

Abstract
This study examined very brief focal ischemia that simulates transient ischemic attacks (TIAs) that occur in humans. Adult rats were subjected to sham operations or 5- minutes, 10- minutes, or 2-hours of middle cerebral artery (MCA) ischemia using the suture (thread) model. Hsp70 protein was induced 24h, 48h and 72h later in neurons throughout the entire MCA territory in many but not all animals. Following 5- and 10-minute MCA occlusions, 9 of 32 animals (28%) had microinfarcts mostly in dorsal lateral striatum. Uncommon Hsp70 stained intracellular cytoplasmic inclusions, some of which co-localized with activated caspase-3, were detected in microglia, macrophages, astrocytes and oligodendrocytes. Hsp70 stained neurons were TUNEL negative at 24h and 48h whereas some Hsp70 stained neurons were TUNEL positive at 72h after reperfusion. Hsp70 positive, activated “bushy” microglia and Hsp70 negative, activated “polarized” or rod-shaped microglia were located outside of the microinfarcts. Thus, experimental focal ischemia simulating TIAs can: induce Hsp70 protein throughout the ischemic vessel territory; produce Hsp70 protein positive glial inclusions; activate Hsp70 positive and negative microglia; and cause microinfarcts in some animals.
Keywords: brain, ischemia, heat shock protein, microglia, infarction, transient ischemic attacks (TIAs)
1. Introduction
The classic definition of a transient ischemic attack (TIA) is a temporary focal neurological deficit caused by brief interruption of local cerebral blood flow (Johnston, 2002). The symptoms of a TIA in the carotid territory typically last 5 to 10 minutes in humans and by definition last less than 24 hours (Pessin et al., 1977). As many as a third of TIA patients eventually go on to have ischemic strokes (Rothwell et al., 2006). Recently, magnetic resonance diffusion-weighted imaging (DWI) has been used to assess cerebral ischemia. DWI measures the Brownian motion of water protons in tissue. When sodium-potassium pumps fail, water moves from the extracellular space to the more restricted intracellular space. This restricted proton motion causes an ischemic lesion to appear bright on DWI (Davis et al., 2006). Though TIAs have not been confirmed pathologically to be associated with cell death in brain, as many as 25-40% of TIA patients have DWI abnormalities on MRI brain scans (Nagura et al., 2003; Rothwell et al., 2006). Animal models show that these DWI changes occur within minutes of the onset of ischemia (Moseley et al. 1990; Li et al., 2000; Tong et al., 2000). These results suggest that some classical TIA patients may have brain infarction. Though TIA is a common cerebrovascular disorder (Johnston et al., 2003; Rothwell et al., 2006), there are few experimental studies directed only at 5-10 minute durations of focal ischemia.
Ischemic brain damage can be prevented or at least significantly reduced when there is a preceding brief ischemic period that does not exceed the “threshold for tissue damage”--a phenomenon termed “ischemic preconditioning” (Sommer, 2008). There are some reports of using 3-20 minutes of focal ischemia, but almost all of these short durations of ischemia are used for the induction of “preconditioning” or “ischemia-induced tolerance” (Chen et al., 1996; Li et al., 2006; Currie et al., 2000; Puisieux et al., 2004; Shimizu et al., 2001; Dhodda et al., 2004; Naylor et al., 2005). Focal ischemia as short as 3 minutes produced preconditioning (Puisieux et al., 2004), with three 10 minute episodes of preconditioning appearing to produce the best protection against focal infarction (Chen et al., 1996; Li et al., 2006).
One study has shown that 10 minutes of focal ischemia induced Hsp70 mRNA 24 hours later in brain (Soriano et al., 1995). Since ischemia produces a translation block that may or may not affect synthesis of heat shock proteins (Vass et al., 1988; Gonzalez et al., 1989; Kinouchi et al., 1993a), this study specifically examined Hsp70 protein expression following very brief focal ischemia.
Given the clinical importance of TIAs and the fact that few studies have studied TIAs experimentally, we initiated these studies. The suture model was used, since it is well suited for producing 5 or 10 minutes of focal ischemia with reperfusion (Longa et al., 1989). We examined Hsp70 protein expression following the brief ischemia, and searched for evidence of infarction using Nissl, NeuN and GFAP staining and searched for evidence of isolated cell death using TUNEL and cleaved, activated Caspase-3 staining.
