

Abstract
Introduction
Plasminogen Activator Inhibitor-1 (PAI-1) is a member of the Serine Protease Inhibitor (SERPIN) gene family and a key regulator of fibrinolysis. PAI-1 is unique among SERPINs in its spontaneous transition to a latent, inactive state, with a half-life of approximately 2 hours under physiologic conditions. The biologic importance of the PAI-1 transition to latency is unknown. This study aimed to engineer transgenic overexpression of a stable murine PAI-1 variant to examine the physiologic effects in vivo from delayed transition of PAI-1 to latency.
Materials and Methods
Ten independent transgenic lines were generated with expression of a stable PAI-1 variant driven by the hybrid CMV/chicken β-actin promoter.
Results
Plasma PAI-1 levels in the transgenic founders ranged from 3.1±0.1 ng/mL to 1268.8±717.0 ng/mL. Quantitative PCR analysis in 3 transgenic lines demonstrated elevated PAI-1 mRNA in multiple tissues, with the highest increases observed in liver, brain, heart, and kidney. The fold-increase in PAI-1 mRNA over wild-type ranged from 2-fold to >2000-fold. Immunohistochemistry showed increased PAI-1 in liver, kidney, heart, spleen, and lung. Histologic examination of transgenic mice showed no evidence of thrombosis. The two founders with the highest plasma PAI-1 levels failed to produce any transgenic offspring that survived to weaning, although genotyping of expired pups revealed successful transmission of the transgene.
Conclusion
These results suggest that high expression of a stable variant of PAI-1 may be lethal in mice, while more moderate expression is generally well tolerated and produces no apparent thrombosis.
Keywords: fibrinolysis, serine protease inhibitor, latency, transgene
PAI-1 is the major physiologic inhibitor of tissue-type Plasminogen Activator (tPA) and urokinase Plasminogen Activator (uPA), enzymes that activate plasminogen to its active form plasmin and lead to fibrinolysis. PAI-1-deficiency in humans results in a mild to moderate bleeding disorder[1-3], while high plasma levels of PAI-1 have been linked to heart disease and diabetes[4-7]. In mice, PAI-1 deficiency results in reduced thrombus formation in models of vascular injury[8,9]. In addition to its role in hemostasis, PAI-1 has been implicated in the pathophysiology of numerous other processes, including atherosclerosis, obesity, tumorigenesis, wound healing, and fibrosis[10].
SERPINs all share a similar structure with 9 alpha-helices, 3 beta-sheets, and a strained reactive center loop. At the molecular level, inhibition occurs when the target protease cleaves the SERPIN at the P1-P1’ peptide bond of the reactive center loop, which subsequently inserts as the fourth strand in beta-sheet A[11-13]. PAI-1 is unique among SERPINs in that this inhibitory activity declines rapidly under physiologic conditions with a half-life of approximately 2 hours. Virtually all plasma PAI-1 is bound to vitronectin, which approximately doubles the functional half-life[14]. The functional instability of PAI-1 is due to a spontaneous conformational change to a more stable yet inactive state[15]. Although similar conformational changes can be induced in other SERPINs[11], only PAI-1 spontaneously transitions to latency. The short functional half-life of PAI-1 suggests that tight control of PAI-1 function may be biologically important. Berkenpas et al identified 14 independent PAI-1 variants, each containing 1 or more amino acid substitutions resulting in enhanced functional stability[16]. The most stable mutant contained 4 amino acid substitutions (N150H, K154T, Q319L, and M354I) and exhibited a functional half-life of 145 hours. This stable human PAI-1 variant was subsequently expressed as a transgene from the preproendothelin-1 promoter in mice to study the effects of enhanced PAI-1 functional stability in vivo[17-19]. The transgenic mice exhibited spontaneous coronary artery thrombosis around 6 months of age. This phenotype had not been observed in previously reported PAI-1 transgenic mice, which include wild-type human PAI-1 overexpressed from the metallothionein-I promoter[20] and wild-type murine PAI-1 overexpressed from either the CMV promoter[21] or the adipocyte aP2 promoter[22]. The metallothionein-I-driven human PAI-1 transgenic mouse demonstrated venous thrombosis of the hind limbs and tail. The murine PAI-1 transgene expressed from the aP2 promoter resulted in adipose hypotrophy, but neither of the murine PAI-1 transgenic mice exhibited any thrombosis. These results raise the possibility of a cross-species difference in PAI-1 function. To address this issue and to examine the physiologic effects in vivo from delayed PAI-1 transition to latency, we generated transgenic mice expressing a stable variant of murine PAI-1.
Materials and Methods
Functional half-life of mutant murine PAI-1
The mutant murine PAI-1 and wild-type human PAI-1 sequences were cloned into pET-3a (Stratagene, Cedar Creek, TX) and expressed in E.coli, strain BL21(DE3)PlysS. Human PAI-1 was purified as previously described[23]. Mutant murine PAI-1 was purified by Molecular Innovations, Inc. (Southfield, MI) as previously described[24]. Murine wild-type PAI-1 was purchased from Molecular Innovations, Inc. PAI-1 protein was diluted to 20ng/μL in assay buffer (150mM NaCl, 50mM Tris pH 7.5, 0.01% Tween 80, 100μg/mL bovine serum albumin) and tested for functional stability against uPA (America Diagnostica, Stamford, CT) using a chromogenic substrate assay as previously described[24]. PAI-1 samples were tested after incubation at 37°C for varying lengths of time, and the remaining inhibitory activity was determined as a percentage of the value at time zero. Final values are averaged from 4 experiments.
Construction of the stable murine PAI-1 transgene
Murine PAI-1 cDNA (from nucleotide 111 to 1538 in Genbank accession number M33960) was cloned into the Sma I restriction site of pALTER-1 (Promega, Madison, WI). Point mutations were introduced into murine PAI-1 cDNA using Promega GeneEditor, according to the manufacturer’s instructions. Codon 177 was mutated from AAG to ACC and codon 342 was mutated from CAG to CTG (Table 1, primers 1 and 2), creating the substitutions K154T and Q319L in the mature peptide. Mutations were confirmed by sequencing at the University of Michigan Sequencing Core.
Table 1.
