

Abstract
Objective
Sepsis impairs the activation of the interleukin (IL)-6 dependent transcription factor signal transducer and activator of transcription (STAT)-3. However, the molecular basis for depressed functionality has not been characterized. In this study we test the hypothesis that altered signal transduction results from a change in the activation state of one or more of the components of the intracellular IL-6-linked pathway.
Design
Randomized prospective experimental study.
Setting
University medical laboratory.
Subjects
Male 6-8 week old C57/Bl6 mice.
Interventions
Cecal ligation and single (CLP) or double (2CLP) puncture were used to model mild and fulminant sepsis respectively. Sham operated and unoperated animals served as controls. All animals were fluid resuscitated at the time of surgery and every 24 hrs thereafter. Surviving animals were euthanized at 3, 6, 16, 24, 48, and 72 hrs, blood samples were obtained and liver tissue was harvested.
Measurements and Main Results
Serum IL-6 levels were elevated in both CLP and 2CLP relative to controls. STAT-3 DNA binding activity and nuclear p-STAT-3 levels were elevated in CLP but decreased abruptly 24 hrs after 2CLP. This 2CLP-induced alteration was associated with attenuated phosphorylation of the key transcellular glycoprotein (gp) 130. Abundance and phosphorylation of the other key component of IL-6 signal transduction pathway, Janus Kinase (JAK)-1, was unchanged following either CLP or 2CLP. 2CLP also did not cause disassociation of the gp130-JAK-1 complex.
Conclusions
Impaired gp130 phosphorylation may be responsible for IL-6 hyporesponsiveness during sepsis.
Keywords: STAT-3, liver, animal models, janus kinase (JAK), signal transduction, cytokines
Introduction
Sepsis is a leading cause of morbidity and mortality in non-coronary intensive care units. The syndrome is associated with approximately 215,000 deaths each year (1). Mortality is typically due to the multiple-organ dysfunction syndrome (MODS). MODS is characterized by dysfunction in nearly all organ systems including the liver (2). This process is mediated, in part, by cytokines such as Interleukin-6 (IL-6). However, the role played by IL-6 in sepsis/MODS is unclear. IL-6 has been reported to be an excellent marker for mortality in both animals and humans but there is debate regarding the importance of this mediator’s contribution to the pathologic process (3). While some believe that IL-6 is most valuable as a biomarker, studies in our laboratory have shown a sepsis-induced decrease in IL-6 activity that parallels the progression of organ dysfunction, especially in the liver. Specifically, previous investigations in a rat model of mild sepsis (cecal ligation and single puncture, CLP) demonstrated a sustained increase in signal transducer and activator of transcription (STAT)-3 DNA binding activity over 72 hours (4). This was accompanied by an increase in the transcription of IL-6-dependent genes. However, we observed an abrupt loss of STAT-3 DNA binding activity 16 hours after the induction of fulminant sepsis via cecal ligation and double puncture (2CLP). This latter finding paralleled both gene expression and mortality (4). Our observations suggested that STAT-3 activation via IL-6 is necessary for survival and that fulminant sepsis is characterized by a loss of cell signaling, a conclusion supported by additional studies in IL-6 -/- mice (5). Because IL-6 levels in this model are known to be elevated (3, 6), it is logical to surmise that there is an abnormality in the IL-6/ STAT-3 signal transduction pathway.
Activation of the IL-6 pathway involves the transmembrane cell surface receptor glycoprotein (gp)130. With IL-6 binding, gp130 dimerizes and induces the phosphorylation of Janus Kinase (JAK)-1. Once phosphorylated, JAK-1 itself acts as a kinase and phosphorylates a number of tyrosine residues on the intra-cytoplasmic tail of gp130. Importantly, gp130 and JAK-1 are tightly linked and this association is believed to be important to both gp130 and JAK-1 kinase activity. gp130 tyrosine residues serve as docking sites for STAT-3 with transfer of a phosphate group to SH-2 sites on the transcription factor. Phosphorylated STAT-3 (p-STAT-3) dimerizes, translocates to the nucleus and induces organ specific gene expression (7). Therefore, we hypothesize that sepsis alters either the abundance or phosphorylation of gp130 or JAK-1 or the association of gp130 with JAK-1. This in turn would result in failed STAT-3 nuclear translocation and DNA binding.
Materials and Methods
Animals
Animal procedures and handling adhered to National Institutes of Health standards and were approved by the Institutional Animal Care and Use Committee. 6-8 week old male C57/Bl6 mice (Charles River, Boston MA) were used in all experiments.
Induction of sepsis
Under isoflurane anesthesia mild, non-fulminant sepsis was induced via cecal ligation and puncture (CLP) with a 23 gauge needle as previously described (8). Fulminant sepsis was performed in a similar manner but the cecum was punctured twice (2CLP). Control animals were either anesthetized only (To) or subjected to sham operation (SO), where the cecum was manipulated but not ligated or punctured. Following surgery, animals were fluid resuscitated with 40 mL/kg of subcutaneously administered normal saline. At 3, 6, 16, 24, 48, and 72 hours following surgery, animals were re-anesthetized with 150 mg/kg pentobarbital sodium. Animals were exsanguinated via the vena cava, with blood being reserved for serum assays.
