured the levels of epitope-I- and epitope-II-specific antibodies in six lots of HCIGIV by ELISA (Fig. 2B). As predicted from the prevalence of antibodies in the patients' plasma, all samples contained both antibodies, with the titer higher for epitope II than for epitope I. The antibody ratio of epitope-II-specific to epitope-I-specific for these HCIGIV lots ranged from 2.0 to 3.8 (Fig. 2B).
We further examined the kinetics of epitope-I- and epitope-II-specific antibody production in patient H, who had established chronic infection (Fig. 2C) (10). ELISA analysis revealed that antibody to epitope II appeared within 51 days of infection. By contrast, antibody to epitope I was not detectable until day 643. At day 5,266, antibody responses to both epitopes were still readily detectable. Notably, the appearance of epitope-I-specific antibody coincided with the presence of elevated levels of epitope-II-specific antibody only in the chronic phase of infection. This suggested that the lack of epitope-I-specific antibody production during the early phase of infection might permit the survival of HCV and that the co-existence of antibodies to epitope II with antibodies to epitope I during the late phase might hamper neutralization mediated by epitope-I-specific antibodies, thereby contributing to HCV persistence.
Neutralizing Activity in Plasma Collected from Patient H.
The fact that HCV persisted in these patients despite the presence of epitope-I-specific antibodies exposed the inadequacy of such antibodies to control HCV infection. Thus the question arose whether the activity of epitope-I-specific antibodies was inhibited by epitope-II-specific antibodies in these patients through antibody interference similar to that which we proposed for HCIGIV (4). We examined neutralizing activity in plasma collected from patient-H on day 5,266 using an HCV cell culture system with a 2a virus (HCVcc/2a) and a 1a/2a chimeric virus (HCVcc/1a; see Materials and Methods). Both were constructed on the backbone of JFH1 (genotype 2a). HCVcc/1a contained the envelope proteins from strain H77, a genotype 1a virus isolated from patient H (10), and HCVcc/2a contained envelope proteins from strain J6, a genotype 2a virus (11, 12). Plasma 5,266, at a dilution of 1:400, neutralized approximately 80% of HCVcc/1a, compared with a mock-treated control (i.e., negative control [NC]; P < 0.01; Fig. 3A). However, it did not significantly reduce the infectivity of HCVcc/2a under the same experimental conditions (Fig. 3A).
Fig. 3.
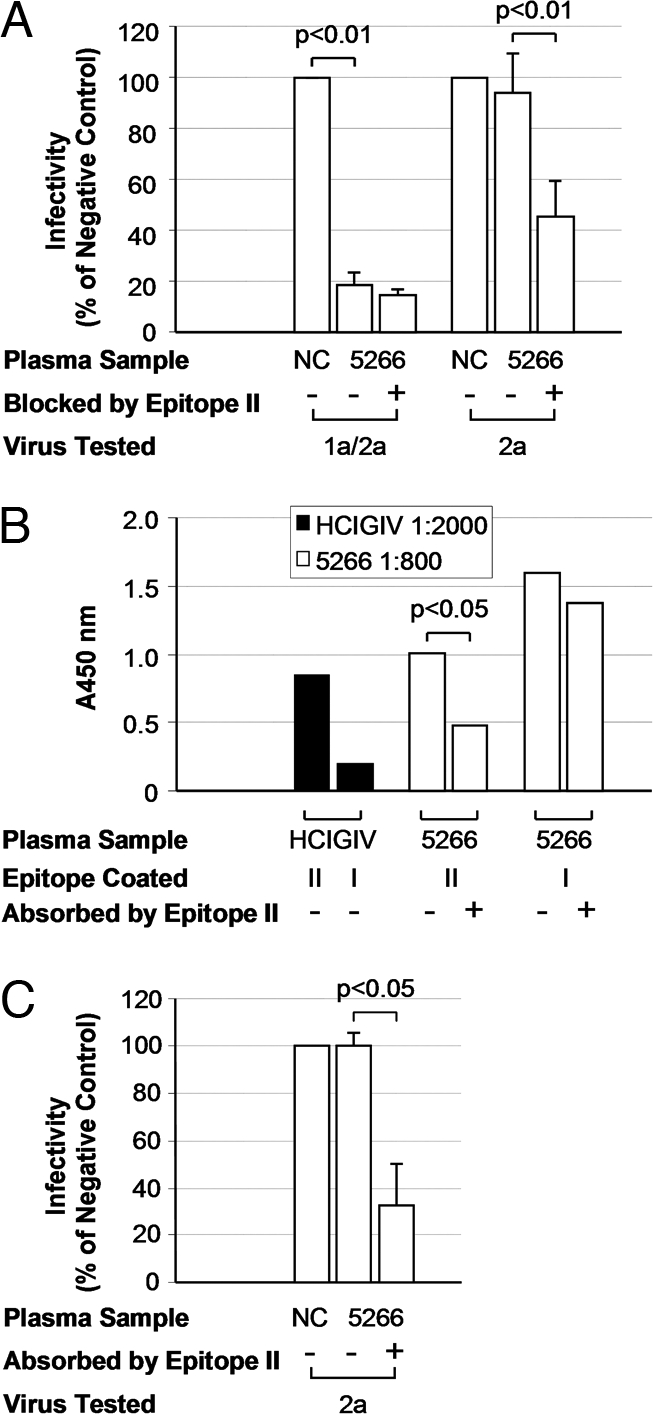