2. Results
2.1 Induction of Hsp70 Protein and Microinfarction Following Brief Focal Ischemia
Hsp70 protein was expressed in the brain of animals subjected to 5 minute, 10 minute and 2h of focal, middle cerebral artery (MCA) ischemia (Fig. 1A-O) and not in animals subjected to sham operations (Fig. 1P-R). Five minutes of focal MCA ischemia induced Hsp70 protein in both striatum and cerebral cortex in much of the MCA territory (Fig.1, A-C). Following 10 minutes of focal MCA ischemia, Hsp70 was similarly expressed in the cortex and striatum in much of the MCA territory (Fig. 1, D-L). The maximal induction of Hsp70 following 10 minutes of ischemia was at 24h (Fig. 1, D-F) with less Hsp70 at 48h (Fig. 1, G-I) and 72h (Fig. 1, J-L). Two hours of MCA ischemia produced infarction of striatum and cortex, with most of the Hsp70 protein expressed at the margins of the infarct (Fig. 1, M-O). At 24h following 5 minutes of focal ischemia Hsp70 protein was expressed primarily in neurons in the cortex (Fig. 1B) and striatum (Fig. 1C). Similarly, following 10 minutes of MCA ischemia Hsp70 protein was expressed primarily in neurons in cortex and striatum at 24h (Fig. 1, E, F), 48h (Fig. 1, H, I) and 72h (Fig. 1, K, L). The Hsp70 positive neurons in cortex were distributed throughout all cortical layers (I-VI) following both 5 and 10 minutes of MCA ischemia at 24 to 72h of reperfusion (Fig. 1, A,D,G,J). Following 2 hours of ischemia with a 24h reperfusion, little Hsp70 protein was expressed in the core of large middle cerebral artery infarctions (Fig. 1M). Hsp70 protein was expressed at the margins of the infarction (Fig. 1M) in microglial cells and in neurons (Fig. 1, N, O).
Fig. 1.
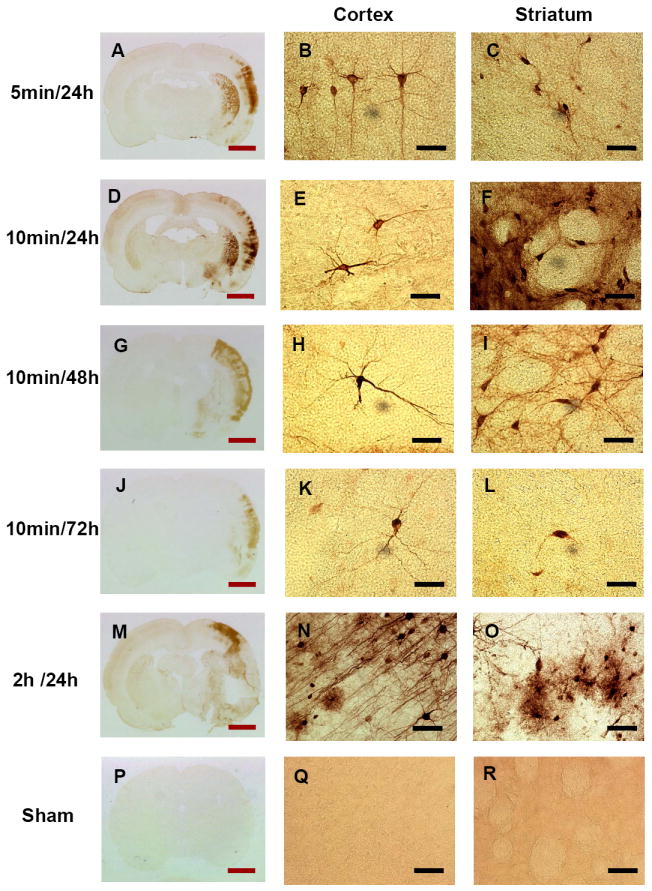
Hsp70 immunostaining following very brief Middle Cerebral Artery Occlusions (MCAO). A, B, C. Five minute MCAO followed by 24h reperfusion. Note Hsp70 immunostaining in cortex and striatum. D, E, F. Ten minute MCAO followed by 24h reperfusion. G, H, I. Ten minute MCAO followed by 48h reperfusion. J, K, L. Ten minute MCAO followed by 72h reperfusion. M, N, O. Two hour MCAO followed by 24h reperfusion. P, Q, R. Sham-operated control. Sham operated control cortex (Q) and control striatum (R) showed no staining. Following MCAO neuronal and glial cells expressed Hsp70 protein in the cortex (B, E, H, K, and N) and striatum (C, F, I, L, and O) following various durations of ischemia (5 minutes, 10 minutes and 2 hours) and reperfusion (24h, 48h and 72h). Red bar=3 mm, black bar=25μm.
