

Abstract
Synaptic glutamate transients resulting from vesicular exocytosis are superimposed on a low baseline concentration of glutamate in the extracellular space. Reported values of baseline glutamate concentrations range up to 4 μm. If glutamate were present tonically at low micromolar concentrations, many receptors, especially the high-affinity NMDA receptors (NMDARs), would be activated or desensitized, altering neuronal excitability. Using NMDARs expressed by CA1 pyramidal cells in acute hippocampal slices to monitor extracellular glutamate, we find that its baseline concentration is much lower, near 25 nm. In addition, superfusion of low micromolar concentrations of glutamate had no effect on neurons, indicating that glutamate transport prevents access to receptors within the slice. However, equipotent concentrations of NMDA, a nontransported agonist, depolarized neurons dramatically. We suggest that ambient concentrations of glutamate in vivo are also in the nanomolar range and are too low to cause significant receptor activation.
Keywords: hippocampus, NMDA receptors, ambient glutamate, glutamate transporter, dihydrokainate, TBOA
Introduction
Glutamate transporters, along with diffusion, terminate excitatory neurotransmission mediated by exocytosis of glutamate-filled vesicles (Isaacson and Nicoll, 1993; Takahashi et al., 1996; Asztely et al., 1997; Diamond and Jahr, 1997; Wadiche and Jahr, 2005) (for review, see Danbolt, 2001). In addition, the densely expressed glutamate transporters maintain baseline levels of extracellular glutamate at concentrations low enough to prevent excitotoxicity (Choi, 1992; Rothstein et al., 1996; Tanaka et al., 1997). Although the thermodynamic coupling of Na+ and K+ gradients to glutamate transport predicts a lower limit of 2 nm extracellular glutamate (Zerangue and Kavanaugh, 1996), the constant efflux of glutamate results in a higher steady-state ambient concentration in the extracellular space (Sah et al., 1989; Rossi and Slater, 1993; Lauri et al., 2006; Le Meur et al., 2007). Microdialysis studies report an in vivo ambient glutamate concentration as high as 1–4 μm (Lerma et al., 1986; Baker et al., 2002; Nyitrai et al., 2006). Given that the EC50 of the NMDA receptor (NMDAR) for glutamate is ∼2 μm (Patneau and Mayer, 1990), this concentration range would have significant effects on neuronal excitability. In contrast to the microdialysis studies, measurements of ambient glutamate in acute brain slice suggest a much lower concentration (Cavelier and Attwell, 2005; Cavelier et al., 2005; Le Meur et al., 2007), although a definitive estimate is lacking, and whether one can extrapolate from brain slice to brain is unclear.
By measuring the tonic current mediated by NMDARs in CA1 pyramidal cells, we estimated the concentration of ambient glutamate. We determined that the tonic spatially and temporally averaged concentration of extracellular glutamate in acute hippocampal slices with intact glutamate transport is ∼25 nm, 100-fold lower than reported previously (Lerma et al., 1986; Baker et al., 2002; Nyitrai et al., 2006). We suggest that this low concentration of extracellular glutamate is controlled by densely expressed glutamate transporters (Garthwaite, 1985; Jabaudon et al., 1999; Danbolt, 2001; Cavelier and Attwell, 2005).
Materials and Methods
Slice preparation.
Postnatal day 15–19 rats were deeply anesthetized with isofluorane and decapitated, as approved by the Oregon Health and Science University Institutional Animal Care and Use Committee. Hippocampi were removed, and transverse slices (300 μm) were cut using a vibroslicer (Leica, Bannockburn, IL) in ice-cold solution containing the following (in mm): 110 choline chloride, 7 MgCl2, 2.5 KCl, 1.25 KH2PO4, 0.5 CaCl2, 25 NaHCO3, 1.3 Na-ascorbate, and 10 glucose (saturated with 95% O2/5% CO2). The slices were incubated for 30–45 min at 34°C and then stored at room temperature in an external solution containing the following (in mm): 119 NaCl, 2.5 KCl, 2.0 CaCl2, 1.3 MgCl2, 1.0 NaH2PO4, 26.2 NaHCO3, and 11 glucose (saturated with 95% O2/5% CO2).
Experimental procedures.
Whole-cell recordings were obtained, using an Axopatch-1B amplifier (Molecular Devices, Union City, CA), from CA1 pyramidal cells visually identified with differential interference contrast optics (Zeiss, Thornwood, NY). Slices were superfused with the above external solution with 2.5 mm CaCl2, 100 μm picrotoxin, 10 μm 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), and 10 μm d-serine, except when noted. Experiments were performed at 32–35°C, with the temperature maintained by an in-line heating device (Warner Instruments, Hamden, CT), unless otherwise noted (see Fig. 3). Patch pipettes (2.0–3.5 MΩ) were filled with an internal solution containing the following (in mm): 135 Cs+ methanesulfonate, 8 NaCl, 10 HEPES, 10 Cs-BAPTA, 4 Mg-ATP, 0.4 Na-GTP, 0.2 verapamil (voltage-clamp experiments) or 135 K+ methanesulfonate, 8 NaCl, 10 HEPES, 10 K-BAPTA, 4 Mg-ATP, 0.4 Na-GTP (current-clamp experiments). Nucleated patch recordings were performed in the standard external solution with 5 mm CaCl2 and 0 mm MgCl2, using patch pipettes of ≤2.0 MΩ resistance. Electrophysiological recordings were acquired using custom software (J. S. Diamond, National Institute of Neurological Disorders and Stroke, Bethesda, MD) written in IgorPro (WaveMetrics, Lake Oswego, OR).