There are two important aspects to the study design. First, our long-used study design required that animals be designated for sacrifice at a specific time point. Therefore, based on previous data, we performed enough procedures to assure that three animals survived to each time point. In effect this constitutes something of an “intention-to-treat” design. To expand, while there were no deaths in the Sham Operation or CLP groups there was significant mortality in the 2CLP group. Therefore, we performed 2CLP in six animals designated to survive for 24 hrs, 12 animals designated to survive to 48 hrs and 30 animals designated to survive to 72 hrs. Because of this design our data have an inherent survival bias. Secondly, all animals to be included in a cohort (defined as one T0, one T3, one T6, one T16, one T24, one T48 and one T72) were designated to be part of that group in advance and the intervention (Sham Operation, CLP, 2CLP) also was designated in advance. Sham operation, CLP or 2CLP was performed on a complete cohort on the same day. This assured that the data obtained from one T0 animal was used to normalize all other animals in the same cohort, permitting us to design our blots to include a full time course of data.
Isolation of Liver Protein Fractions
Liver tissue was harvested immediately following exsanguination and both nuclear and cytosolic protein fractions were isolated. All isolation procedures were performed on ice or at 4°C and in the presence of the protease inhibitors antipain, aprotinin, bestatin, and leupeptin and the phosphatase inhibitors phenylmethylsulfonyl fluoride (PMSF), NaF, Na2MoO4 and Na3VO4. Nuclear and cytosolic protein fractions were harvested and homogenized as previously described (9,10). Briefly, samples were centrifuged at 83,000g (26,000 rpm) for 50 min. to separate the cytosolic fraction (supernatant) from intact nuclei (pellet). Nuclear proteins then were isolated from the pellet.
Immunoprecipitation from the cytosolic protein fraction
500 μg of cytosolic protein were diluted to a final concentration of 1 μg/μL with PBS pH 7.4. For gp130 immunoprecipitation, samples were pre-cleared for 30 minutes at 4°C with 50 μL of a 50% Protein G sepharose bead slurry. The beads subsequently were removed by centrifugation. 1.6 μg of gp130 rabbit polyclonal IgG (Santa Cruz Biotechnology Inc, Santa Cruz CA) were incubated overnight at 4°C using circular rotation. Following this, 100 μL of 50% Protein G sepharose beads in PBS were added and each sample was placed in a circular rotator and incubated overnight. After centrifugation, the supernatant was discarded and the bead pellet was saved. The beads were washed three times with a modified RIPA buffer (50 mM Tris-HCl and 150 mM NaCl pH to 7.4, 10% NP-40, 10% Na-deoxylate, 0.5 M EDTA + protease and phosphatase inhibitors). For JAK-1 immunoprecipitation the same procedure was followed but a 50% slurry of Protein A sepharose beads and 1.6 μg of JAK-1 IgG rabbit polyclonal (Santa Cruz) were used. After the final wash, 75 μL of 2 X Laemmli buffer was added to the beads and the mixture boiled for 5 minutes. The resultant supernatant was subjected to 10% SDS-PAGE.
Immunoblotting
All procedures were performed at room temperature. 8% SDS-PAGE gels containing 20μg of sample/well were transferred to PVDF (BioRad, Hercules CA) using electroblotting. After transfer, membranes were rinsed and blocked for 2 hours at room temperature in a 5% (w/v) blocking solution (non-fat dry milk in Tris-buffered saline pH 7.6 with 0.001% Tween-20). After blocking, membranes were probed for two hours with antibodies directed at either gp130, phosphorylated gp130, JAK-1, phosphorylated JAK-1, STAT-3, or tyrosine-phosphorylated STAT-3 (Santa Cruz) using a 1:1000 dilution. Membranes next were incubated for one hour with species specific secondary antibodies (Santa Cruz) added in a 1:2000 dilution. Protein bands were visualized using enhanced chemi-luminescence (Amersham, Piscataway, NJ). Abundance was quantified using densitometry.
Electrophoretic Mobility Shift Assay (EMSA) for STAT-3
Nuclear protein extracts (5 μg) were incubated at room temperature in an equal volume of reaction buffer (10 mM HEPES/KOH pH 7.8, 50 mM KCl, 1 mM EDTA, 5 mM MgCl2, 5 mM DTT and 10% glycerol) and 1 μL of poly (dI-dC) for 15 minutes. For super shift (SS) assays, 1 μL of anti-mouse monoclonal p-STAT-3 antibody (Santa Cruz) was added to the incubation mixture for one hour. One μL of a radiolabeled (100,000 cpm) oligonucleotide that consisted of the sequence for the STAT-3 DNA binding site was added to each reaction mixture and incubated at room temperature for 15 minutes. For cold competition (CC) 3μl of unlabelled oligonucleotide was added at the time of incubation. Samples were loaded on a 5% non-denaturing TBE-polyacrylamide gel for electrophoresis (300 V, 2 hours, 4°C). The gels were dried and autoradiography was performed (exposure time 6 hrs). Abundance was quantified using densitometry.
Determination of IL-6 levels in Vena Caval Blood
Determinations of IL-6 serum concentrations were performed using a BD™ Cytometric Bead Array (CBA) Mouse Inflammation Kit (BD Biosciences, San Jose CA) via flow cytometry. IL-6 concentrations were determined from a standard curve. The lower limit for IL-6 detection was 20 pg/ml
Statistics
Statistical significance (P < 0.05) was determined using ANOVA for repeated measures with the Bonferroni test for post hoc significance.