Of the 8 animals subjected to 5 minutes of ischemia, 5 (63%) showed Hsp70 staining 24h later. Of the 8 animals subjected to 2h of ischemia, 6 (75%) showed Hsp70 staining 24h later. Following 10 minutes of ischemia 87.5%, 75.0%, and 50.0% of animals (n=8 at each time) expressed Hsp70 protein at 24h, 48h, and 72h later, respectively.
Western blot analysis (Fig. 2A) confirmed that the maximal induction of Hsp70 protein at 24h of reperfusion occurred following 10 minute and 120 minute (2h) MCA occlusions (Fig. 2A,B). Following 10 min MCA ischemia, the expression of Hsp70 protein was maximal at 24h and decreased at 48h and 72h reperfusion (Fig. 2A,C).
Fig. 2.
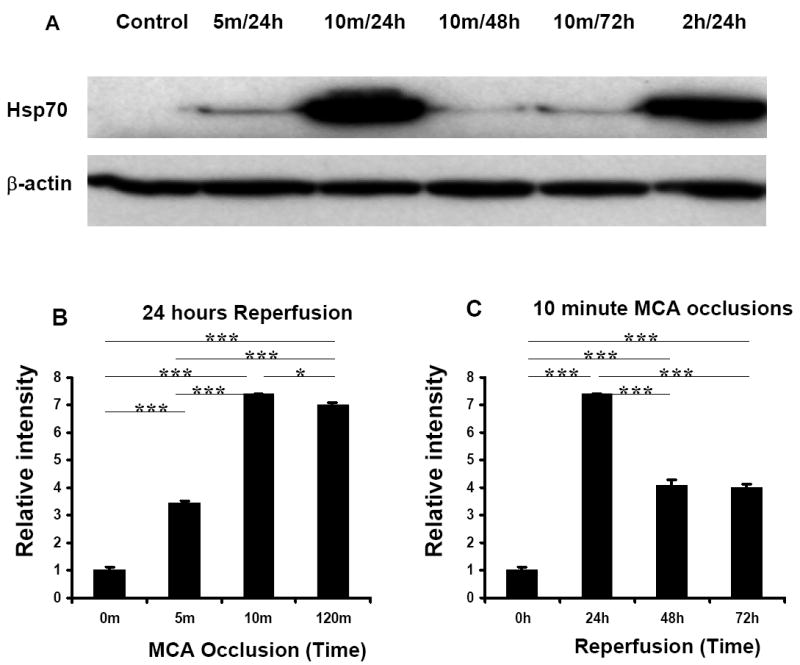
Western Blot Analysis of Hsp70 protein in brains following 5 minute, 10 minute, or 2h MCAO. β-actin served as a loading control. A. Hsp70 protein was induced following 5 minute, 10 minute, or 2h of focal ischemia. B. Relative Intensities (Y axis) of the Hsp70 protein bands following 5 minute, 10 minute, and 120 minute MCAO at 24h after ischemia compared to control. Note that the maximal induction of Hsp70 protein was following the 10 minute-MCAO relative to control. C. Relative Intensities (Y axis) of the Hsp70 protein bands following 10-minute MCAO after 0h, 24h, 48h, or 72h of reperfusion (X axis). Note that the maximal induction of Hsp70 protein was at 24h after 10 minute-MCAO. n=3 in all groups. *p<0.05; *** p<0.001.
Microinfarcts, not visible with the naked eye, were noted in 9 of the 32 rats (28%) subjected to 5 or 10 minutes of ischemia (Figs. 3B,D,F). Following 5 minutes of focal ischemia 2/8 of the animals had microinfarcts at 24h. The areas of microinfarction were limited to the dorsal lateral striatum. Following 10 minutes of focal ischemia 3/8 of the animals had microinfarcts at 24h, 2/8 at 48h, and 2/8 at 72h. These microinfarcts following 10 minutes of focal ischemia mainly involved the dorsal lateral striatum but occasionally involved adjacent cortex. Microinfarcts in ischemic striatum showed no NeuN staining of neuronal nuclei (Fig. 3B) compared to normal NeuN staining of neuronal nuclei in the contralateral non-ischemic striatum (Fig. 3A). Microinfarcts in ischemic striatum showed no GFAP stained astrocytes (Fig. 3D) compared to normal GFAP stained astrocytes in the non-ischemic, contralateral striatum (Fig. 3C). These same regions in dorsal lateral striatum showed disintegration of cells in the microinfarcts with Nissl staining (Fig. 3F) compared to normal Nissl staining in the contralateral non-ischemic striatum (Fig. 3E). Similar findings were observed in the scattered cortical microinfarcts following 10 minutes of focal ischemia (not shown).