Primers
| Primer | Sequence 5’→3’ |
|---|---|
| 1 | CAATGACTTACTGGCCACCGGGGCTGTAGACGA |
| 2 | CAGCTCTCTGTAGCACTGGCACTGCAAAAGGTC |
| 3 | ATTGCGGCTGCAGTGGGCACTGGGCAGGTAAG |
| 4 | AGTGGAGCATGCCTGTAATTGAACTGGGAGTGGAC |
| 5 | CCATCGTAACGCAGGGCTGTAAGTCTGTG |
| 6 | GCTAGGCGTCGACGTCATACATTGATGAGTTTGG |
| 7 | GCGTGGCGTGAAGCTTATGTTACCCCTCCGAGAATC |
| 8 | CGCCGCAGCCGAACGACCGAG |
| 9 | GACCGATCCTTTCTCTTTGT |
| 10 | TCTTTTCCCTTCAAGAGTCC |
| 11 | GGTATGATCAGTGACTTACTGGC |
| 12 | CCATCATGGGCACAGAGAC |
| 13 | ACCCAGAAGACTGTGGATGG |
| 14 | GGAGACAACCTGGTCCTCAG |
The table lists the primers used for mutagenesis, cloning, genotyping, copy number estimation, and qPCR. Restriction sites used in cloning are shown in italics.
The complete transgenic construct is shown in Figure 1A and contains from 5’ to 3’: the CAG promoter consisting of the CMV immediate-early enhancer and the chicken β-actin promoter, a chimeric intron, murine PAI-1 cDNA mutated as described above, and the SV40 early polyadenylation signal. The CAG promoter was initially reported by Niwa et al. [25], and was subsequently cloned into pGEM-3Z (Promega, Madison, WI) to create pCAG3Z[26](gift of Sally Camper). The CAG promoter contains not only the promoter region from chicken β-actin, but also the transcription start site and a portion of the 5’ UTR, as indicated in Figure 1. A chimeric intron from cloning vector pCI (Promega Corporation) was amplified (Table 1, primers 3 and 4) from nucleotide 845 to nucleotide 1014 and cloned into the PstI and SphI restriction sites downstream of the CAG promoter in the pCAG3Z vector.
Figure 1. Stable PAI-1 transgenic construct.

A. The transgenic construct is drawn to scale. Approximate locations of the transcription start site, the first ATG, and the 2 substitutions are indicated. The 5’ and 3’ limits of the included PAI-1 cDNA sequence are shown. CMV IE = Cytomegalovirus Immediate-Early Enhancer. UTR = Untranslated Region. SV40 PolyA = Simian Virus 40 Polyadenylation signal.
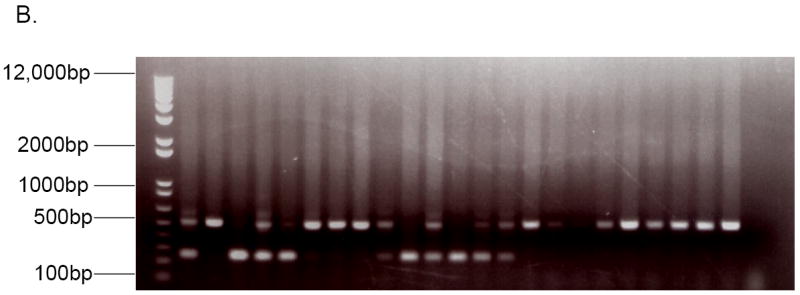
B. PCR across PAI-1 intron 8 using genomic DNA from founder mice and wild-type littermates. Amplification of the intron served as an internal positive control and gave a 520 base-pair band, while amplification of a 255 base-pair band lacking the intron indicated the presence of the PAI-1 cDNA transgene.
The SV40 early polyadenylation signal from the pEGFP-1 cloning vector (Clontech, Mountain View, CA) was amplified (Table 1, primers 5 and 6) from nucleotide 826 to nucleotide 1056 and cloned into the XbaI and SalI restriction sites downstream of the mutant PAI-1 cDNA in pALTER-1. Then a fragment containing both the mutant PAI-1 and the SV40 polyA sequence was amplified (Table 1, primers 7 and 8) and cloned into the HindIII restriction site downstream of the CAG promoter and chimeric intron in pCAG3Z.
The resultant transcript from the completed transgenic construct should contain a portion of 5’UTR from chicken β-actin, followed by the artificial UTR surrounding the chimeric intron, followed by a portion of murine PAI-1 5’UTR and the murine PAI-1 coding sequence. The content of the completed construct was confirmed by sequencing at the University of Michigan Sequencing Core.
Generation of transgenic mice
The purification of transgene construct DNA and subsequent generation of transgenic mice were performed by the University of Michigan Transgenic Animal Core. Briefly, the transgene construct was linearized with PvuII and purified from low-melting agarose using the Nucleopsin Extract Kit (Clontech, Mountain View, CA). Purified DNA was microinjected into fertilized eggs obtained by mating (C57BL/6 X SJL)F1 female mice with (C57BL/6 X SJL)F1 male mice. Pronuclear microinjection was performed as described[27]. Genomic DNA was purified from tail biopsies of founder mice at 2 weeks of age, and genotyping performed by PCR across PAI-1 intron 8 (Table 1, primers 9 and 10). The PCR reactions were incubated at 94 °C for 5 minutes, followed by 35 cycles of 94 °C for 1 minute, 54 °C for 1 minute, and 72 °C for 1 minute, followed by 72 °C for 10 minutes. Amplification of the intron served as an internal positive control and gave a 520 base-pair band, while amplification of a 255 base-pair band lacking the intron indicated the presence of the PAI-1 cDNA transgene (Fig 1B). Founder mice were (C57BL/6 X SJL)F2 and were backcrossed to C57BL/6J mice. The first generation of offspring from founders crossed to C57BL/6J will be referred to as N1.
All animals received care in compliance with the American Convention on Animal Care. This study was approved by the University Committee on Use and Care of Animals.
Estimating transgene copy number
A 172bp fragment of PAI-1 containing codon 154, which was mutated from AAG to ACC in the transgene, was amplified by PCR and sequenced (Table 1, primers 11 and 12). The area under the curve on the chromatogram for both the endogenous and transgenic alleles were calculated using PolyPhred [28], and the ratio of transgene to endogenous allele determined from a standard curve constructed with defined ratios of transgene construct DNA to mouse genomic DNA (Fig S1). The values obtained for the standards were subject to linear regression using Microsoft Excel.
Measuring plasma PAI-1 levels
Blood samples were obtained by tail snip or by terminal cardiac puncture in each founder and in 1-4 transgenic offspring from each transmitting line. Nontransgenic controls included 2 founder littermates and 6 F1 littermates from various lines. Blood was collected in tubes with 4% sodium citrate at a final dilution of 1:9. Whole blood was separated by centrifugation at 8000g for 10 minutes. Plasma was collected and stored at -80°C. Plasma PAI-1 levels were determined by ELISA using the total mouse PAI-1 antigen assay from Molecular Innovations, Inc. (Southfield, MI), according to the manufacturer’s instructions. Plasma samples were diluted 1:5 and compared to standardized concentrations of purified PAI-1 protein. All PAI-1 ELISAs were performed by the University of Michigan Thrombosis Program Mouse Coagulation Laboratory.