Results
Outcome Following 2CLP is Different from that following Sham Operation or CLP
As noted, our study involved an intention-to-treat design. Therefore we performed Sham Operation and CLP on three animals at each of seven time points. In this study there was no mortality following either sham operation or CLP. However, because previous investigations indicated that 2CLP was often fatal after 16 hrs, we performed 2CLP on a larger number of animals. These data reveal no mortality at 3, 6, or 16 hrs following 2CLP. However, mortality following 2CLP was 50% (3/6) at 24 hrs, 66.7% (8/12) at 48 hrs and 86.67% (26/30) at 72 hrs. These data allow us to correlate IL-6 signaling data with outcome.
CLP and 2CLP Induce Different Changes in STAT-3 DNA Binding Activity
Previous investigations in rats had demonstrated a failure to activate STAT-3, the primary IL-6-dependent transcription factor, following 2CLP (3). Therefore, we examined the effects of CLP and 2CLP on the DNA binding activity of STAT-3 in our mouse model. Data in mice are detailed in Fig. 1. No STAT-3 DNA binding activity was detected at baseline or following Sham Operation. As noted previously (3), STAT-3 DNA binding peaked around 3 hours after CLP and decreased afterwards but never reached baseline levels. In 2CLP, we found a less pronounced rise in STAT-3 DNA binding than that observed following CLP at 3, 6 and 16 hrs increases initially were similar to those observed following CLP. However, STAT-3 DNA binding activity became virtually undetectable between 16 and 24 hours after 2CLP. This indicates an alteration in some phase of the activation of the IL-6 signal transduction pathway.
Figure 1. Electrophoretic Mobility Shift Assay (EMSA) of STAT-3 DNA Binding Activity following Sham Operation (SO), Single Puncture CLP (CLP) and Double Puncture CLP (2CLP).

Top: Autoradiogram of representative EMSA detailing STAT-3 DNA binding activity following SO, CLP or 2CLP. Subscript indicates time (hrs) following intervention. CC - cold competition, SS - supershift. Bottom: quantification of densitometrically-determined relative STAT-3 DNA Binding Activity of STAT-3 following SO, CLP or 2CLP. Each data point represents mean ± SD of values from 3 different animals. Time following intervention on x-axis. * - value significantly different from To. ^ - value significantly different from value for SO at same time point. # - value for 2CLP significantly different from value for CLP at same time point.
CLP and 2CLP Induce Different Changes in the Nnuclear Abundance of Phosphorylated STAT-3
Binding of STAT-3 to DNA and initiation of IL-6-dependent transcription requires that STAT-3 be phosphorylated and form homodimers that translocate into the nucleus. One possible contribution to failed IL-6 signal transduction is impaired nuclear translocation of phosphorylated (p)-STAT-3. This would result in low intra-nuclear p-STAT-3 abundance. We examined this following Sham Operation, CLP and 2CLP. Results are detailed in Fig. 2. Intranuclear p-STAT-3 abundance was unchanged following Sham Operation. CLP was associated with increased abundance of STAT-3 in the nuclear fraction at 3, 6, 16, 24 and 48 hours, peaking at the six hour timepoint. Levels at 72 hrs were statistically indistinguishable from those observed at T0. Abundance increased at three and six hours following 2CLP but returned to basal levels by sixteen hours. This is consistent with, and might explain, the observed loss of DNA binding activity.
Figure 2. Nuclear p-STAT-3 Abundance Following Sham Operation (Sham), Single Puncture CLP (CLP) and Double Puncture CLP (2CLP).
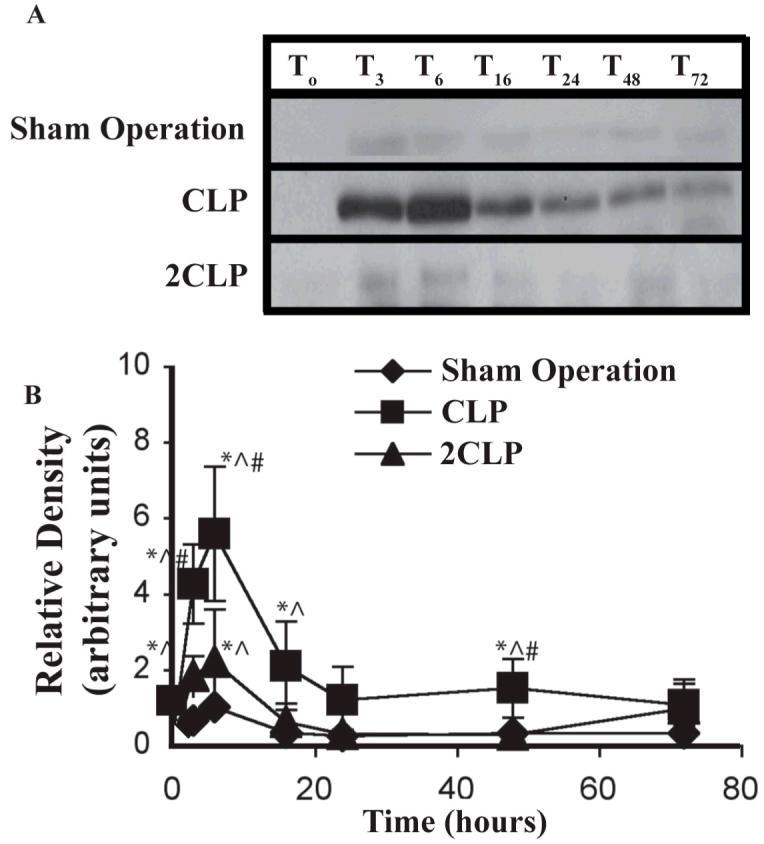
A: autoradiogram of representative immunoblot detailing abundance of phosphorylated (p)-STAT-3 in the nuclear fraction following Sham, CLP and 2CLP. B: quantification of densitometrically-determined relative abundance of nuclear p-STAT-3 following intervention. Each data point represents mean ± SD of values from 3 different animals. Mean value at To set equal to unity; values at other time points normalized to this value. Time following intervention is shown on x-axis. * - value significantly different from To. ^ - value significantly different from value for Sham at same time point. # - value for 2CLP significantly different from value for CLP at same time point.