Fig. 3.
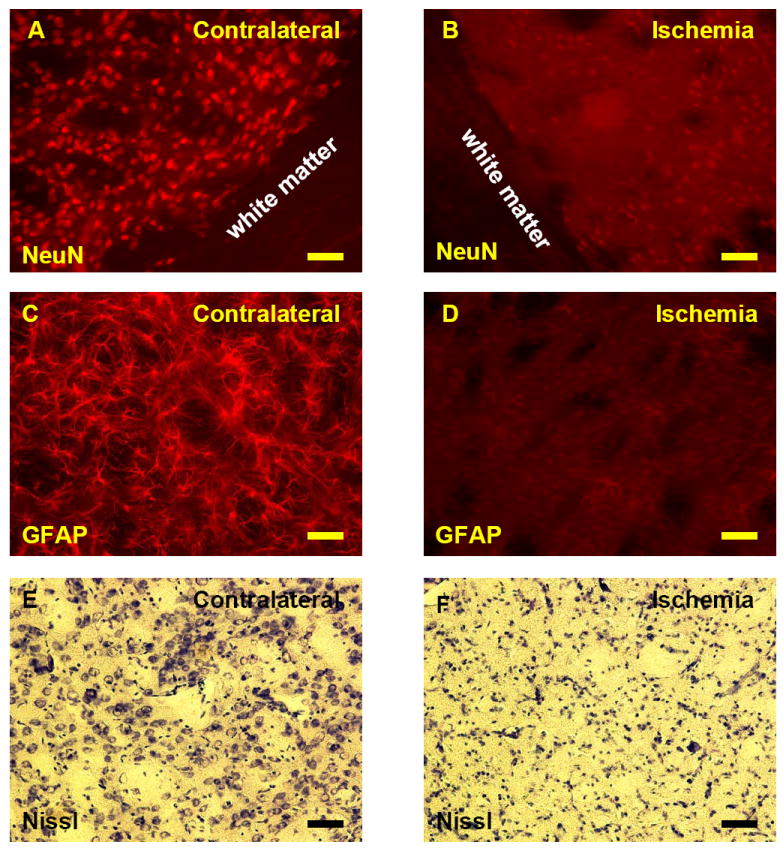
(A-F) NeuN, GFAP and Nissl stained microinfarcts (A-F) in dorsal lateral striatum following 10 minute MCAO and 72h reperfusion. (A) Normal NeuN stained neuronal nuclei of non-ischemic striatum contralateral to the MCAO. (B) NeuN stained neuronal nuclei of ischemic striatum ipsilateral to the MCAO are completely absent. (C) Normal GFAP stained astrocytes in the non-ischemic striatum contralateral to the MCAO. Note stained astrocytes throughout the normal dorsal lateral striatum. (D) Absent GFAP stained astrocytes in the striatum in the microinfarct ipsilateral to the MCAO. (E) Nissl staining of non-ischemic dorsal lateral striatum contralateral to the MCAO showing Nissl stained neurons and glia interspersed with relatively cell free normal white matter tracts. (F) Nissl staining of ischemic dorsal lateral striatum showing a microinfarct with disintegration and atrophic changes of cells ipsilateral to the 10 minute MCAO following 72h of reperfusion.
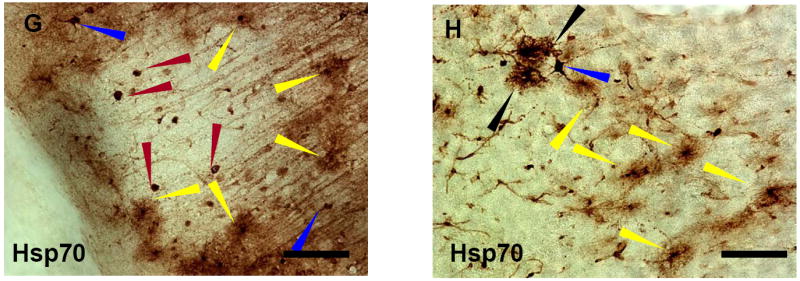
(G-H) Hsp70 stained cells surrounding a microinfarct in cortex following 2h of MCAO and 24h reperfusion. G. Hsp70-positive microglia (yellow arrows) encircled the microinfarct. Hsp70 stained macrophages (red arrows) were located within infarct. Hsp70 stained neurons (blue arrows) were located outside the infarct. H. In regions of Hsp70 stained microglia (yellow arrows) were rare Hsp70 stained cells that were negative for all glial stains used (black arrows). Yellow arrows – microglia; Red arrows- macrophages; Blue arrows – neurons; Black arrows - unidentified cells. Microglia were identified using OX42 double labeling, macrophages were identified using ED1 double labeling, and neurons were identified using NeuN double labeling (not shown). Bar=25μm.