Quantification of PAI-1 mRNA
Mice were anesthetized with pentobarbital. Blood was collected by terminal cardiac puncture and animals were perfused with 5 mL phosphate-buffered saline (137mM NaCl, 10mM phosphates, 2.7mM KCl, pH 7.5). Tissues were rapidly collected from 3 transgenic mice at 2-4 months of age from each of the 3 lines and 1 wild-type littermate from each of lines 1 and 2, snap frozen in liquid nitrogen, and stored at -80°. Thirty-milligram tissue samples were homogenized using a handheld rotor-stator homogenizer (Pellet Pestle from Kontes, Vineland, NJ). RNA extraction was performed using the RNeasy Mini Kit (Qiagen, Valencia, CA). After RNA purification, cDNA was prepared from approximately 2μg of each RNA sample using M-MLV Reverse Transcriptase (Promega, Madison, WI) and oligodT primers (Invitrogen, Carlsbad, CA). Subsequently, PAI-1 cDNA was assayed by quantitative PCR with SYBR Green PCR Master Mix and the 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA). A 172bp fragment of PAI-1 was amplified (Table 1, primers 11 and 12), and results were normalized using qPCR of a 300bp fragment of GAPDH cDNA (Glyceraldehyde-3-phosphate dehydrogenase, Table 1, primers 13 and 14). Reaction volume was 20μL with each primer at 100nM. Forty cycles of 95 ° for 15 seconds and 60° for 1 minute were performed. PCR was performed in triplicate on each sample for each set of primers.
Histology and immunohistochemistry
Tissues were collected at 2-4 months of age from 1 to 2 transgenic mice and 1 wild-type littermate from each of lines 1, 2, and 7. Tissues examined included brain, heart, lung, liver, spleen, intestine, kidney, muscle, and adipose. From 2 mice affected with tail autoamputation, 1 from each of lines 1 and 7, tail samples less than 1 cm were collected from the distal remaining tail. Samples were rapidly collected and fixed in Z-fix (Anatech Ltd., Hayward, CA) for 4 hours at room temperature and overnight at 4°C. Samples were embedded in paraffin, sectioned, and hematoxylin and eosin staining were performed by the University of Michigan Comprehensive Cancer Center Tissue Core Research Histology and Immunoperoxidase Laboratory. Immunohistochemistry was performed following a previously established protocol[29]. Briefly, tissue sections were deparaffinized in Xylene, treated with 0.3% hydrogen peroxide and suppressor sera before incubation with primary antibodies for 4 hr at 25°C, either: anti-mouse PAI-1 antibody (Abcam, Cambridge, MA) diluted 1:2,000, or anti-mouse fibrinogen antibody (Vector, Burlingame, CA) diluted 1:10,000. The sections were then rinsed with PBS/Tween 20/Brij, incubated serially in ABC solution (Vector, Burlingame, CA) and DAB chromogen (Vector, Burlingame, CA), and counterstained with hematoxylin.
Results and Discussion
Generation of stable PAI-1 transgenic mice
Murine PAI-1 cDNA was mutated to encode 2 of the 4 amino acid substitutions found in the previously reported stable human PAI-1 mutant[16]. These 2 substitutions, K154T and Q319L, more than tripled the functional half-life of murine PAI-1 (8.7±1.2 hr compared to 2.5±0.6 hr for wild-type; Fig 2). The other 2 substitutions from the stable human PAI-1 are M354I and N150H. Murine PAI-1 already contains isoleucine at position 354 and contains aspartate at position 150, which also stabilizes human PAI-1[30]. These latter 2 amino acids may contribute to the longer functional half-life of murine PAI-1 compared to human PAI-1 (2.52±0.55hr versus 1.6±0.23hr; Fig 2). Human PAI-1 containing the 3 substitutions K154T, Q319L, and M354I displays a functional half-life of 91 hours[16]. The mutant murine PAI-1 in this study has the same amino acids at these positions but displays a much lower functional half-life of 8.7 hours, likely due to other amino acid differences between murine and human PAI-1 at positions that interact with these mutations and affect overall stability. To determine the effects in vivo from delayed PAI-1 latency, a transgene was engineered encoding the stable mutant murine PAI-1 downstream of the CAG promoter, a hybrid regulatory element containing the CMV immediate-early enhancer and the Chicken β-actin promoter[25]. Ten independent transgenic founders were generated. Transgene copy number (estimated by competitive PCR across the K154T mutation) ranged from 3 to 154 (Fig S1, Table 2).
Figure 2. Functional half-life of mutant PAI-1.
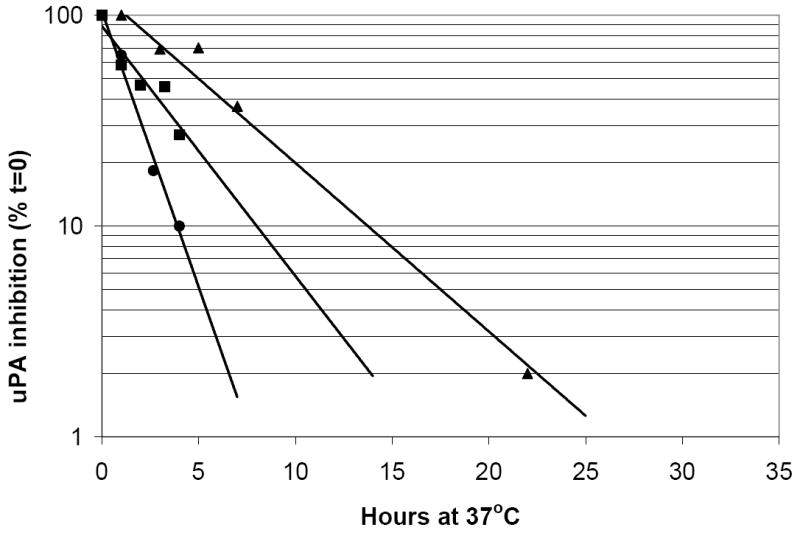
Wild-type murine PAI-1 (■), wild-type human PAI-1 (●) and mutant murine PAI-1 with substitutions K154T and Q319L (▲) were tested for inhibitory activity against human uPA after incubation at 37°C for the indicated numbers of hours. Values are plotted as the percentage of inhibition at time zero and represent the average of 4 independent experiments.
Table 2.