Figure 3.

Solution exchange efficiency with flow-pipe applications. A, Currents evoked by 5 μm NMDA in control or in the presence of 70 μm DAA (Vh of +40mV; room temperature). Top bars indicate bath and bottom bars indicate flow-pipe applications. B, Flow-pipe or bath-evoked currents blocked by DAA as a percentage of the control flow-pipe current (n = 4).
Agonists and antagonists were applied using a custom-built flow-pipe perfusion apparatus with a flow rate of ∼0.1 ml/min positioned above the slice (Fig. 1A). Flow-pipe solutions were continuously bubbled with 95% O2/5% CO2.
Figure 1.
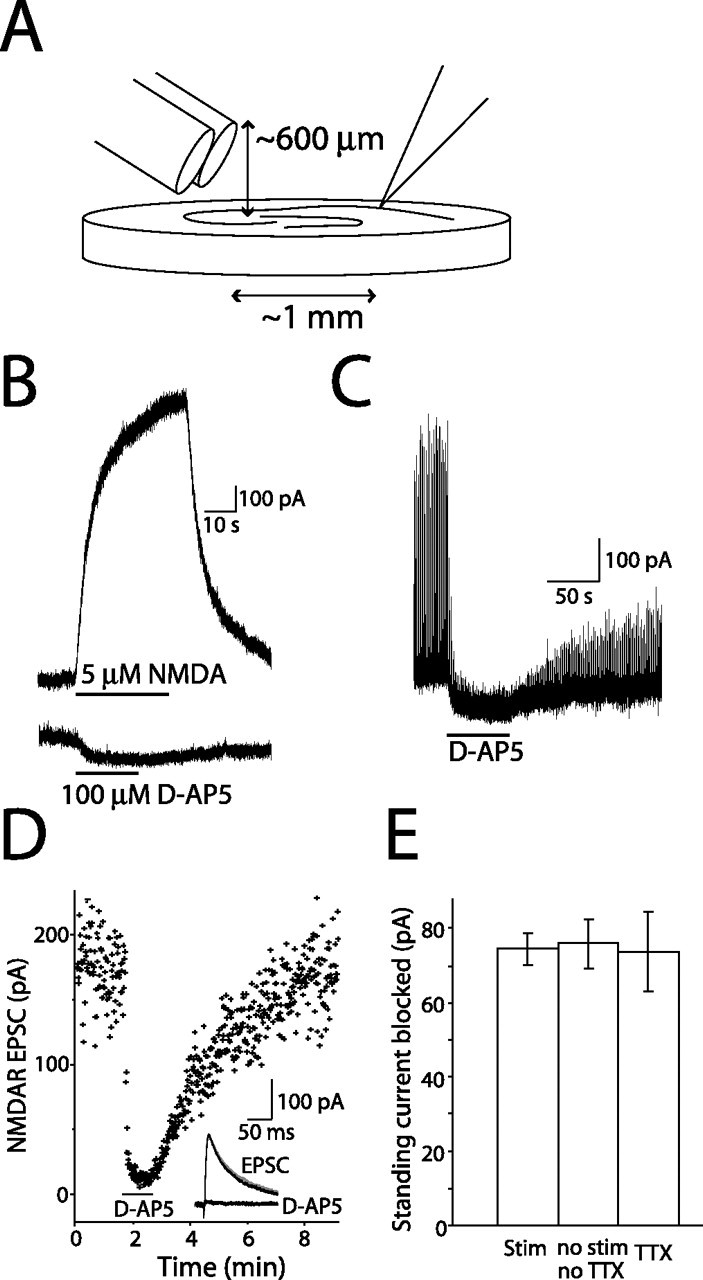
NMDAR-mediated currents in CA1 pyramidal cells. A, Schematic of flow-pipe configuration. Barrels were positioned at ∼30° and ∼1 mm from the recording pipette. B, Currents elicited from a CA1 pyramidal neuron by 5 μm NMDA and 100 μm d-AP-5 (Vh of +40 mV). Bars indicate flow-pipe application of NMDA and d-AP-5. Baseline currents have been offset. C, d-AP-5 (100 μm) applied to CA1 pyramidal neuron during 1 Hz stimulation of Schaffer collaterals. D, Effect of d-AP-5 on NMDAR EPSC amplitude. Bar indicates d-AP-5 application. Inset, Average traces from control, d-AP-5 treatment, and washout (gray) periods. E, Average amplitude of NMDAR current with 1 Hz stimulation (n = 7), without stimulation or TTX (n = 14), or 0.5 μm TTX (control; n = 7).
Data analysis and statistics.