The animals with 2h occlusions had large MCA infarcts involving the striatum and cortex as well as microinfarcts (Fig. 3G) involving the cortex. Microinfarcts were often observed around the large infarcts following 2h MCA occlusion. Macrophages expressed Hsp70 protein in the core of the microinfarct (Fig. 3G, red arrows). Hsp70 stained microglia surrounded the infarct and had a “bushy” appearance (Fig. 3G,H, yellow arrows). Some Hsp70 stained neurons were intermixed with the Hsp70 stained microglia (Figs. 3G, H, blue arrows) at the margins of the infarct. Neurons were identified using NeuN double labeling, microglia were identified using OX42 double labeling, and macrophages were identified using ED1 double labeling (not shown). Hsp70 stained glial-like cells that did not co-localize with any cell markers used in the study were occasionally found at the margins of infarcts either after 5 or 10 minute ischemia or 2h ischemia (Fig. 3H, black arrows).
2.2 Localization of Hsp70 in neurons and glia
Double-label immunocytochemistry confirmed the Hsp70 protein staining of individual cell types. Following 5-minute or 10-minute (Fig. 4, A1, A2, A3) MCA occlusion, Hsp70 protein was localized mainly to cytoplasm of neurons. Following 2h MCA occlusion and 24h reperfusion, Hsp70 protein was localized to cytoplasm and occasionally to nuclei (Fig. 4, B1, B3) of NeuN stained neurons (Fig. 4, B2, B3).
Fig. 4.

Double-label immunostaining of neurons (A3, B3) for Hsp70 (A1, B1) and NeuN (A2, B2). A. A ten minute MCAO followed by 24h reperfusion (A1,A2,A3) shows Hsp70 stained protein (A1, A3) was localized mainly to cytoplasm of NeuN positive (A2, A3) neurons (blue arrows). B. Two hour MCAO followed by 24h reperfusion shows Hsp70 stained protein (B1, B3) was localized to cytoplasm and occasionally to nuclei of NeuN positive (B2, B3) neurons (blue arrows). Bar=25μm.
To confirm the types of glial cells that expressed Hsp70, tissues were stained for Hsp70 protein and OX42, ED1, RIP, or GFAP following 5-minute, 10-minute or 2h MCA occlusions. Hsp70 was co-localized with OX42 (Fig. 5, A2, A3), ED1 (Fig. 5, B2, B3), RIP (Fig. 5C2, C3) and GFAP (Fig. 5, D2, D3). The Hsp70 stained, OX42 double-labeled microglia were localized predominantly at the margins of infarcts following 5 minute, 10 minute or 2h MCA ischemia. The Hsp70 stained, ED1 double-labeled macrophages were localized primarily within the core of infarcts and adjacent to blood vessels, the pia mater and the ependymal cell layer (not shown). Hsp70 stained, double-labeled GFAP stained astrocytes were very sparse and found either near infarcts or at the cortical surface. Hsp70 stained, RIP double-labeled oligodendrocytes were sparse and located near Hsp70 positive neurons and microglia (not shown).
Fig. 5.

Hsp70 co-localizes with glial cells and macrophages following 10-minute MCAO and 48h reperfusion. A. OX42 stained microglia (A2) that co-localized with Hsp70 (A1, A3) were located at the margins of the microinfarcts following brief focal ischemia. B. ED1 positive macrophages (B2) that co-localized with Hsp70 (B1, B3) were most often located in the core of the microinfarcts. C. RIP stained oligodendrocytes (C2) rarely stained for Hsp70 (C1, C3) and were located adjacent to microinfarcts. D. Only occasional GFAP stained astrocytes (D2) were Hsp70 positive (D1,D3) at the margins of the microinfarcts. Hsp70 positive inclusions (A1, B1, C1, D1, arrows) were occasionally observed in OX42 stained microglia (A2, A3), ED1 stained macrophages (B2, B3), RIP stained oligodendrocytes (C2, C3) and GFAP stained astrocytes (D2, D3). Microglia - yellow arrows; Macrophages - red arrows; Astrocytes - orange arrows; Oligodendrocytes - white arrows. Bar=25μm.