Transgene copy number estimates and plasma PAI-1 levels
| Transgenic Founder (gender) | Copy# using G/C* ratio | Copy# using G/T* ratio | Avg. Copy# | Founder Plasma PAI-1 (ng/mL)† | Offspring Plasma PAI-1 (ng/mL)† |
|---|---|---|---|---|---|
| 1 (M) | 6.7 | 9.8 | 8 | 218.0±237.0 | 682.8 |
| 2 (F) | 158.3 | 150.8 | 154 | 30.7±17.4 | 5.5±2.3 |
| 3 (F) | 31.4 | 25.9 | 29 | 1268.8±717.0 | N/A |
| 4 (F) | 37.1 | 35.8 | 36 | 3.1±0.1 | 8.5±4.2 |
| 5 (M) | 5.8 | 10.9 | 8 | 80.4±53.2 | 5.9±1.1 |
| 6 (F) | 20.3 | 20.9 | 21 | 284.6±328.6 | N/A |
| 7 (M) | 4.8 | 6.0 | 5 | 280.2±323.5 | 402.8±111.6 |
| 8 (M) | 122.9 | 110.2 | 117 | 14.2±8.7 | 6.0 |
| 9 (F) | 9.7 | 9.4 | 10 | 15.2±14.7 | 5.7±1.1 |
| 10 (F) | 4.9 | 1.4 | 3 | 237.8±330.0 | N/A |
| Wild-type | - | - | - | 4.3±1.8 | |
Indicates the two point mutations introduced by the PAI-1 K154T substitution that were used independently for copy number determination. The values in the 2 columns correlate with r2=0.996.
Expressed as mean ± SD, n=2 (Founder) & n=1-4 (Offspring). The 2 columns correlate with r2=0.759.
Characterization of transgene expression
Plasma PAI-1 levels were determined by ELISA in each founder and in 1-4 transgenic progeny (≥N1) at 2-4 months of age from each line that produced viable transgenic offspring (Table 2). Plasma PAI-1 levels ranged from 3 to 1269 ng/mL in the founders and from 6 to 683 ng/mL in the offspring, compared to ~4 ng/mL in wild-type mice. Individual measurements in wild-type mice were all less than 6.5 ng/ml, consistent with previous reports[31,32]. Thus, except for line 4, plasma PAI-1 levels in all mice carrying a transgene markedly exceeds the reported level of tPA and uPA in mouse plasma [31] and should result in no residual plasma activity for these target proteases.
Similar PAI-1 levels were observed in founders and offspring (r2=0.759). Mosaicism in the founders could result in lower plasma PAI-1 levels in some founders than in their offspring, potentially accounting for some of the variability in levels, such as in lines 1 and 7 (Table 2). Founders 3, 6, and 10, and lines 1 and 7 exhibit higher plasma PAI-1 levels than noted in any of the previously reported PAI-1 transgenic mice[20,21,33,34]. Although plasma PAI-1 levels were not described for the human PAI-1 transgenic mouse reported by Erickson et al [20], murine PAI-1 transgenes driven by the CMV and aP2 promoters produced levels of 108±17 and 24±3.8 ng/mL, respectively[32,35], with levels of 23±12 ng/mL reported for a human PAI-1 transgene driven by the preproendothelin-1 promoter[36].
Of the 10 founders generated in the current report, 3 failed to transmit the transgene to viable offspring (Table 3). Of the remaining 7 founders from which lines could be established, lines 1 and 7 with the highest plasma PAI-1 levels and line 2 exhibiting a lower level similar to wild-type were chosen for further study. PAI-1 mRNA levels were determined by quantitative PCR in a panel of tissues (Fig 3). The fold-increase in PAI-1 mRNA over wild-type ranged from 22 to 1087-fold in line 1, from 2 to 303-fold in line 2, and from 15 to 2245-fold in line 7. Although PAI-1 mRNA was not quantified in the previous transgene reports, the metallothionein promoter human PAI-1 transgene produced increases in PAI-1 antigen ranging from 10 to 75-fold across several tissues[20], and the murine adipocyte aP2 PAI-1 transgene produced protein levels elevated from 1 to 8-fold across tissues[37]. Thus, the levels of PAI-1 expression seen here with the CAG promoter appear significantly higher than observed in most or all previous reports.
Table 3.
Numbers of transgenic and nontransgenic mice at weaning in each line
| Line | Founder plasma PAI-1† | Generation | Tg+/- mice | Tg-/- mice | % Tg+/- | p value |
|---|---|---|---|---|---|---|
| 1 | 218.0±237.0 | N1 | 17 | 51 | 25% | p<0.0001 |
| N2-5 | 14 | 23 | 38% | p=0.1390 | ||
| 2 | 30.7±17.4 | N1 | 14 | 20 | 41% | p=0.3034 |
| N2-4 | 26 | 37 | 41% | p=0.1657 | ||
| 3 | 1268.8±717.0 | N1 | 0 | 15 | 0% | p<0.0005 |
| 4 | 3.1±0.1 | N1 | 7 | 8 | 47% | p=0.7958 |
| 5 | 80.4±53.2 | N1 | 9 | 15 | 38% | p=0.2207 |
| N2 | 4 | 2 | 67% | p=0.6667 | ||
| 6 | 284.6±328.6 | N1 | 0 | 18 | 0% | p<0.0001 |
| 7 | 280.2±323.5 | N1 | 3 | 38 | 7% | p<0.0001 |
| N2-4 | 31 | 31 | 50% | p=1.0000 | ||
| 8 | 14.2±8.7 | N1 | 13 | 15 | 46% | p=0.7053 |
| N2 | 17 | 27 | 39% | p=0.1316 | ||
| 9 | 15.2±14.7 | N1 | 9 | 14 | 39% | p=0.2971 |
| 10 | 237.8±330.0 | N1 | 0 | 0 | - | - |
| Total | N1 | 72 | 194 | 27% | p<0.0001 | |
| ≥N2 | 92 | 120 | 43% | p=0.0545 |
Significant and suggestive p values are shown in bold.
Shown in ng/mL, expressed as mean + SD.
Figure 3. PAI-1 mRNA levels in transgenic tissues.

RNA levels determined by qPCR from 3 transgenic mice (≥N1) in each of lines 1 (light grey bars), 2 (dark grey bars), and 7 (black bars), and in 2 wild-type mice (white bars). The exception is skin, where RNA was from 1 transgenic mouse in line 1, and 2 transgenic mice in each of lines 2 and 7. “Intestine” is from the small intestine, “fat” is from the gonadal fat pad, and “muscle” is from the quadriceps. Values are shown as a fold-increase over wild-type liver, which had the lowest PAI-1 RNA level of any sample and was arbitrarily set to one. This allows comparison of PAI-1 RNA levels between tissues as well as between transgenic and wild-type mice. The asterisks indicate a failed reaction (line 2, skin, 1 mouse; and line 7, skin, both mice). Values are expressed as mean ± SD.