Recovery of cross-genotype neutralizing activity from patient H plasma. (A) epitope-II-specific antibodies in plasma 5266 were specifically blocked by epitope II peptide or left untreated as a control. These samples were then tested for their abilities to neutralize genotype 1a/2a or 2a virus in HCV cell culture. A normal IGIV at 1:400 dilution was used as NC. The x axis indicates the plasma sample used in this assay at 1:400 dilution. The y axis indicates the infectivity expressed as percentage of NC. Statistical significance of difference in infectivity is indicated. (B) Biotin-conjugated epitope II and epitope I peptides were added to streptavidin-coated 96-well plates (200 ng/well). Plasma 5266, before and after 3 rounds of absorption with epitope II peptide, was diluted at 1:800 and used as primary antibody in ELISA. HCIGIV lot A, at 1:2,000 dilution, was used as the positive control. The y axis indicates absorbance at 450 nm, representing specific binding of a given plasma sample to each individual peptide. (C) Plasma 5266 was absorbed with epitope II peptide to deplete epitope-II-specific antibodies. These absorbed samples, along with those left untreated, were diluted at 1:400 and assayed for their abilities to neutralize the 2a virus in HCV cell culture. A normal IGIV at 1:400 dilution was used as NC. The neutralizing activity was expressed as the relative infectivity of the virus, i.e., percent of NC. Statistical significance of difference in infectivity is indicated.
Depletion of Epitope-II-Specific Antibodies Reveals Cross-Genotype Neutralization.
Knowing that epitope I is highly conserved among genotypes, the failure of the H77 plasma to cross-neutralize HCVcc/2a indicated a potential disruption of epitope-I-specific antibodies, possibly by epitope-II-specific antibodies. We thus tested whether blocking epitope-II-specific antibodies would lead to neutralization of HCVcc/2a. Indeed, when epitope-II-specific antibodies in the plasma were blocked by an epitope-II-specific peptide (Fig. 3A; see Materials and Methods), the plasma exhibited detectable neutralizing activity against HCVcc/2a. Approximately 50% reduction of infectivity was observed (P < 0.01; Fig. 3A). We determined the end-point titer that would give 50% neutralization (ID50) (13) and found that the ID50 for untreated H77 plasma was 1:1,238 against HCVcc/1a and <1:200 against HCVcc/2a. After epitope-II-specific antibodies were blocked, the ID50 increased to >1:1,600 against HCVcc/1a and to 1:531 against HCVcc/2a. These results demonstrate that both genotype-specific neutralization and cross-genotype neutralization could be enhanced or revealed by using genotype-specific plasma in which interfering antibodies are blocked.
We asked whether removal, instead of blocking, of epitope-II-specific antibodies from the plasma could also reveal the neutralization of HCVcc/2a. Epitope-II-specific antibodies were thus depleted from plasma 5266 (Fig. 2B; see Materials and Methods). As expected, the plasma with a reduced level of epitope-II-specific antibodies neutralized HCVcc/2a (P < 0.05; Fig. 3C). Therefore, cross-neutralization of the 2a virus could be realized when interfering antibodies were removed from a genotype 1a-specific plasma, even when the removal was less than complete.