2CLP is not associated with p-STAT-3 Retention in the Cytoplasm
One possible explanation of the low abundance of p-STAT-3 in the nucleus following 2CLP is retention of p-STAT-3 in the cytoplasm. This could reflect a failure of p-STAT-3 to migrate or an active expulsion of this protein from the nucleus back into the cytoplasm. Therefore we examined cytoplasmic p-STAT-3 abundance following Sham Operation, CLP and 2CLP. No p-STAT-3 was detectable under any condition (data not shown). This reflects the limited activation observed after Sham Operation and the high migration into the nucleus that likely accompanied CLP. However, the low levels observed both in the cytoplasm and the nucleus following 2CLP can be explained only by an absence of p-STAT-3. This in turn must reflect either failed phosphorylation or active degradation.
2CLP is Associated with Increased STAT-3 Abundance in the Cytoplasm
One possible explanation for low p-STAT-3 in both the nucleus and the cytoplasm is a depletion of STAT-3. This could result either from failed synthesis or active degradation. To investigate this we isolated cytoplasmic fractions and measured STAT-3 abundance following Sham Operation, CLP and 2CLP. These data are detailed in Fig. 3. STAT-3 abundance was decreased significantly at 3, 6, and 16 hours following Sham Operation. Cytoplasmic abundance of STAT-3 was similarly decreased following CLP. These latter findings paralleled increases in the intra-nuclear compartment. Abundance began to return to baseline 24 hrs after CLP but never fully achieved basal levels. Following 2CLP we observed an early decrease in cytoplasmic STAT-3 abundance similar to that observed in SO and CLP. However, by 24 hrs after 2CLP, abundance had returned to baseline and became elevated significantly at 48 and 72 hours. As with Sham Operation and CLP, these findings are reciprocal to intra-nuclear levels and STAT-3 DNA binding activity. Of importance, observed immunoblot bands had a molecular weight of ∼92 kDa, indicating that they reflect STAT-3 monomers. Therefore, failed intra-nuclear translocation of STAT-3 did not reflect absent STAT-3 but likely was due to failed STAT-3 nuclear transmigration. Further, the intracytoplasmic retention of STAT-3 monomer coupled with the absence of p-STAT-3 noted above indicates a failure of dimerization that is consistent with failed phosphorylation.
Figure 3. Cytoplasmic STAT-3 Abundance Following Sham Operation (Sham), Single Puncture CLP (CLP) and Double Puncture CLP (2CLP).
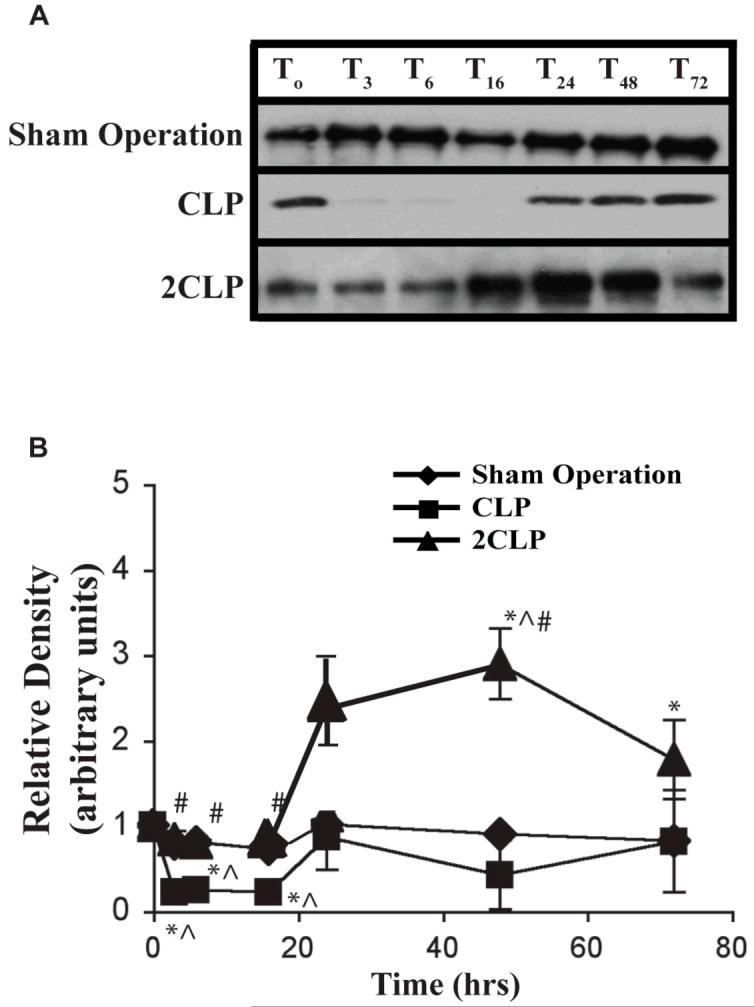
A: autoradiogram of representative immunoblot detailing relative abundance of cytoplasmic STAT-3 following Sham, CLP and 2CLP. B: quantification of densitometrically-determined relative abundance of cytoplasmic STAT-3 following intervention. Each data point represents ± SD of values from 3 different animals. Mean value at To set equal to unity; values at other time points normalized to this value. Time following intervention is shown on x-axis. * - value significantly different from To. ^ - value significantly different from value for Sham at same time point. # - value for 2CLP significantly different from value for CLP at same time point.