The distribution of PAI-1 protein expression within tissues was ascertained by immunohistochemistry. Transgenic mice (≥N1) from line 1 showed prominent PAI-1 protein in liver, kidney, heart, spleen, and lung. Line 7 showed a similar PAI-1 distribution, with weaker staining in heart and spleen. In line 2, PAI-1 staining was strongest in the heart, but otherwise undetectable except at low levels in lung. Interestingly, no PAI-1 protein was detected in the brains of either transgenic or wild-type mice (data not shown), despite high mRNA expression, as detected by qPCR (Fig 3). Expression patterns were similar in different organs across multiple transgenic lines: in the kidney, most PAI-1 protein concentrated in the glomeruli; in the liver, PAI-1 was consistently observed in sinusoidal endothelial cells, although focal hepatocytic expression was also prominent; in the lung, the majority of PAI-1 positive cells were alveolar epithelial cells (Figs 4 & 5). PAI-1 was undetectable in wild-type tissues. These consistent patterns across lines likely reflect in large part the specific expression program of the CAG promoter. Tissue specific variation in the level of PAI-1 binding proteins such as vitronectin may also contribute to this distribution. PAI-1 mRNA upregulation was observed in more tissues than PAI-1 antigen. Both PAI-1 mRNA and antigen were observed at higher levels across more tissues in lines 1 and 7 than in line 2, which is also consistent with the higher plasma PAI-1 levels in the former 2 lines. In line 2, both PAI-1 mRNA and antigen were highest in the heart, with only low levels detected in other tissues.
Figure 4. Immunodetection of PAI-1 in mouse tissues.
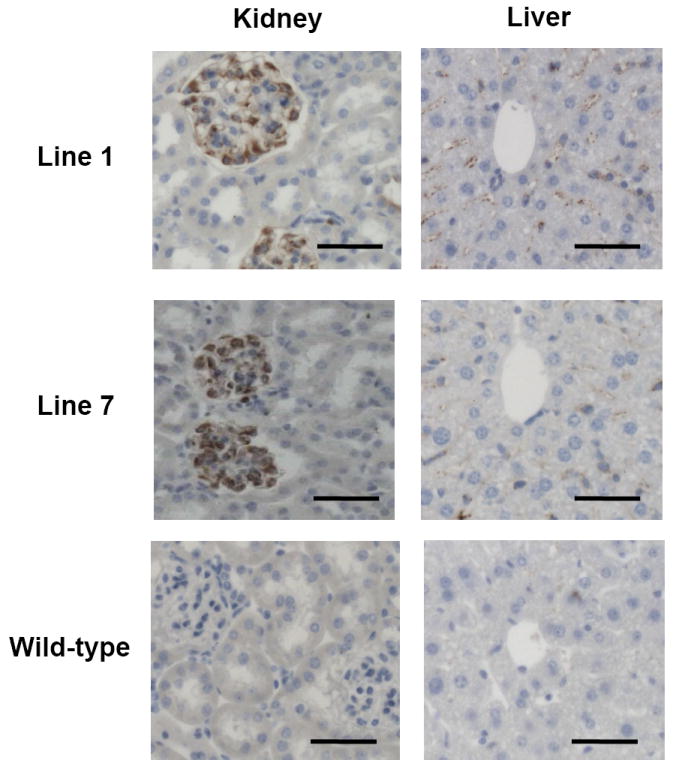
PAI-1 immunohistochemistry on sections from liver and kidney of one N1 transgenic mouse from each of lines 1 and 7, and a wild-type littermate from line 1, shown above at 20X magnification. Bars indicate 50 μm.
Figure 5. Histology of transgenic pulmonary tissue.
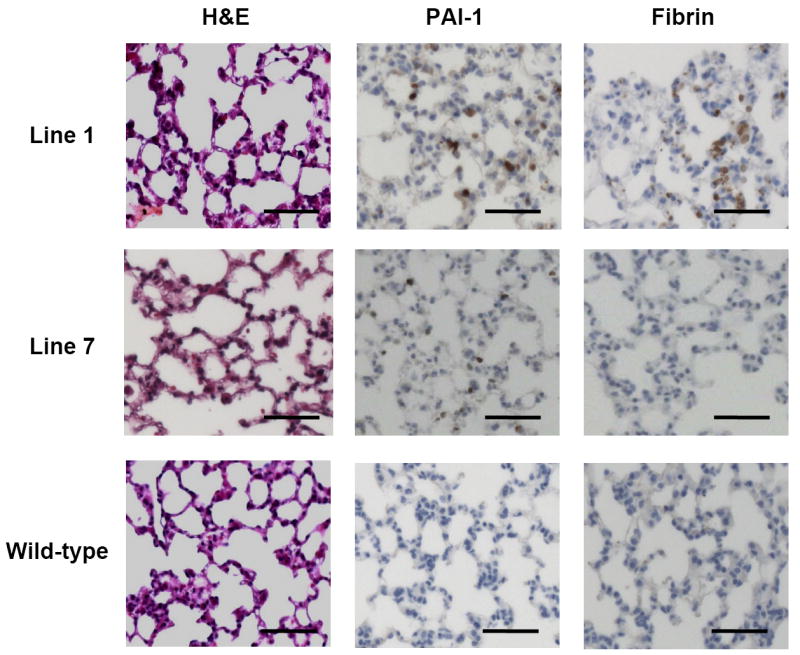
Hematoxylin and eosin staining, fibrin immunohistochemistry, and PAI-1 immunohistochemistry are shown for pulmonary tissue from one N1 transgenic mouse from each of lines 1 and 7, and a wild-type mouse from line 1, at 20X magnification. Bars indicate 50 μm.
The CAG promoter has been reported to direct strong, ubiquitous transgene expression in mice[38-40], with high level expression documented in brain, liver, kidney, adrenal, testis, lung, muscle, heart, intestine, adipose, thymus, and spleen[38,39]. Expression has been inconsistent in some tissues, including erythrocytes and hair[38,39]. The PAI-1 immunohistochemistry data suggest that transgene expression in these mice is more limited than previous reports of the CAG promoter, perhaps due to transgene insertion effects. In addition, PAI-1 protein stability is dependent on binding to vitronectin [14], and differences in levels of vitronectin and other binding proteins may contribute to the apparent discrepancies between the PAI-1 antigen detected in the transgenic mice versus the relatively more widespread PAI-1 mRNA distribution and the more ubiquitous expression pattern previously reported for the CAG promoter[38-40].