This conclusion was further substantiated by generation of cross-genotype neutralizing activity in the plasma of vaccinated chimpanzees (Fig. 4). Plasma from 2 chimpanzees, Ch1587 and Ch1601, was collected 2 weeks after vaccination with rE1E2 protein (14). The sequence of rE1E2 used for vaccination was identical to that of HCVcc/1a. We found that plasma from Ch1587 contained antibodies capable of reducing the infectivity of HCVcc/1a to approximately 40% of the NC at a 1:400 dilution (Fig. 4A). By contrast, the plasma showed enhanced neutralizing activity when epitope-II-specific antibody was blocked, reducing virus infectivity to 10% of the NC. Similarly, only after epitope-II-specific antibodies were blocked by peptide did the plasma show neutralizing activity against a second chimeric virus HCVcc/1b, which was constructed on the backbone of JFH1 containing the core and envelope regions of a 1b isolate. Such a cross-genotype neutralization was observed when HCVcc/2a was investigated under the same experimental condition (Fig. 4A). These findings were confirmed when the plasma from the second vaccinated chimpanzee, Ch1601, was studied for its ability to neutralize the infectivity of HCVcc/1a and 2a (Fig. 4B). These data demonstrated that cross-genotype neutralizing activity was induced following vaccination with a monotypic antigen, which not only represents a single genotype, but also consists of a single protein sequence. However, the cross-genotype neutralizing ability was revealed only when interfering antibodies directed against epitope II were blocked.
Fig. 4.
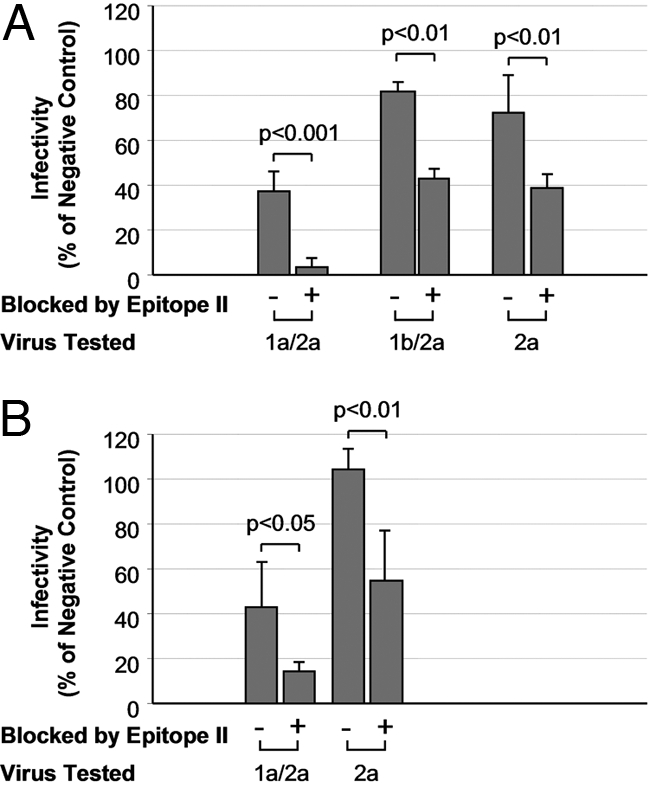
Recovery of cross-genotype neutralizing activity from plasma of rE1/E2 vaccinated chimpanzees. Epitope II peptide (500 ng/mL) was incubated with the plasma of 2 vaccinated chimpanzees, Ch1587 and Ch1601, to block epitope-II-specific antibodies wherein. These samples, at 1:400 dilution, were then tested for their abilities to neutralize the 1a/2a, 1b/2a, or 2a virus in HCV cell culture. (A) Neutralization with plasma from Ch1587 and (B) neutralization with plasma from Ch1601. For comparisons, control plasma samples and virus stocks were mock treated before the neutralization assay. A normal IGIV at 1:400 dilution was used as NC. The neutralizing activity was expressed as the relative infectivity of the virus, i.e., percent of NC. Statistical significance of difference in infectivity is indicated.