AxoGraph X software (AxoGraph Scientific, Sydney, New South Wales, Australia) was used for analysis. Whole-cell recordings were excluded from analysis if series resistance was ≥10 MΩ or changed ≥15% during the course of an experiment. Current amplitudes were measured at peak deflection relative to baseline. The current blocked by application of 100 μm d-(−)-2-amino-5-phosphonopentanoic acid (d-AP-5) (Tocris Bioscience, Ellisville, MO) was normalized by the amplitude of the current evoked by 5 μm NMDA (Tocris Bioscience) for cells recorded in the presence of TTX. Currents recorded from hippocampal astrocytes were normalized to the plateau of the test pulse, to scale for proportional rundown of the synaptic transporter current with increase in access resistance (Diamond et al., 1998). Statistical analysis was performed using Excel (Microsoft, Seattle, WA) and Instat (GraphPad Software, San Diego, CA). Error bars on graphs correspond to the SEM. Significance was determined using ANOVA (Dunnett's or Tukey's post hoc) or Student's t test. Nonlinear regression analyses and EC50 estimates for dose–response data were performed with Prism (GraphPad Software).
In every whole-cell recording in which the standing NMDAR-mediated current was determined by block with d-AP-5, the current evoked by 5 μm NMDA was also measured (see Results). To estimate the concentration of NMDA that would evoke a current the size of the standing current, each current evoked by 5 μm NMDA was set to 0.073, the fraction of the maximal response (1 mm NMDA) evoked by 5 μm NMDA in nucleated patches. The standing current was scaled by the factor required to scale the 5 μm NMDA current to 0.073. Because the ratio of standing current to 5 μm NMDA current amplitude was 0.09, the standing current was 0.0065, i.e., <1% of the maximal current evoked by saturating NMDA. The concentration of NMDA necessary to evoke such a current was found by reading it off the abscissa of the dose–response curve at the point at which the current was 0.65% of maximal.
Results
Ambient glutamate generates a standing NMDAR current in hippocampus
Ambient extracellular glutamate activates an NMDAR-mediated current in hippocampal pyramidal cells (Sah et al., 1989; Cavelier and Attwell, 2005; Le Meur et al., 2007), which we used to estimate the concentration of glutamate in acute hippocampal slices. NMDAR currents were recorded in the presence of d-serine (10 μm), NBQX (10 μm), TTX (0.5 μm), and picrotoxin (100 μm) at +40 mV. To determine the amplitude of the standing NMDAR current, we applied the competitive NMDAR antagonist d-AP-5 (100 μm) via a flow pipe to CA1 pyramidal neurons (Fig. 1A). d-AP-5 application decreased the holding current by 73.7 ± 11.2 pA (Fig. 1B) (n = 7), blocking the NMDAR-mediated standing current generated by ambient glutamate. For comparison across cells, we divided this current amplitude by the current evoked by 5 μm NMDA (Fig. 1B) (831 ± 74.5 pA; n = 7), a nontransported agonist (Garthwaite, 1985; Dowd et al., 1996). The resulting measure of the standing current was 0.090 ± 0.01 (n = 7), a value that was used to quantify the ambient glutamate concentration.
To confirm that the current evoked by 5 μm NMDA application was a direct effect of the agonist and not contaminated with depolarization-induced glutamate release from other cells in the slice, we recorded the action of 5 μm NMDA on CA1 pyramidal neurons in current clamp in the same conditions. These cells were depolarized by 2.56 ± 0.9 mV during a 90 s application of 5 μm NMDA (n = 6). It seems unlikely that this depolarization would release enough glutamate to influence the evoked current.
It is possible that the ambient glutamate concentration is altered with changes in the level of synaptic activity. To test this, we investigated the magnitude of the NMDAR-mediated standing current while altering neuronal activity. The size of the current blocked by d-AP-5 was not different in the presence or absence of TTX (Fig. 1E) (n = 7) nor with continuous 1 Hz stimulation of Schaffer collaterals, which evoked NMDAR EPSCs (Fig. 1C–E) (n = 7). This suggests that the concentration of ambient glutamate is unaltered by activity, which is consistent with the results of other groups that vesicular release of glutamate does not contribute to the ambient extracellular concentration (Jabaudon et al., 1999; Cavelier and Attwell, 2005).
The ambient glutamate concentration is submicromolar
To estimate the average extracellular glutamate concentration, we first constructed dose–response relationships for NMDA and glutamate to which we could compare the normalized current blocked by d-AP-5 (Fig. 2). An accurate dose–response curve for glutamate could not be determined in the slice, because of the powerful uptake system present in the tissue (Garthwaite, 1985; Danbolt, 2001). Therefore, responses of pyramidal cells to the nontransported agonist NMDA (1–10 μm) (Dowd et al., 1996) were recorded in acute slices (Fig. 2A) (n = 6). These NMDA concentrations define only the foot of the dose–response curve (Patneau and Mayer, 1990). The entire dose–response curve could not be determined with whole-cell recordings, because the currents evoked with higher concentrations of NMDA were so large that we were concerned about the adequacy of the voltage clamp. Therefore, the complete dose–response curves for both NMDA and glutamate were generated with nucleated patches from CA1 pyramidal cell somata (Fig. 2B) (n = 7). A semi-log plot was constructed, and the patch data were fitted with the Hill equation yielding an EC50 of 37.7 μm for NMDA and 1.8 μm for glutamate (Fig. 2C), which are values close to those reported from dispersed neurons or neurons in primary culture at room temperature (Garthwaite, 1985; Patneau and Mayer, 1990). The whole-cell currents activated by 5 μm NMDA were scaled to the NMDA patch dose–response curve at the 5 μm point. The whole-cell measurements for 1–10 μm NMDA were well described by the Hill equation fit of the patch responses to NMDA (Fig. 2C, inset). When the whole-cell data alone were fitted with the Hill equation, assuming a maximal response at 1 mm, the resulting fit was nearly identical (EC50 of 39 μm). From the Hill fit, a concentration of 0.790 ± 0.069 μm NMDA would be necessary to produce a current equivalent to the normalized d-AP-5-blocked standing current (see Materials and Methods).