Histopathology of Transgenic Mice
Histopathology was examined in a panel of tissues from transgenic mice in lines 1, 2, and 7. No histological abnormalities were observed in sections stained with hematoxylin and eosin. Transgenic mice appeared to have increased fibrin deposition, assessed by immunohistochemistry, compared to wild-type, especially in the liver and lung in line 1, and the liver and heart in lines 2 and 7. H&E, PAI-1 expression, and fibrin deposition in the pulmonary tissues of transgenic and wild-type mice are illustrated in Fig 5. No evidence of thrombosis was detected in any of the mice.
An unusual autoamputation of the tail was observed in ~30% of transgenic mice in both lines 1 and 7 (5 of 17 and 1 of 3 N1 offspring, respectively), but in 0% of nontransgenic mice in these lines (0 of 51 and 0 of 38 N1 offspring, respectively). Furthermore, tail autoamputation was not observed in lines 2, 5, or 8. Tails of affected mice appeared normal at birth, with autoamputation occurring prior to weaning (Fig 6). Histological examination in 2 affected mice failed to reveal any evidence of thrombosis or other vascular or developmental abnormality. In addition, approximately 40% of line 1 transgenic mice (7 of 17 N1 offspring) displayed a disheveled coat, which was not observed in any line 1 nontransgenic mice (0 of 51 N1 offspring), or in lines 2, 5, 7, or 8. In addition, occasionally transgenic pups from lines 1, 3, and 6 displayed stunted growth, lack of fur, and wrinkled skin, and died prior to weaning. The latter nonspecific phenotypes could be coincidental and cannot be definitively attributed to the transgene.
Figure 6. Transgene-associated tail autoamputation.

A 5-week wild-type male from line 1 (top) and a transgenic female littermate (bottom) demonstrating tail autoamputation in the latter.
The occasional tail autoamputation observed in the transgenic mice in this study is reminiscent of a previously generated mouse transgenic for human PAI-1 expressed from the metallothionein-I promoter, which exhibited both tail necrosis and swelling of the hind limbs due to venous thrombosis[20]. However, no thrombi were detected in the present study, and a different underlying pathology could be responsible for the tail autoamputation reported here. As discussed previously, thrombosis was observed in both reports of mice transgenic for human PAI-1[20,41], but not in any of the mice transgenic for murine PAI-1[21,42], including the mice reported here. These results may reflect differences between human and murine PAI-1 and potential cross-species interactions between human PAI-1 and nonphysiologic targets in the mouse.
Under-representation of transgenic offspring
In the absence of mosaicism, 50% of the offspring (N1) from each transgenic founder are expected to carry the transgene [43,44]. As shown in Table 3, only 27% of the N1 offspring from all 10 lines carried the transgene (p<0.0001). Individually, lines 1, 3, 6, and 7 show a statistically significant under-representation of N1 transgenic offspring. However, for subsequent generations of transgenics produced from the N1 mice, the difference between the numbers of transgenic and nontransgenic offspring is no longer statistically significant. These data suggest a surprisingly high level of mosaicism in the transgenic founders. Though not statistically significant, the trend toward continued under-representation of transgenics among ≥ N2 offspring (92 transgenic versus 120 nontransgenic, p=0.0545) suggests a negative effect of the transgene on survival and perhaps selection for mosaicism among the original founders. However, we can’t rule out a modifying effect of genetic background on transgenic survival, with further backcross into the C57BL/6J strain.
Transgene mosaicism results from transgene integration into the founder genome after the 1-cell stage, and depending on the contribution to the germline, leads to reduced transgene transmission to N1 offspring. Mosaicism is observed in 20-30% of transgenic founders[43,44], consistent with the reduced transgene transmission observed for 4 of 10 founders in this study. Germinal mosaicism seems particularly likely in founder 7, with transgene transmission observed to only 7% of N1 offspring, with a 50% transmission frequency for subsequent generations.
Founder 10 produced several litters but no offspring survived to weaning. Founders 3 and 6 produced no transgenic offspring that survived to weaning (0 out of 15 and 0 out of 18, respectively), although genotyping of pups that expired prior to weaning revealed that both founders transmitted the transgene (5 out of 6 and 5 out of 9, respectively). The high frequency of the transgenic genotype among expired pups suggests that the absence of viable transgenic offspring for these two lines is not due solely to founder mosaicism. However, the cause of death for these transgenic neonates was not readily apparent. Of note, founders 3 and 6 also showed the highest plasma PAI-1 levels among the original 10 founders. Taken together, these data strongly suggest that high expression of this stable murine PAI-1 variant is lethal in mice. This lethality is not necessarily due to elevated plasma PAI-1, which could simply be an indirect marker of high PAI-1 expression in another critical tissue. The occurrence of this apparent lethality in at least 2 independent lines makes it unlikely that this is a secondary effect due to a transgene insertion site-specific expression pattern or interruption of an adjacent gene. The survival of founders 3 and 6 could be due to mosaicism for the transgene, leading to lower PAI-1 expression levels than in their nonmosaic offspring. Such lethality may also have selected for a high degree of mosaicism among the founders. Of note, both founders 3 and 6 are female (Table 2), suggesting the possibility that the lethality in their transgenic offspring might have resulted from high maternal PAI-1 and high fetal PAI-1 acting in concert.
If PAI-1 functioned solely to inhibit tPA and uPA, then overexpression of PAI-1 should be no more severe than combined deficiency of the target proteases. However, though mice doubly deficient for tPA and uPA exhibit widespread fibrin deposition and a reduced life span, survival to weaning is normal [45]. Thus, the neonatal lethality we observed from transgenic PAI-1 expression suggests inhibition of another protease, or an alternative nonprotease-related function, such as an effect on cell adhesion or migration mediated through PAI-1 interaction with vitronectin.
Supplementary Material
In each founder, the K154T mutation was amplified and sequenced (noncoding strand) and the area under the curve on the chromatogram was compared for the endogenous allele (CTT) and the transgenic allele (GGT). The ratio of G:C area under the curve was determined in a series of copy-number PCR standards (squares, line fit by linear regression) and in each transgenic founder (triangles) to estimate transgene copy number. A similar analysis was perfomed for the G:T at the second position of the codon (see Table 2).