Sepsis Does Not Alter the Abundance of the Key IL-6-Linked Signal Transduction Proteins gp130 or JAK-1
The signal transduction complex that links extracellular IL-6 with the cytoplasmic protein STAT-3 consists of two proteins, the transmembrane protein gp130 and the associated protein JAK-1. These are covalently linked. Decreased abundance of either could explain failed STAT-3 phosphorylation and thus the low levels of IL-6 signal transduction following 2CLP. Therefore, we used immunoblotting to examine the abundance of gp130 and JAK-1 at different time points following Sham Operation, CLP and 2CLP. These data demonstrated that the abundance of both gp130 and JAK-1 was unchanged by Sham Operation, CLP, or 2CLP (data not shown).
Sepsis Does Not Alter Phosphorylation of JAK-1
gp130 contains both transmembrane and intra-cytoplasmic components. The intra-cytoplasmic portion closest to the cell membrane is closely associated with JAK-1. Upon association with IL-6, gp130 dimerizes and undergoes a conformational change that catalyzes the phosphorylation of JAK-1. This in turn enables JAK-1 kinase activity, an action that results in phosphorylation and activation of gp130. Therefore, we used immunoprecipitation and immunoblotting to examine phosphorylation of JAK-1 following Sham Operation, CLP and 2CLP. While phospho-JAK-1 was detected in all three inteventions and at all time points, no change in the intra-cytoplasmic abundance of p-JAK-1 was observed following sham operation, CLP or 2CLP (Fig. 4B). Therefore, failed STAT-3 phosphorylation cannot be explained by impairment of JAK-1 phosphorylation.
Figure 4. Abundance of p-gp130 and p-JAK-1 Following Sham Operation (SO), Single Puncture CLP (CLP) and Double Puncture CLP (2CLP).

A: (top) autoradiogram of representative immunoblots for p-gp130 following SO, CLP and 2CLP. (bottom) quantification of densitometrically-determined relative abundance of cytoplasmic p-gp130 following SO, CLP and 2CLP. Each data point represents ± SD of values from 3 different animals. Mean value at To set equal to unity; values at other time points normalized to this value. Time following intervention is shown on x-axis. * - value significantly different from To. ^ - value significantly different from value for SO at same time point. # - value for 2CLP significantly different from value for CLP at same time point. B: (top) autoradiogram of representative immunoblots of p-JAK-1 following sham operation, single puncture CLP and double puncture CLP. (bottom) quantification of densitometrically-determined relative abundance of cytoplasmic p-JAK-1 following SO, CLP and 2CLP.
Sepsis Alters Phosphorylation of gp130
IL-6 mediated dimerization and activation of the gp130-JAK-1 complex results in JAK-1-mediated phosphorylation of gp130 at several specific tyrosine residues. These provide the phosphorylation site as well as the phosphate group that is transferred to STAT-3. While the phosphorylation of JAK-1 is not altered, kinase activity might be impaired following 2CLP. This would prevent tyrosine phosphorylation of gp130 and thus of STAT-3. Therefore, we immunoprecipitated gp130 and determined the abundance of the phosphorylated form of the protein following Sham Operation, CLP and 2CLP (Fig. 4A). Sham Operation induced a minimal change in the phosphorylation state of gp130. CLP dramatically increased p-gp130 abundance. This was apparent almost immediately following the induction of sepsis, peaked at six hours and retuned towards, but never reached, baseline. In contrast, we observed a peak increase in p-gp130 six hours after 2CLP. However, levels of p-gp130 were virtually undetectable at all later times post-2CLP. This is consistent with the time course of decreased STAT-3 activation and IL-6-dependent gene expression. Thus, failed gp130 phosphorylation represents a logical explanation of 2CLP-associated impairment of IL-6 activity.
Sepsis does not cause disassociation of gp130 and JAK-1
gp130 and Jak-1 must be associated to induce proper phosphorylation. Thus, disassociation of the gp130-Jak-1 complex would explain the improper phosphorylation of gp130. To test this, we used an antibody directed at gp130 to immunoprecipitate and performed immunoblotting with an antibody to JAK-1. For completeness, we also performed the reverse - immunoprecipitating with an antibody to JAK-1 and immunoblotting with an antibody to gp130. These investigations showed that the association between gp130 and JAK-1 was unaffected by either CLP or 2CLP (Fig 5). Therefore, dissociation of gp130 and JAK-1 and a resultant failure of the kinase activity of either cannot explain the loss of gp130 phosphorylation following 2CLP.
Fig. 5. Association of JAK-1 and gp130 following Sham Operation, CLP and 2CLP.