Figure 2.
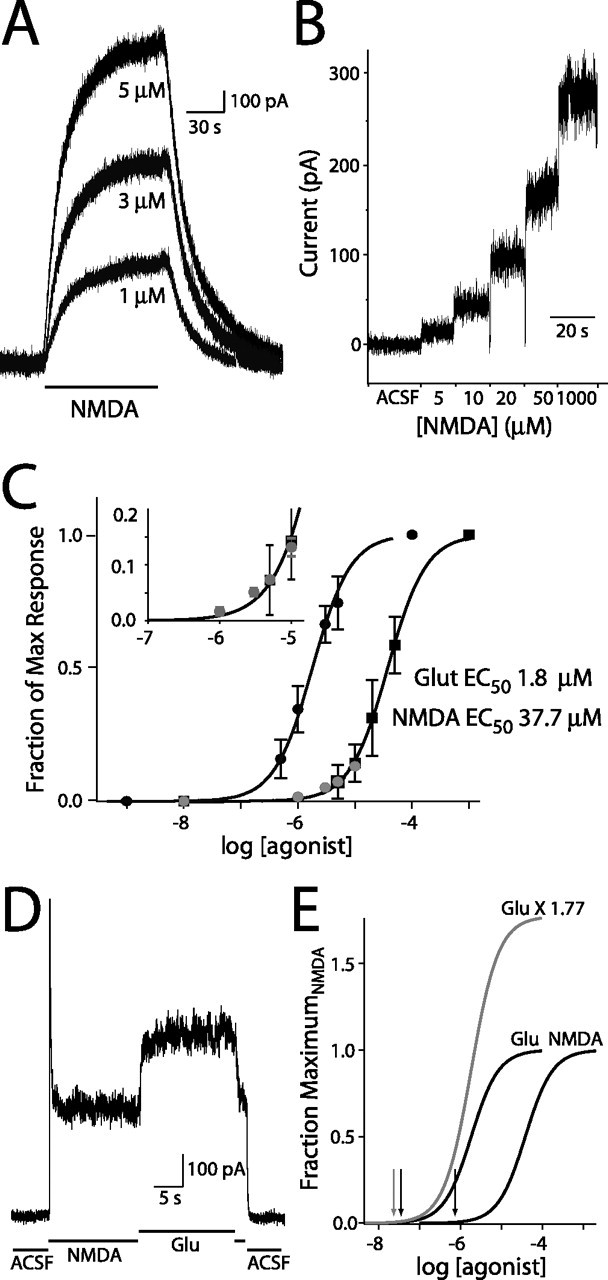
Estimation of ambient glutamate concentration. A, NMDA-evoked currents in a CA1 pyramidal neuron (Vh of +40 mV). Bar indicates applications of 1, 3, and 5 μm NMDA. Baseline currents have been zeroed. B, Nucleated patch currents produced by flow-pipe application of 5, 10, 20, 50, and 1000 μm NMDA (Vh of +40 mV). C, Semi-log plot of whole-cell (n = 6; gray circles) and nucleated patch (n = 6) current responses from application of NMDA (black squares) and glutamate (Glut; black circles; 0.5, 1, 3, 5, 100 μm; n = 6). Nucleated patch response were normalized to the maximum response. Whole-cell responses were scaled by the mean nucleated patch response to 5 μm NMDA. Line through NMDA responses is the nonlinear regression fit with Hill equation of nucleated patch dose responses (EC50 of 37.7 μm; nH = 1.3). Line through glutamate responses is the NMDA fit shifted by EC50Glut/EC50NMDA. Inset, Expansion of low concentration portion of NMDA dose–response curve. D, Nucleated patch responses to saturating concentrations of glutamate (Glu; 100 μm) and NMDA (1 mm). E, Dose–response curves from C, with glutamate-fit curve scaled by the efficacy ratio of 1.77. Arrows indicate the concentrations of glutamate and NMDA required to evoke currents of the same amplitude as that induced by ambient glutamate.
The concentration of glutamate required to activate a current of the same size as that blocked by d-AP-5 was estimated by comparing the dose–response curves for NMDA and glutamate. Comparison of the EC50 values resulted in an NMDA to glutamate conversion factor of 0.048. Multiplying the NMDA dose–response regression fit by this conversion factor results in a good fit of glutamate responses (Fig. 2C). Using this conversion factor, the ambient glutamate concentration necessary to produce the standing current in pyramidal cells is 37.9 ± 10.8 nm.