Acknowledgments
We acknowledge Wanda Filipiak, Galina Gavrilina, and Maggie Van Keuren for preparation of transgenic mice and the Transgenic Animal Model Core of the University of Michigan’s Biomedical Research Core Facilities. Core support was provided by the University of Michigan Cancer Center, NIH grant number CA46592, the University of Michigan Multipurpose Arthritis Center, NIH grant number AR20557, and the University of Michigan Center for Organogenesis. We acknowledge Dr. Robert Lyons and the University of Michigan Sequencing Core. We acknowledge Nancy McAnsh, Alan Burgess, Reena Pathak, and Dr. Thomas Giordano from the University of Michigan Comprehensive Cancer Center Tissue Core Research Histology and Immunoperoxidase Laboratory for performing paraffin embedding, sectioning, and hematoxylin and eosin staining. We acknowledge Mark Warnock and Dr. Daniel Lawrence from the Mouse Coagulation Laboratory of the University of Michigan Thrombosis Program Project Grant for performing the PAI-1 ELISA. We acknowledge Duane Day and Molecular Innovations, Inc. for purification of the mutant PAI-1 protein. We acknowledge Dr. David Gordon for reviewing slides. We acknowledge Xixi Wang for maintaining the transgenic lines. We acknowledge support from the National Institutes of Health to D.G. (PO1 HL 057346 and R37 HL 036963). D.G. is an investigator of the Howard Hughes Medical Institute.
Abbreviations
- PAI-1
Plasminogen Activator Inhibitor-1
- SERPIN
Serine Protease Inhibitor
- tPA
tissue Plasminogen Activator
- uPA
urokinase Plasminogen Activator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Diéval J, Nguyen G, Gross S, Delobel J, Kruithof EKO. A lifelong bleeding disorder associated with a deficiency of plasminogen activator inhibitor type I. Blood. 1991;77:528–32. [PubMed] [Google Scholar]
- 2.Fay WP, Shapiro AD, Shih JL, Schleef RR, Ginsburg D. Complete deficiency of plasminogen-activator inhibitor type 1 due to a frame-shift mutation. N Engl J Med. 1992;327:1729–33. doi: 10.1056/NEJM199212103272406. [DOI] [PubMed] [Google Scholar]
- 3.Fay WP, Parker AC, Condrey LR, Shapiro AD. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: Characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. 1997;90:204–8. [PubMed] [Google Scholar]
- 4.Hamsten A, Wiman B, de Faire U, Blombäck M. Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N Engl J Med. 1985;313:1557–63. doi: 10.1056/NEJM198512193132501. [DOI] [PubMed] [Google Scholar]
- 5.Hamsten A, de Faire U, Walldius G, Dahlen G, Szamosi A, Landou C, Blombäck M, Wiman B. Plasminogen activator inhibitor in plasma: risk factor for recurrent myocardial infarction. Lancet. 1987;2:3–9. doi: 10.1016/s0140-6736(87)93050-9. [DOI] [PubMed] [Google Scholar]
- 6.De Taeye B, Smith LH, Vaughan DE. Plasminogen activator inhibitor-1: a common denominator in obesity, diabetes and cardiovascular disease. Curr Opin Pharmacol. 2005;5:149–54. doi: 10.1016/j.coph.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 7.Gough SC, Rice PJ, McCormack L, Chapman C, Grant PJ. The relationship between plasminogen activator inhibitor-1 and insulin resistance in newly diagnosed type 2 diabetes mellitus. Diabet Med. 1993;10:638–42. doi: 10.1111/j.1464-5491.1993.tb00137.x. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P, Stassen JM, Schoonjans L, Ream B, van den Oord JJ, De Mol M, Mulligan RC, Collen D. Plasminogen activator inhibitor-1 gene-deficient mice. II. Effects on hemostasis, thrombosis, and thrombolysis. J Clin Invest. 1993;92:2756–60. doi: 10.1172/JCI116893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eitzman DT, Westrick RJ, Nabel EG, Ginsburg D. Plasminogen activator inhibitor-1 and vitronectin promote vascular thrombosis in mice. Blood. 2000;95:577–80. [PubMed] [Google Scholar]
- 10.Lijnen HR. Pleiotropic functions of plasminogen activator inhibitor-1. J Thromb Haemost. 2005;3:35–45. doi: 10.1111/j.1538-7836.2004.00827.x. [DOI] [PubMed] [Google Scholar]
- 11.Carrell RW, Evans DL, Stein PE. Mobile reactive centre of serpins and the control of thrombosis. Nature. 1991;353:576–8. doi: 10.1038/353576a0. [DOI] [PubMed] [Google Scholar]
- 12.Lawrence DA, Olson ST, Palaniappan S, Ginsburg D. Serpin reactive-center loop mobility is required for inhibitor function but not for enzyme recognition. J Biol Chem. 1994;269:27657–62. [PubMed] [Google Scholar]
- 13.Potempa J, Korzus E, Travis J. The serpin superfamily of proteinase inhibitors: structure, function, and regulation. J Biol Chem. 1994;269:15957–60. [PubMed] [Google Scholar]
- 14.Declerck PJ, De Mol M, Alessi MC, Baudner S, Pâques E-P, Preissner KT, Müller-Berghaus G, Collen D. Purification and characterization of a plasminogen activator inhibitor 1 binding protein from human plasma. Identification as a multimeric form of S protein. J Biol Chem. 1988;263:15454–61. [PubMed] [Google Scholar]
- 15.Mottonen J, Strand A, Symersky J, Sweet RM, Danley DE, Geoghegan KF, Gerard RD, Goldsmith EJ. Structural basis of latency in plasminogen activator inhibitor-1. Nature. 1992;355:270–3. doi: 10.1038/355270a0. [DOI] [PubMed] [Google Scholar]
- 16.Berkenpas MB, Lawrence DA, Ginsburg D. Molecular evolution of plasminogen activator inhibitor-1 functional stability. EMBO J. 1995;14:2969–77. doi: 10.1002/j.1460-2075.1995.tb07299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. 2002;106:491–6. doi: 10.1161/01.cir.0000023186.60090.fb. [DOI] [PubMed] [Google Scholar]
- 18.Devin JK, Johnson JE, Eren M, Gleaves LA, Bradham WS, Bloodworth JRJ, Vaughan DE. Transgenic overexpression of plasminogen activator inhibitor-1 promotes the development of polycystic ovarian changes in female mice. J Mol Endocrinol. 2007;39:9–16. doi: 10.1677/JME-06-0057. [DOI] [PubMed] [Google Scholar]
- 19.Nordstrom SM, Carleton SM, Carson WL, Eren M, Phillips CL, Vaughan DE. Transgenic over-expression of plasminogen activator inhibitor-1 results in age-dependent and gender-specific increases in bone strength and mineralization. Bone. 2007;41:995–1004. doi: 10.1016/j.bone.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erickson LA, Fici GJ, Lund JE, Boyle TP, Polites HG, Marotti KR. Development of venous occlusions in mice transgenic for the plasminogen activator inhibitor-1 gene. Nature. 1990;346:74–6. doi: 10.1038/346074a0. [DOI] [PubMed] [Google Scholar]