Discussion
Our data provide evidence of a general concept that survival of virus by escaping from antibody neutralization does not necessarily require mutations occurring at a neutralization epitope. Instead, there appears to be an additional escape mechanism whereby non-neutralizing antibody binding to virus interferes with the binding of antibodies with neutralizing capability. Interestingly, the epitope I region is reportedly recognized by at least 3 mAbs (Ap33, 3/11, and e137) that have been shown to neutralize HCV in vitro (7, 8, 15, 16). However, these mAbs appear to contact different sets of amino acids, including those in epitope I. For example, residues L413, I414, T416, G418, W420, and H421 were mapped for AP33 binding; T416, W420, W529, and G530 for 3/11; and T416, W420, W529, G530, and D535 for e137. In addition, a recent study revealed a conformational neutralization epitope that contains at least 3 segments at residues 396–424, 436–447, and 523–540 (8). Notably, the first 2 segments overlap with epitope I and epitope II, respectively (5, 6, 17–19). Recognition of the difference in antibody specificity raises the possibility that the approach used for obtaining the NAb, i.e., either direct purification from the plasma of infected patients or immunization with recombinant proteins, affects the breadth of neutralization. In this connection, antibodies obtained directly by elution after absorption of HIV-1-positive sera with specific viral antigen can neutralize diverse strains of HIV-1 that are partially or fully resistant to mAbs (20).
Because epitope II can be relatively heterogeneous, different binding capacities of antibodies to epitope II are expected in patients infected with distinct HCV genotypes. In essence, a difference of this nature can be viewed as an acquired characteristic of the virus to tune the inhibition of neutralization by the interfering antibody, whereby the state of disease could be modified to an extent depending on both the genotype(s) of the virus and the binding strength of the interfering antibody.
Indeed, by changing the ratio of interfering/NAbs in plasma of a chronically HCV-infected patient and vaccinated chimpanzees, we were able to recover otherwise undetectable, cross-genotype neutralizing activity. Based on these findings, we propose that both the successful generation of an HCIGIV product and the development of an effective HCV vaccine may be feasible by eliciting strong NAbs against epitope I while avoiding the production of antibodies against epitope II.
Finally, the operation of a mechanism of viral escape mediated by interfering antibody should not be thought of as limited to HCV. It should be considered in the development of strategies for prophylaxis and treatment of other infections in humans, especially those with highly heterogeneous viruses such as HIV.
Materials and Methods
Immune Globulins.
Several independent lots of HCIGIV (A-F) were made from the pooled plasma of anti-HCV (EIA-2)-positive donors who otherwise met the requirements for normal plasma donations, i.e., negative for both anti-HIV and hepatitis B surface antigen and without elevated levels of alanine aminotransferase. These preparations had been treated by a solvent–detergent process to inactivate potential contaminating viruses. Neutralization of HCV infection by HCIGIV lot A was demonstrated previously in both a pseudo-particle system and a chimpanzee model (3). A commercial 5% immune globulin IV (IGIV) solution, which was manufactured from anti-HCV (EIA-2)-negative plasma donations, was used as NC. This IGIV preparation was also virally inactivated by a solvent–detergent treatment.
Plasma Samples.
Nine human plasma samples were obtained at the National Institutes of Health (NIH) Clinical Center from patients, including patient H, who were chronically infected with HCV and randomly selected for this study. All samples were collected according to protocols approved by the NIH institutional review board. Chimpanzee plasma samples were collected from 2 chimpanzees, one naive (Ch1601) and one recovered from HCV acute infection (Ch1587), both of which were vaccinated with recombinant envelope glycoproteins (E1E2) derived from a genotype 1a sequence isolated from patient H. High ELISA antibody titers to E1E2 after immunization had been previously demonstrated (14).
Phage Display.
Selection of peptides from a random peptide phage display library (PhD-12; New England Biolabs,) was described previously (21). Briefly, approximately 1010 phages were incubated with an individual Ig fraction/protein-G mixture for 20 min at room temperature. After 8 washings with 10 mM Tris-HCl buffer (pH 7.5) containing 0.02% Tween-20, the phages were eluted from the complex with 0.1 M HCl for 8 min at room temperature. The eluted phages were then amplified in the host strain ER2738. Amplified phages were subjected to 3 additional rounds of selection by the same Ig fraction. After selection, collected phages were grown on LB-agar plates. DNA from each single phage plaque was sequenced, and the corresponding peptide sequence was then deduced from the DNA sequence.