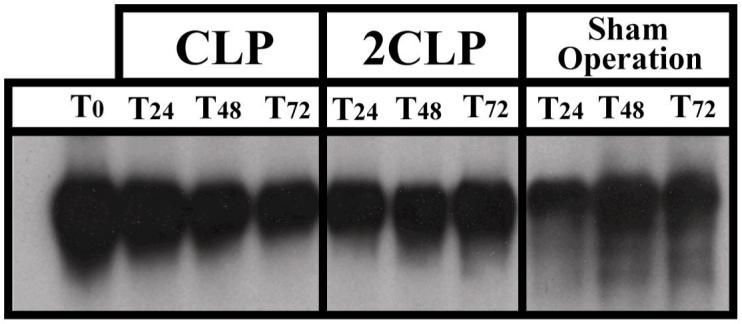
Autoradiograms of representative immunoblots following Sham Operatopn, CLP and 2CLP. Cytosolic protein extract immunoprecipitated with an antibody to either gp130 or JAK-1, and subjected to 10% SDS-PAGE. After transfer blot probed with antibody to whichever protein had not been immunoprecipitated. The above autoradiogram reflects immunoprecipitation with an antibody to JAK-1 and detection with an antibody to gp130. Results were the same when we immunoprecipitated with an antibody to gp130 and probed with an antibody to JAK-1. Data from 24, 48 and 72hrs post procedure as these were the time points at which IL-6 signaling was impaired.
CLP/ 2CLP Increase IL-6 Serum Levels
Finally, the sepsis-induced alteration in IL-6 signal transduction might result from a decrease in the extracellular concentration of IL-6. To investigate this possibility, we examined serum IL-6 levels. Data are detailed in Fig 6. IL-6 levels did not change following Sham Operation. They were significantly (50-fold increase over Sham Operation) elevated six and sixteen hrs after CLP. Increased serum levels also were noted at six and sixteen following 2CLP. However, these elevations were substantially more pronounced than those observed following CLP. Indeed, in agreement with the findings of others, IL-6 levels increased nearly 1400-fold (2) and remain somewhat elevated throughout the time course studied. These data indicate that the absence of extracellular IL-6 cannot explain the decreased signal transduction following 2CLP.
Fig 6. Serum IL-6 Levels following Sham Operation, CLP and 2CLP.

Serum IL-6 levels (pg/mL) determined using flow cytometry with BD™ Cytometric Bead Array (CBA) Mouse Inflammation Kit (BD Biosciences, San Jose CA). Time following intervention on x-axis. * - value significantly different from To. ^ - value significantly different from value for SO at same time point. # - value for 2CLP significantly different from value for CLP at same time point.
Discussion
The data presented here demonstrate impaired IL-6 signal transduction in a well-validated, well-accepted and reproducible model of intra-peritoneal sepsis in mice. Specifically, we found a late decrease in STAT-3 DNA binding activity and of phosphorylated STAT-3 in the nucleus following 2CLP, a model of fulminant sepsis. This occurred in a setting where serum IL-6 levels were markedly elevated, a finding that indicates attenuation of the intra-cellular IL-6 signal transduction pathway. Further, the study revealed nuclear STAT3 levels that were qualitatively reciprocal to those in the cytoplasm. Our data. suggest that altered gp130 phosphorylation is of primary importance in the observed alteration in IL-6 activity.
The late loss of IL-6 signaling in a setting of elevated serum levels is a finding we have observed previously following 2CLP in rats (4). Therefore we conducted a systematic, step-by-step investigation of the key elements involved in the intracellular IL-6 pathway—STAT-3, JAK-1, and gp130. Abundance of these elements was not altered. This indicates that the observed transcriptional defect in proteins of hepatic origin that we have noted previously is not an underlying cause (8,11,12). Altered gp130 phosphorylation represents the most upstream abnormality we observed and therefore we presume it is of primary importance. Certainly, impaired gp130 phosphorylation is consistent with our finding of attenuated nuclear p-STAT-3 levels and STAT-3 binding activity. The latter, in turn, is known to parallel mortality in sepsis (4). Sepsis-associated impairment of phosphorylation has been noted in other pathways (13, 14, 15) including our own unpublished work on hepatic NF-κB. Of potential importance, Ling et. al. found that endotoxemia inhibited JAK-1 phosphorylation. They also found impaired phosphorylation of gp130 but attributed this to loss of JAK-1 kinase activity (15). However, LPS induces toxic changes in the liver that differ significantly from those induced by sepsis. Therefore, our observation of an alteration in gp130 phosphorylation represents a unique, previously unreported change during sepsis or any other disease. This indicates that the approaches designed to maintain or transfer phosphate groups may have therapeutic potential.