- 21.Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest. 1996;97:232–7. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lijnen HR, Maquoi E, Morange P, Voros G, Van Hoef B, Kopp F, Collen D, Juhan-Vague I, Alessi MC. Nutritionally induced obesity is attenuated in transgenic mice overexpressing plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. 2003;23:78–84. doi: 10.1161/01.atv.0000044457.60665.dd. [DOI] [PubMed] [Google Scholar]
- 23.Sherman PM, Lawrence DA, Yang AY, Vandenberg ET, Paielli D, Olson ST, Shore JD, Ginsburg D. Saturation mutagenesis of the plasminogen activator inhibitor-1 reactive center. J Biol Chem. 1992;267:7588–95. [PubMed] [Google Scholar]
- 24.Lawrence D, Strandberg L, Grundström T, Ny T. Purification of active human plasminogen activator inhibitor 1 from Escherichia coli. Comparison with natural and recombinant forms purified from eucaryotic cells. Eur J Biochem. 1989;186:523–33. doi: 10.1111/j.1432-1033.1989.tb15238.x. [DOI] [PubMed] [Google Scholar]
- 25.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–9. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 26.Cushman LJ, Burrows HL, Seasholtz AF, Lewandoski M, Muzyczka N, Camper SA. Cre-mediated recombination in the pituitary gland. Genesis. 2000;28:167–74. doi: 10.1002/1526-968x(200011/12)28:3/4<167::aid-gene120>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 27.Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo: A Laboratory Manual. New York: Cold Spring Harbor Press; 2003. [Google Scholar]
- 28.Nickerson DA, Tobe VO, Taylor SL. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25:2745–51. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Olszewski B, Rosebury W, Robertson A, Keiser JA, Gordon D. Imapired angiogenesis in SHR is associated with decreased KDR and MT1-MMP expression. BBRC. 2003;315:363–8. doi: 10.1016/j.bbrc.2004.01.059. [DOI] [PubMed] [Google Scholar]
- 30.Stoop AA, Eldering E, Dafforn TR, Read RJ, Pannekoek H. Different structural requirements for plasminogen activator inhibitor 1 (PAI-1) during latency transition and proteinase inhibition as evidenced by phage-displayed hypermutated PAI-1 libraries. Journal of Molecular Biology. 2001;305:773–83. doi: 10.1006/jmbi.2000.4356. [DOI] [PubMed] [Google Scholar]
- 31.Declerck PJ, Verstreken M, Collen D. Immunoassay of murine t-PA, u-PA and PAI-1 using monoclonal antibodies raised in gene-inactivated mice. Thromb Haemost. 1995;74:1305–9. [PubMed] [Google Scholar]
- 32.Eitzman DT, Krauss JC, Shen T, Cui J, Ginsburg D. Lack of plasminogen activator inhibitor-1 effect in a transgenic mouse model of metastatic melanoma. Blood. 1996;87:4718–22. [PubMed] [Google Scholar]
- 33.Lijnen HR, Maquoi E, Morange P, Voros G, Van Hoef B, Kopp F, Collen D, Juhan-Vague I, Alessi MC. Nutritionally induced obesity is attenuated in transgenic mice overexpressing plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. 2003;23:78–84. doi: 10.1161/01.atv.0000044457.60665.dd. [DOI] [PubMed] [Google Scholar]
- 34.Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. 2002;106:491–6. doi: 10.1161/01.cir.0000023186.60090.fb. [DOI] [PubMed] [Google Scholar]
- 35.Lijnen HR, Maquoi E, Morange P, Voros G, Van Hoef B, Kopp F, Collen D, Juhan-Vague I, Alessi MC. Nutritionally induced obesity is attenuated in transgenic mice overexpressing plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. 2003;23:78–84. doi: 10.1161/01.atv.0000044457.60665.dd. [DOI] [PubMed] [Google Scholar]
- 36.Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. 2002;106:491–6. doi: 10.1161/01.cir.0000023186.60090.fb. [DOI] [PubMed] [Google Scholar]
- 37.Lijnen HR, Maquoi E, Morange P, Voros G, Van Hoef B, Kopp F, Collen D, Juhan-Vague I, Alessi MC. Nutritionally induced obesity is attenuated in transgenic mice overexpressing plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. 2003;23:78–84. doi: 10.1161/01.atv.0000044457.60665.dd. [DOI] [PubMed] [Google Scholar]
- 38.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Letters. 1997;407:313–9. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 39.Sawicki JA, Morris RJ, Monks B, Sakai K, Miyazaki J. A composite CMV-IE enhancer/b-Actin promoter is ubiquitously expressed in mouse cutaneous epithelium. Exp Cell Res. 1998;244:367–9. doi: 10.1006/excr.1998.4175. [DOI] [PubMed] [Google Scholar]
- 40.Kato M, Yamanouchi K, Ikawa M, Okabe M, Naito K, Tojo H. Efficient selection of transgenic mouse embryos using EGFP as a marker gene. Mol Reprod Dev. 1999;54:43–8. doi: 10.1002/(SICI)1098-2795(199909)54:1<43::AID-MRD6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 41.Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. 2002;106:491–6. doi: 10.1161/01.cir.0000023186.60090.fb. [DOI] [PubMed] [Google Scholar]
- 42.Lijnen HR, Maquoi E, Morange P, Voros G, Van Hoef B, Kopp F, Collen D, Juhan-Vague I, Alessi MC. Nutritionally induced obesity is attenuated in transgenic mice overexpressing plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. 2003;23:78–84. doi: 10.1161/01.atv.0000044457.60665.dd. [DOI] [PubMed] [Google Scholar]
- 43.Wilkie TM, Brinster RL, Palmiter RD. Germline and somatic mosaicism in transgenic mice. Dev Biol. 1986;118:9–18. doi: 10.1016/0012-1606(86)90068-0. [DOI] [PubMed] [Google Scholar]
- 44.Whitelaw CBA, Springbett AJ, Webster J, Clark AJ. The majority of G0 transgenic mice are derived from mosaic embryos. Transgenic Res. 1993;2:29–32. doi: 10.1007/BF01977678. [DOI] [PubMed] [Google Scholar]
- 45.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen JL, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–24. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In each founder, the K154T mutation was amplified and sequenced (noncoding strand) and the area under the curve on the chromatogram was compared for the endogenous allele (CTT) and the transgenic allele (GGT). The ratio of G:C area under the curve was determined in a series of copy-number PCR standards (squares, line fit by linear regression) and in each transgenic founder (triangles) to estimate transgene copy number. A similar analysis was perfomed for the G:T at the second position of the codon (see Table 2).