Peptide Synthesis.
All peptides were synthesized by the Core Laboratory of the Center for Biologics Evaluation and Research at the US Food and Drug Administration with an Applied Biosystems model 433A peptide synthesizer using standard FastMoc chemistry (22). Synthesis of biotinylated peptides was carried out with Fmoc-Lys (Biotin-LC)-Wang resin (AnaSpec). The crude peptides were precipitated, washed with butyl methyl ether, dried under vacuum, purified by RP HPLC on a DeltaPak C-18 reversed-phase column (Waters), and analyzed by MALDI-TOF MS with a Voyager DE-RP MALDI-TOF mass spectrometer (PE Biosystems).
ELISA.
Streptavidin-coated 96-well plates were used for ELISA according to the manufacturer's instructions (Pierce). Biotinylated peptides (200 ng/well) were added to streptavidin-coated wells and incubated at room temperature for 30 min in SuperBlocker blocking buffer (Thermo Scientific). The wells were then blocked with SuperBlocker at 37 °C for 1 h. After washings with PBS buffer containing 0.05% Tween-20, antibodies were added to the wells and incubated for 1 h at 37 °C. After removal of unbound antibodies by washings with PBS buffer containing 0.05% Tween-20, a goat anti-human peroxidase-conjugated IgG (Sigma-Aldrich) at 1:5,000 dilution was added to the wells and incubated at 37 °C for 30 min. After washings, the plates were incubated in the dark for 10 min with 100 μL/well of 1-Step Ultra TMB-ELISA (Thermo Scientific). The reaction was stopped by adding 100 μL/well 4 N H2SO4. The absorbance of each well was measured at 450 nm with a microtiter plate reader (Optimax; Molecular Devices).
Peptide-Blocking and Affinity Depletion of Epitope-II-Specific Antibodies.
For peptide-blocking of epitope-II-specific antibodies in the plasma, epitope II peptide corresponding to the amino acid sequence of HCV genotype 1a (H77) was synthesized. One microgram of peptide was added to the diluted plasma and incubated at 37 °C for 60 min. Treated samples were subsequently used in the neutralization assay. For affinity depletion of epitope-II-specific antibodies, biotinylated epitope II peptide (500 ng/well) was added to streptavidin-coated wells and incubated at room temperature for 30 min in Tris-HCl buffer (pH 7.5) containing 0.02% Tween-20. A diluted plasma was added to the well and incubated for 30 min at room temperature for absorption. The unbound portion was collected. ELISA analysis was performed to monitor the levels of the antibodies during affinity depletion.
Neutralization Assay.
Virus stock was prepared by transfecting HCV RNA derived from genotype 1a/2a or 1b/2a chimeras or 2a (J6/JFH1) into Huh 7.5 cells, respectively, according to the procedures described previously (4, 12, 13). Huh 7.5 cells were seeded at a density of 4–5 × 103 cells/well in 96-well plates to obtain 50%–60% confluence in 24 h. Virus stock was diluted in DMEM supplemented with 10% FBS/1% penicillin/streptomycin/2 mM glutamine to yield approximately 100 infected foci per well in the absence of NAbs. To test neutralization capacity, an antibody, in parallel with a positive (HCIGIV) and an NC, was mixed with the virus stock before addition to the cells. After incubation at 37 °C for 1 h, the supernatants containing the virus/antibody mixture were removed by washing with PBS solution and complete DMEM was added to each well. The cells were continuously cultured in DMEM for 3 d. To count infected foci, the cells were fixed with cold methanol and stained with a monoclonal antibody for the HCV core antigen that recognized both the 1a and 2a sequences. This was followed by peroxidase staining, and positive cells were visualized with diaminobenzidine tetra-hydrochloride. Positive foci were counted. Infectivity was expressed as percent of NC, i.e., numbers of infected foci with a given antibody divided by numbers of foci with a NC antibody, and the quotient multiplied by 100%.
Statistical Analysis.
JMP software (v.7.0; SAS Institute) was used for analyzing data. Means were compared between 2 samples by using the Student t test. For an overall comparison of means, the Tukey-Kramer HSD test was used. Statistical significance was set at an α of 0.05. A positive test value generated between 2 means is indicative of a significant difference.
Acknowledgments.