Agonists for the NMDAR can have different efficacies as well as different affinities (Lester and Jahr, 1992). With heterologously expressed NMDARs, the maximum response to glutamate is 1.2- to 1.5-fold larger than that of NMDA (Priestley et al., 1995). We tested this in our system by applying 100 μm glutamate and 1 mm NMDA, both saturating concentrations, to nucleated patches from CA1 pyramidal neurons. Glutamate produced a current that was 1.77 ± 0.08-fold larger than NMDA (Fig. 2D,E) (n = 5). Scaling the glutamate dose–response curve by this factor decreased our estimate of ambient glutamate to ∼25 nm (Fig. 2E).
Flow-pipe and bath-applied drugs have comparable access to receptors
Flow-pipe drug applications are often used for patches and cultures but not for acute brain slice. We performed control experiments to examine the extent to which drugs applied via flow pipe penetrate the slice tissue and affect receptors on the recorded cell (Figs. 1, 3). Flow-pipe application of 100 μm d-AP-5 blocked the NMDAR-mediated EPSC by 94.4 ± 0.9% (87 ± 13% recovery with washout; n = 8) (Fig. 1C,D). In addition, we used the NMDAR-mediated current to investigate the concentration of agonist that reached the receptors (Fig. 3). To do so, we compared the extent to which d-aminoadipic acid (DAA), a competitive low-affinity NMDAR antagonist, would block currents evoked with bath or flow-pipe applications of 5 μm NMDA. If bath-applied NMDA penetrated the slice better than flow-pipe applied NMDA, DAA should block the current evoked by bath application to a lesser extent. On the contrary, there was no difference in the block of currents evoked by flow-pipe and bath-applied NMDA (69.4 ± 8.8 and 66.8 ± 8.7% of control, respectively) (Fig. 3A, B), suggesting that drug applied by flow pipe reaches the receptors at a similar concentration as drug applied by bath.
Ambient glutamate estimate is not an artifact of washout
The ambient glutamate value of 25 nm could be an underestimate if endogenous extracellular glutamate were washed out of the slice with bath perfusion. We addressed this concern by comparing the magnitude of the standing NMDAR current blocked by 100 μm d-AP-5 in control and while incubating the slice in 2 μm glutamate, a concentration in the range found by microdialysis (Lerma et al., 1986; Baker et al., 2002; Nyitrai et al., 2006). We found that the currents blocked by d-AP-5 with 2 μm glutamate in the bath were not different from controls (103.8 ± 7.21% of control; n = 6) (Fig. 4A). As we would expect, a half-maximal activation of NMDARs with this glutamate concentration (equivalent to ∼40 μm NMDA), yet the standing current was not increased, we suggest that glutamate uptake is capable of maintaining extracellular glutamate at very low levels even in the presence of an endless supply of exogenous glutamate (Garthwaite, 1985).
Figure 4.
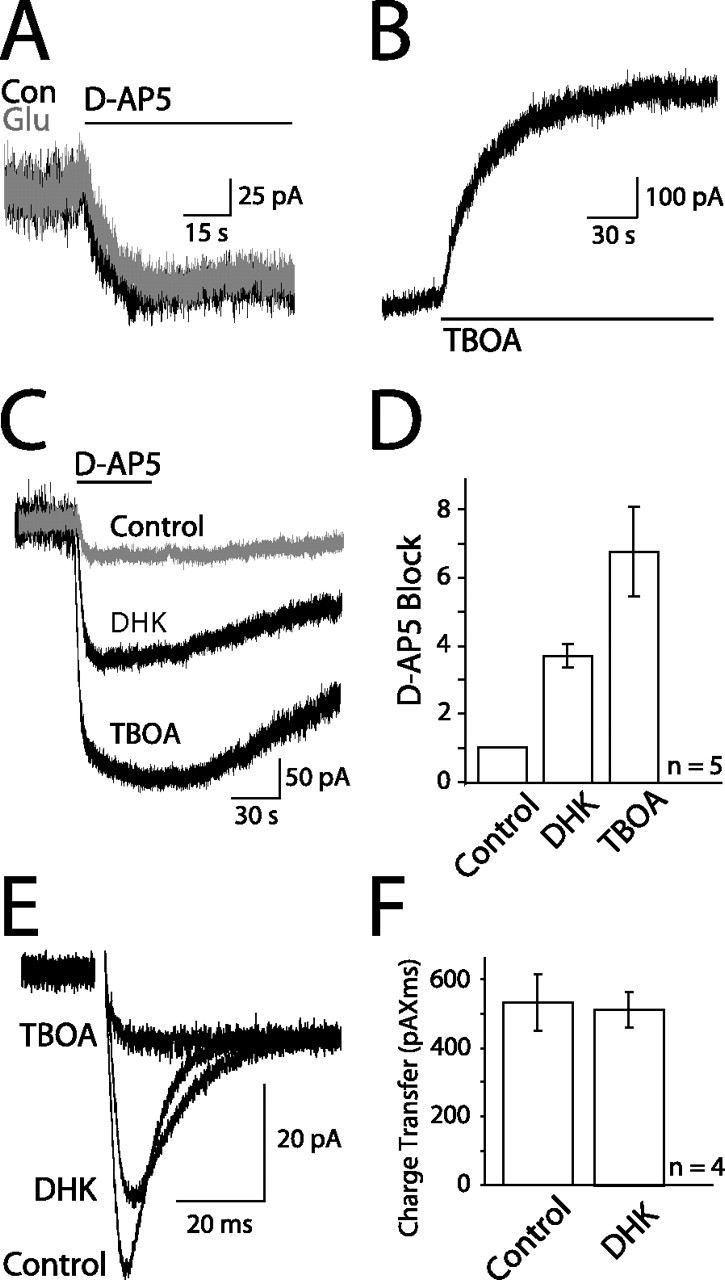
Glutamate transport maintains endogenous ambient glutamate. A, Standing current blocked by 100 μm d-AP-5 in control (Con; black) or with 2 μm glutamate (Glu; gray) in the bath. B, Current evoked by flow-pipe application of 200 μm TBOA. C, Standing currents blocked by 100 μm d-AP-5 in control (gray), 100 μm DHK, or 100 μm TBOA in the bath. Baseline currents have been zeroed. D, Averaged amplitudes of currents in control, DHK, or TBOA normalized to the control current for each cell (n = 5). DHK and TBOA significantly increase the magnitude of the current blocked (p < 0.05 and p < 0.01, respectively). E, Schaffer collateral-evoked transporter current in a hippocampal astrocyte in control, 100 μm DHK, or 100 μm TBOA, in addition to 10 μm NBQX, 100 μm picrotoxin, and 50 μm d-AP-5. All traces normalized to the plateau of the test pulse (see Materials and Methods). F, Average charge transfer of evoked transporter currents recorded in control and 100 μm DHK.