There are several possible explanations for altered gp130 phosphorylation. Indeed, the observed defect may represent an imbalance in the normal regulatory/counter-regulatory activity of kinases and phosphatases. One simple explanation is an energy defect that limits phosphate transfer. We (16) and others (17, 18) have demonstrated impaired mitochondrial function in sepsis. This would result in a reduced energy supply for phosphorylation. Similarly, Ince et. al. have invoked a defect in the microcirculation that would limit ATP generation via attenuated oxygen delivery (19). Sepsis-induced attenuation of gp130 phosphorylation also may be due to failed JAK-1 kinase activity. Activity assays of this process involve complex experimental techniques that require significant refinement in current approaches to isolation of liver tissues. In addition, we did not directly examine gp130 kinase activity. This requires isolation of the enzymatic complex, a process that so far has not been accomplished in vivo. Alternatively, phosphate groups might be removed at an accelerated rate. Liver is rich in phosphatases. For example, the (SH2)-containing tyrosine phosphatase SHP-2 interacts specifically with JAK-1 and gp130 tyrosine residues (20, 21). In addition, suppressors of cytokine signaling (SOCS)-3 binds to gp130 (as well as JAK-1) with high affinity (21) and specifically effect tyrosine phosphorylation (22, 23). This interaction occurs without inhibition of JAK-1 activity (23). Another SOCS family member, SOCS-1, reduces tyrosine-phosphorylation of both gp130 and STAT-3 (25).
Importantly, gp130 is a ligand for a number of other extracellular mediators. These include, among others, leukemia inhibitory factor (LIF), IL-11, oncostatin and neuropoetin. However, there is little in the literature regarding the importance of these in sepsis-induced hepatic dysfunction. Conversely, IL-6 is universally regarded as a key mediator of septic effects. None-the-less, our data might reflect an impaired response to one of these other cytokines along with IL-6.
Failed IL-6 signal transduction may represent yet another manifestation of well-described phenomenon of sepsis-induced immunosuppression. Hotchkiss et. al. have shown that sepsis causes early apoptosis of B and CD4+ T lymphocytes that correlates with mortality (26, 27). The peak increase in percentage of apoptotic lymphocytes occurs at 24 hours. This parallels the observed decrease in STAT-3 DNA binding activity reported here and elsewhere (28). Indeed, the two findings may be related as STAT-3 positively regulates transcription of the anti-apoptotic Bcl-xL proteins (29).
Another finding of key importance is the lack of a hepatocellular response to elevated serum IL-6 levels during sepsis. Our findings of elevated IL-6 levels parallel those reported by Remick and Coopersmith (3, 6). Indeed, Remick has shown a clear correlation between IL-6 elevations and mortality in this model of sepsis. However, up until now the biology underlying the elevations in serum IL-6 levels has been obscure. Our data support the hypothesis that a failed intracellular response to increased serum IL-6 levels stimulates ongoing and enhanced IL-6 production. This hypothesis has important diagnostic and therapeutic ramifications. If correct, this hypothesis would indicate that changes in serum levels must be interpreted with caution. Further, it would strongly argue against attempts aimed at exogenous reduction in serum IL-6 levels. Indeed, there has been extensive discussion regarding the balance of the pro-/anti-inflammation in sepsis. If extracellular cytokines do not provoke an intracellular response this debate is moot. In support of these postulates, anti-cytokine therapeutic interventions for TNF-α, IL-1, and IL-6 have failed to significantly alleviate the septic response or improve survival (30).
In summary, our data indicate a sepsis-induced defect in intracellular IL-6 signal transduction that appears to originate with impaired gp130 tyrosine phosphorylation. This has important mechanistic, diagnostic and therapeutic implications. Current studies into the exact cause of this attenuation and potential approaches to its reversal are underway.
Acknowledgments
Supported in part By NIGMS Grant 5-R01-GM059930-08 (CSD), NIGMS Grant 5T32GM007612-30 (KMA, CSD) and the Stavropoulos Sepsis Research Program (AA, CSD).
Financial Support: Supported in part by NIGMS Grant 5R01GM059930-08 (CSD), NIGMS Grant 5T32GM007612-30 (KMA, CSD) and the Stavropoulos Sepsis Research Program (AA, CSD).
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clemont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Deitch EA. Multiple organ failure: pathophysiology and potential future therapy. Ann Surg. 1991;216:117–134. doi: 10.1097/00000658-199208000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock. 2002;17(6):463–467. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Andrejko KM, Chen J, Deutschman CS. Intrahepatic STAT3 activation and acute phase gene expression predict outcome after CLP sepsis in the rat. Am J Physiol. 1998;275:G1423–G1429. doi: 10.1152/ajpgi.1998.275.6.G1423. [DOI] [PubMed] [Google Scholar]
- 5.Deutschman CS, Cereda M, Ochroch A, Raj NR. Sepsis-induced cholestasis, steatosis, hepatocellular injury, and impaired hepatocellular regeneration are enhanced in interleukin-6 -/- mice. Crit Care Med. 2006;34(10):2613–2620. doi: 10.1097/01.CCM.0000240229.98275.07. [DOI] [PubMed] [Google Scholar]