We thank Drs. John Finlayson and Mahmood Farshid for comments on the manuscript; Dr. Basil Golding for interest and support; the Core Laboratory of the Center for Biologics Evaluation and Research for peptide synthesis and DNA sequencing; and Dr. Mei-ying Yu and Nabi Biopharmaceuticals for providing experimental HCIGIV preparations for this study.
Footnotes
The authors declare no conflict of interest.
References
- 1.Alter HJ, Seeff LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin Liver Dis. 2000;20:17–35. doi: 10.1055/s-2000-9505. [DOI] [PubMed] [Google Scholar]
- 2.Davis GL. Hepatitis C immune globulin to prevent HCV recurrence after liver transplantation: chasing windmills? Liver Transpl. 2006;12:1317–1319. doi: 10.1002/lt.20889. [DOI] [PubMed] [Google Scholar]
- 3.Yu MW, et al. Neutralizing antibodies to hepatitis C virus (HCV) in immune globulins derived from anti-HCV-positive plasma. Proc Natl Acad Sci USA. 2004;101:7705–7710. doi: 10.1073/pnas.0402458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang P, et al. Hepatitis C virus epitope-specific neutralizing antibodies in Igs prepared from human plasma. Proc Natl Acad Sci USA. 2007;104:8449–8454. doi: 10.1073/pnas.0703039104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Triyatni M, et al. Structural features of envelope proteins on hepatitis C virus-like particles as determined by anti-envelope monoclonal antibodies and CD81 binding. Virology. 2002;298:124–132. doi: 10.1006/viro.2002.1463. [DOI] [PubMed] [Google Scholar]
- 6.Hsu M, et al. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci USA. 2003;100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarr AW, et al. Determination of the human antibody response to the epitope defined by the hepatitis C virus-neutralizing monoclonal antibody AP33. J Gen Virol. 2007;88:2991–3001. doi: 10.1099/vir.0.83065-0. [DOI] [PubMed] [Google Scholar]
- 8.Perotti M, et al. Identification of a broadly cross-reacting and neutralizing human monoclonal antibody directed against the hepatitis C virus E2 protein. J Virol. 2008;82:1047–1052. doi: 10.1128/JVI.01986-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Law M, et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med. 2008;14:25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 10.Logvinoff C, et al. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci USA. 2004;101:10149–10154. doi: 10.1073/pnas.0403519101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato T, et al. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology. 2003;125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 12.Lindenbach BD, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 13.Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 14.Puig M, Major ME, Mihalik K, Feinstone SM. Immunization of chimpanzees with an envelope protein-based vaccine enhances specific humoral and cellular immune responses that delay hepatitis C virus infection. Vaccine. 2004;22:991–1000. doi: 10.1016/j.vaccine.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Tarr AW, et al. Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology. 2006;43:592–601. doi: 10.1002/hep.21088. [DOI] [PubMed] [Google Scholar]
- 16.Flint M, et al. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J Virol. 1999;73:6235–6244. doi: 10.1128/jvi.73.8.6235-6244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owsianka A, Clayton RF, Loomis-Price LD, McKeating JA, Patel AH. Functional analysis of hepatitis C virus E2 glycoproteins and virus-like particles reveals structural dissimilarities between different forms of E2. J Gen Virol. 2001;82:1877–1883. doi: 10.1099/0022-1317-82-8-1877. [DOI] [PubMed] [Google Scholar]
- 18.Clayton RF, et al. Analysis of antigenicity and topology of E2 glycoprotein present on recombinant hepatitis C virus-like particles. J Virol. 2002;76:7672–7682. doi: 10.1128/JVI.76.15.7672-7682.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owsianka AM, et al. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J Virol. 2006;80:8695–8704. doi: 10.1128/JVI.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, et al. Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat Med. 2007;9:1032–1034. doi: 10.1038/nm1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang P, Yu MW, Venable R, Alter HJ, Shih JW. Neutralization epitope responsible for the hepatitis B virus subtype-specific protection in chimpanzees. Proc Natl Acad Sci USA. 2006;103:9214–9219. doi: 10.1073/pnas.0603316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barany G, Merrifield RB. Solid-phase peptide syntheses. In: Gross E, Meienhofer J, editors. The peptides: analysis, synthesis and biology. New York, NY: Academic; 1980. pp. 1–284. [Google Scholar]