If uptake is an important mechanism for maintaining low ambient glutamate levels, transporter antagonists should increase the standing current. As others have shown (Jabaudon et al., 1999; Baker et al., 2002; Cavelier and Attwell, 2005), application of the glutamate transporter competitive antagonist dl-threo-benzyloxyaspartic acid (TBOA) (200 μm; n = 8) (Fig. 4B) caused a rapid increase in current (τ1 = 14.1 ± 2.85 s; τ2 = 65.3 ± 9.03 s), but the current did not reach steady state during a 2.5 min application, suggesting a continual buildup of glutamate. The rising phase of this current reflects both the rate at which transporters are blocked as TBOA enters the slice and the rate at which glutamate is being released into the extracellular space. This current was mediated by NMDARs because it was blocked by d-AP-5 (Fig. 4C,D). Of the transporters expressed in the hippocampus, the astrocytic glutamate transporter 1 (GLT-1) subtype accounts for ∼80% of uptake (Lehre and Danbolt, 1998; Danbolt, 2001). Blocking GLT-1 transporters with dihydrokainate (DHK) (100 μm) (Fiacco et al., 2007) increased the standing current almost fourfold, whereas blocking the remaining 20% of uptake with TBOA caused an additional doubling of the current (Fig. 4C,D). This suggests that the ambient glutamate concentration is controlled by transporters in a nonlinear manner. In contrast, although DHK decreased the amplitude of transporter currents evoked in hippocampal astrocytes by Schaffer collateral stimulation, it did not decrease the amount of glutamate taken up, as measured by the integral of the transporter current (Fig. 4E,F) (Diamond and Jahr, 2000). TBOA, conversely, blocked the transporter current leaving only the astrocytic response to elevated extracellular potassium (Fig. 4E) (Bergles and Jahr, 1997). These results suggest glutamate transporters play a dual role in the homeostasis of extracellular glutamate. First, they maintain low levels of ambient glutamate, and, second, they rapidly curtail the fast transients of glutamate after exocytotic release.
Diffusion of ambient glutamate from the surface of the slice into the bath inevitably occurs. However, given the sensitivity of extracellular glutamate concentration to transporter impairment and its resistance to change with exogenous glutamate in the bath, we suggest that tonic glutamate release and uptake are much more important in controlling the extracellular glutamate concentration in acute brain slice than diffusion into the bath.
NMDAR activation dramatically alters neuronal excitability
If extracellular glutamate were as high as reported previously (1–4 μm), what effect would this have on neuronal excitability? To determine this, we applied NMDA at concentrations equivalent to this range of glutamate to cells recorded in current clamp in the absence of receptor antagonists. Using the conversion factor generated by the EC50 values of the NMDAR for glutamate and NMDA, 15, 30, and 60 μm NMDA should mimic effects of 0.75, 1.5, and 3 μm glutamate, respectively. All concentrations of NMDA produced profound depolarization and spiking (Fig. 5). A 30 s application of 15 μm NMDA produced an average depolarization of 24.9 ± 10.1 mV, which was increased to 47.4 ± 22.9 mV with a 60 s application (Fig. 5A,C). Application of 30 and 60 μm NMDA for 30 s produced more rapid and larger depolarizations (60.6 ± 6.60 and 60.9 ± 5.77 mV, respectively) accompanied by spike accommodation (Fig. 5B,C). These results indicate that neurons in healthy acute slices are not normally enveloped by micromolar concentrations of glutamate. Were ambient glutamate to rise into the micromolar range, synaptic transmission, regenerative spiking, and neuronal health would be compromised. This also suggests that, even in vivo, ambient glutamate levels must be in the nanomolar range.