- 6.Vyas D, Javadi P, DiPasco PJ, Buchman TG, Hotchkiss RS, Coopersmith CM. Early antibiotic administration but not antibody therapy directed against IL-6 improves survival in septic mice predicted to die on basis of high IL-6 levels. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1048–R1053. doi: 10.1152/ajpregu.00312.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinrich PC, Behrman I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin-6 type cytokine signaling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deutschman CS, Andrejko KM, Haber BA, Harrison R, Elenko E, Taub R. Sepsis-induced expression of rat glucose-6-phosphatase gene expression and activity. Am J Physiol. 1997;42:R1709–R1718. doi: 10.1152/ajpregu.1997.273.5.R1709. [DOI] [PubMed] [Google Scholar]
- 9.Kim PK, Andrejko KM, Chen J, Raj N, Deutschman CS. Intraabdominal sepsis downregulates transcription of sodium taurocholate cotransporter and multidrug resistance-associated protein in rats. Shock. 2000;14:176–181. doi: 10.1097/00024382-200014020-00017. [DOI] [PubMed] [Google Scholar]
- 10.Haaxma CA, Kim PK, Andrejko KM, Raj NR, Deutschman CS. Trancription factor C/EBP-α and HNF-1α are associated with decreased expression of liver-specific genes in sepsis. Shock. 2003;19(1):45–49. doi: 10.1097/00024382-200301000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Deutchman CS, DeMaio A, Buchman TG, Clemens MG. Sepsis-induced alterations in phosphoenolpyruvate carboxylkinase expression: the role of insulin and glucagons. Circulatory Shock. 1993;40:295–302. [PubMed] [Google Scholar]
- 12.Andrejko KM, Deutschman CS. Altered hepatic gene expression in fecal peritonitis: changes in trancription of gluconeogenic β-oxidative, and ureagenic genes. Shock. 1997;7:164–167. [PubMed] [Google Scholar]
- 13.Kilpatrick LE, Sun S, Mackie D, Basik F, Li H, Korchak HM. Regulation of TNF mediated antiapoptotic signaling in human neutrophils: role of δ-PKC and ERK1/2. J Leukoc Biol. 2006;80:1512–1521. doi: 10.1189/jlb.0406284. [DOI] [PubMed] [Google Scholar]
- 14.Deaciuc IV, Spitzer JA. Protein phosphorylation in isolated hepatocytes of septic and endotoxemic rats. Am J Physiol Regul Integr Comp Physiol. 1989;257:R1232–R1240. doi: 10.1152/ajpregu.1989.257.5.R1232. [DOI] [PubMed] [Google Scholar]
- 15.Ling P, Smith RJ, Mueller C, Mao Y, Bistrian Inhibition of interleukin-6-activated janus kinases/ signal transducers and activators of transcription but not mitogen-activated protein kinase signaling in liver endotoxin-treated rats. Crit Care Med. 2002;30(1):202–211. doi: 10.1097/00003246-200201000-00029. [DOI] [PubMed] [Google Scholar]
- 16.Levy RJ, Vijayasarathy C, Raj NR, Avadhani NG, Deutschman CS. Competitive and noncompetitive inhibition of myocardial cytochrome C oxidase in sepsis. Shock. 2004;21(2):110–114. doi: 10.1097/01.shk.0000108400.56565.ab. [DOI] [PubMed] [Google Scholar]
- 17.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies, Cooper, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- 18.Crouser ED. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion. 2004;4:729–741. doi: 10.1016/j.mito.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 19.Ince C, Sinaasappel M. Microcirculatory oxygenation and shunting in sepsis and shock. Crit Care Med. 1999;27(7):1369–1377. doi: 10.1097/00003246-199907000-00031. [DOI] [PubMed] [Google Scholar]
- 20.Schaper F, Gendo C, Eck M, Schmitz J, Grimm C, Anhuf D, Kerr IM, Heinrich PC. Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6-signal tranducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem J. 1998;335:557–565. doi: 10.1042/bj3350557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Souza D, Fabri LJ, Nash A, Hilton DJ, Nicola NA, Baca M. SH2 domains from suppressor of cytokine signaling-3 and protein tyrosine phosphatase SHP-2 have similar binding specificities. Biochemistry. 2002;41:9229–9236. doi: 10.1021/bi0259507. [DOI] [PubMed] [Google Scholar]
- 22.Schmitz J, Weissenbach M, Haan S, Heinrich PC, Schaper F. SOCS3 exerts its inhibitory function on interleukin-6-signal transduction through the SHP2 recruitment site of gp130. J Biol Chem. 2000;275:12848–12856. doi: 10.1074/jbc.275.17.12848. [DOI] [PubMed] [Google Scholar]
- 23.Nicholson SE, De Souza D, Fabri LJ, Corbin J, Willson TA, Zhang JG, Silva A, Asimakis M, Farley A, Nash AD, Metcalf D, Hilton DJ, Nicola NA, Baca M. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2 binding site on the shared cytokine receptor subunit gp130. Biochemistry. 2000;97(12):6493–6498. doi: 10.1073/pnas.100135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, Alexander WS, Metcalf D, Hilton DJ, Nicola NA. Mutational analsyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. 1999;18:375–385. doi: 10.1093/emboj/18.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Tetsuya T, Yoshizaki K, Akira S, Kishimoto T. Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997;387:924–929. doi: 10.1038/43219. [DOI] [PubMed] [Google Scholar]
- 26.Hotchkiss RS, Tinsley KW, Swanson PE, Shmieg RE, Jr., Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP, Buchman TG, Karl IE. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. 2001;166:6952–6963. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 27.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci USA. 1999;96(25):14541–14546. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Starkel P, De Sauger C, Leclercq I, Strain A, Horsmans Y. Deficient STAT3 DNA-binding is associated with high Pias3 expression and a positive anti-apoptotic balance in human end-stage alcoholic and hepatitis C cirrhosis. J Hepatol. 2005;43:687–695. doi: 10.1016/j.jhep.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 29.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, Dalton WS, Jove R. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 30.Remick DG. Pathophysiology of sepsis. Am J Pathol. 2007;170:1435–1444. doi: 10.2353/ajpath.2007.060872. [DOI] [PMC free article] [PubMed] [Google Scholar]