Figure 5.

NMDA application causes depolarization and spiking. Applications of 15 μm (A) or 30 μm (B) NMDA to a CA1 pyramidal neuron in current clamp in the absence of antagonists. The inset shows first spike burst on an expanded timescale indicated by the asterisk (A). C, Average change in membrane potential with 30 s (n = 6) or 60 s (n = 5) application of 15 μm NMDA and 30 s applications of 30 μm (n = 6) or 60 μm NMDA (n = 5).
Discussion
Ambient glutamate is in the submicromolar range
To avoid excitotoxicity, glutamate transporters must maintain extracellular glutamate at a low concentration. However, previous studies report ambient glutamate concentrations of 1–4 μm in vivo (Lerma et al., 1986; Baker et al., 2002; Nyitrai et al., 2006). We propose that an ambient glutamate concentration in this range would not only have deleterious effects on neurons and synaptic transmission but is also unrealistic given the efficacy of the glutamate uptake system. In the present study, we determined that the ambient glutamate concentration in hippocampal brain slice is much lower than previously described. Our estimated value of 25 nm would have negligible actions on most glutamate receptors (Trussell and Fischbach, 1989; Patneau and Mayer, 1990; Conn and Pin, 1997), would not compromise neuronal excitability, and is in better agreement with the theoretical minimum concentration of glutamate (2 nm) (Zerangue and Kavanaugh, 1996; Levy et al., 1998).
Were ambient glutamate in the micromolar range, NMDARs in vivo would be at half-maximum activation, because the EC50 of glutamate for the NMDAR is ∼2 μm (Garthwaite, 1985; Patneau and Mayer, 1990). Additionally, because low micromolar concentrations of glutamate can also cause desensitization of AMPA receptors (Trussell and Fischbach, 1989) and may activate high-affinity metabotropic glutamate receptors (Conn and Pin, 1997), synaptic transmission may be affected. We show that application of NMDA to acute brain slice at concentrations equivalent to 1–4 μm glutamate results in neuronal depolarization to nearly 0 mV and, as a consequence, complete spike accommodation. Because of these findings and considerations, we suggest that ambient glutamate levels in vivo are also in the nanomolar range.
Glutamate transport is highly efficient
The discrepancy in our estimate of the ambient glutamate concentration and that estimated by microdialysis may result from the exceedingly high concentration of glutamate transporters expressed in the CNS (Danbolt, 2001). The significant volume of tissue damage surrounding the microdialysis probe (Clapp-Lilly et al., 1999) could dramatically increase the distance between the probe tip and undamaged, transporter-expressing astrocytic membrane. We speculate that this transporter-free space surrounding the microdialysis probe, along with damage to the blood–brain barrier (Westergren et al., 1995), allows for buildup of extracellular glutamate and results in an artificially high ambient glutamate estimate. Our data support this speculation by showing that acutely blocking transport with TBOA increased the ambient glutamate concentration, even in conditions in which neuronal activity was blocked.
It is unlikely that the discrepancy between our results and previous in vivo studies result from (1) flow-pipe applied drugs not sufficiently blocking or activating NMDARs or (2) ambient glutamate washing out of the slice by diffusion into the bath. We addressed the first possibility by showing that flow-pipe applied d-AP-5 reversibly blocked the NMDAR-mediated EPSC and that NMDA-evoked currents were not differentially blocked by the low-affinity antagonist DAA when NMDA was applied by flow pipe or bath. We suggest that ambient glutamate in the slice is not lowered by diffusion into the bath, because the standing NMDAR-mediated current in the presence of 2 μm glutamate was not different from control and blocking transport with TBOA resulted in a rapid increase in standing NMDAR-mediated current, indicating that glutamate release is relatively rapid. These results indicate that tonic release and uptake, rather than diffusion out of the slice, control the extracellular glutamate concentration. We suggest that, even in vivo, transporters have the capacity to keep extracellular glutamate at nanomolar concentrations. However, ambient glutamate concentrations may not be uniform throughout the CNS, or even in the same structures across development, because the expression levels of transporters can vary.
Two roles for glutamate transport: synaptic and nonsynaptic uptake
As we and others (Jabaudon et al., 1999; Cavelier and Attwell, 2005) have shown, ambient levels of extracellular glutamate are not dependent on or altered by vesicular release but rather require a different mechanism of efflux, possibly from glia (Jabaudon et al., 1999; Cavelier and Attwell, 2005; Le Meur et al., 2007). Because synaptic release does not change ambient glutamate levels, the densely expressed transporters (Lehre et al., 1995; Lehre and Danbolt, 1998) must be capable of sequestering synaptically released glutamate very rapidly (Diamond, 2005; Wadiche et al., 2006) to prevent protracted diffusion. Thus, glutamate transporters appear to have distinct actions: rapid binding and uptake of vesicular release, which prevents or diminishes spillover, and a tonic clearance mode that maintains the average extracellular glutamate concentration at very low levels.
Footnotes
This work was supported by National Institutes of Health Grants MH074989 (C.E.J.) and T32 DK07680 (M.A.H.). We thank Vanessa Bender, Jason Christie, Ko Matsui, Richard Piet, Henrique von Gersdorff, and Jacques Wadiche for valuable comments and suggestions.
References
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus; dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol (Lond) 2005;564:397–410. doi: 10.1113/jphysiol.2004.082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelier P, Hamann M, Rossi D, Mobbs P, Attwell D. Tonic excitation and inhibition of neurons: ambient transmitter sources and computational consequences. Prog Biophys Mol Biol. 2005;87:3–16. doi: 10.1016/j.pbiomolbio.2004.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Clapp-Lilly KL, Roberts RC, Duffy LK, Irons KP, Hu Y, Drew KL. An ultrastructural analysis of tissue surrounding a microdialysis probe. J Neurosci Methods. 1999;90:129–142. doi: 10.1016/s0165-0270(99)00064-3. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25:2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 2000;83:2835–2843. doi: 10.1152/jn.2000.83.5.2835. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Bergles DE, Jahr CE. Glutamate release monitored with astrocyte transporter currents during LTP. Neuron. 1998;21:425–433. doi: 10.1016/s0896-6273(00)80551-6. [DOI] [PubMed] [Google Scholar]
- Dowd LA, Coyle AJ, Rothstein JD, Pritchett DB, Robinson MB. Comparison of Na+-dependent glutamate transport activity in synaptosomes, C6 glioma, and Xenopus oocytes expressing excitatory amino acid carrier 1 (EAAC1) Mol Pharmacol. 1996;49:465–473. [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J, McCarthy KD. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron. 2007;54:611–626. doi: 10.1016/j.neuron.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Cellular uptake disguises action of l-glutamate on N-methyl-d-aspartate receptors. With an appendix: diffusion of amino acids into brain slices. Br J Pharmacol. 1985;85:297–307. doi: 10.1111/j.1476-5381.1985.tb08860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Nicoll RA. The uptake inhibitor l-trans-PDC enhances responses to glutamate but fails to alter the kinetics of excitatory synaptic currents in the hippocampus. J Neurophysiol. 1993;70:2187–2191. doi: 10.1152/jn.1993.70.5.2187. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gähwiler BH, Gerber U. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci USA. 1999;96:8733–8738. doi: 10.1073/pnas.96.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Vesikansa A, Segerstrale M, Collinridge GL, Isaac JT, Taira T. Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron. 2006;50:415–429. doi: 10.1016/j.neuron.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol (Lond) 2007;580:373–383. doi: 10.1113/jphysiol.2006.123570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J, Herranz AS, Herreras O, Abraira V, Martin Del Rio R. In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res. 1986;384:145–155. doi: 10.1016/0006-8993(86)91230-8. [DOI] [PubMed] [Google Scholar]
- Lester RAJ, Jahr CE. NMDA channel behavior depends on agonist affinity. J Neurosci. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Attwell D, Hoover F, Ash JF, Bjoras M, Danbolt NC. Inducible expression of the GLT-1 glutamate transporter in a CHO cell line selected for low endogenous glutamate uptake. FEBS Lett. 1998;422:339–342. doi: 10.1016/s0014-5793(98)00036-2. [DOI] [PubMed] [Google Scholar]
- Nyitrai G, Kékesi KA, Juhász G. Extracellular level of GABA and Glu: in vivo microdialysis-HPLC measurements. Curr Top Med Chem. 2006;6:935–940. doi: 10.2174/156802606777323674. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Structure–activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priestley T, Laughton P, Myers J, Le Bourdelles B, Kerby J, Whiting PJ. Pharmacological properties of recombinant human N-methyl-d-aspartate receptors comprising NR1a/NR2A and NR1a/NR2B subunit assemblies expressed in permanently transfected mouse fibroblast cell. Mol Pharmacol. 1995;48:841–848. [PubMed] [Google Scholar]
- Rossi DJ, Slater NT. The developmental onset of NMDA receptor-channel activity during neuronal migration. Neuropharmacology. 1993;32:1239–1248. doi: 10.1016/0028-3908(93)90018-x. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Briston LA, Jin L, Kunci RW, Kanai Y, Hediger MA, Wang Y, Scheilke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Sah P, Hestrin S, Nicoll RA. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science. 1989;246:815–818. doi: 10.1126/science.2573153. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Sarantis M, Attwell D. Postsynaptic glutamate uptake in rat cerebellar Purkinje cells. J Physiol (Lond) 1996;497:523–530. doi: 10.1113/jphysiol.1996.sp021785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Fischbach GD. Glutamate receptor desensitization and its role in synaptic transmission. Neuron. 1989;3:209–218. doi: 10.1016/0896-6273(89)90034-2. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Patterned expression of Purkinje cell glutamate transporters controls synaptic plasticity. Nat Neurosci. 2005;8:1329–1334. doi: 10.1038/nn1539. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Tzingounis AV, Jahr CE. Intrinsic kinetics determine the time course of neuronal synaptic transporter currents. Proc Natl Acad Sci USA. 2006;103:1083–1087. doi: 10.1073/pnas.0510476103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergren L, Nystrom B, Hamberger A, Johansson BB. Intracerebral dialysis and the blood brain barrier. J Neurochem. 1995;64:229–234. doi: 10.1046/j.1471-4159.1995.64010229.x. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh M. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]