Following 10 minutes of ischemia and 48h and 72h of reperfusion, some of the Hsp70 (Fig. 5A1, B1, C1, D1) stained microglia (Fig. 5A2), macrophages (Fig. 5B2), oligodendrocytes (Fig. 5C2) and astrocytes (Fig. 5D2) also had Hsp70 protein stained, cytoplasmic inclusions within the cells (Figs. 5A1-D3, arrows). Hsp70 positive inclusions were not observed following 5 and 10 minutes of ischemia and 24h reperfusion, but were observed following 2h ischemia and 24h reperfusion.
2.3 Bushy and Polarized microglia and Glial Inclusions
Morphological criteria for classifying ramified (resting), hypertrophied and bushy microglia were used in this study (Soltys et al., 2001; Ziaja et al., 1999; Davis et al., 1994). Ramified microglia are characterized by small cell bodies with a large nucleus, minimal perinuclear cytoplasm and highly branched, thin processes. Hypertrophied microglia have larger cell bodies and thicker processes than resting microglial forms. Bushy microglia have numerous but short and poorly ramified processes of different diameters forming thick bundles around their enlarged cell bodies. Macrophages have large, round or oval cell bodies with no processes. Cells were identified by a combination of morphology and immunostaining, including Hsp70 and OX42 (or Iba-1) stained hypertrophied microglia (Fig. 6A), Hsp70 and OX42 (or Iba-1) stained bushy microglia (Fig. 6B) and Hsp70 and ED1 positive macrophages (Fig. 6C).
Fig. 6.
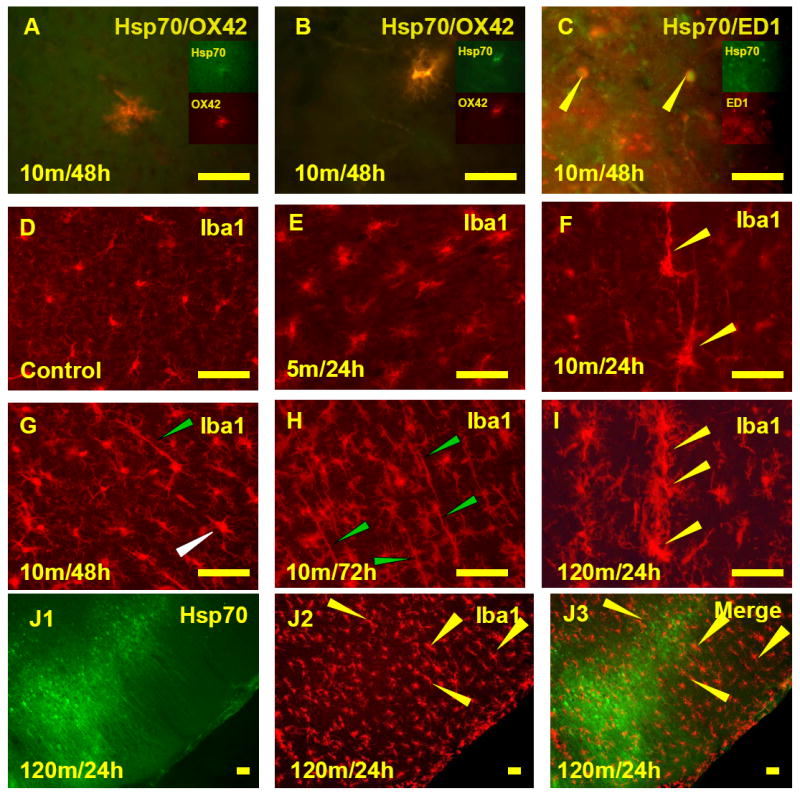
Activated Hsp70 positive microglia and activated Hsp70 negative microglia following MCAO. A. Co-localization of Hsp70 protein and OX42 in a hypertrophied microglia. B. Co-localization of Hsp70 protein and OX42 in a bushy microglia. C. Co-localization of Hsp70 protein and ED1 in the Macrophages. D. Ramified microglia stained with Iba-1 were regularly spaced and not activated in cortex of sham animals. E and F. Iba-1 positive microglia became activated following 5 minute or 10 minute MCAO and 24h reperfusion. Microglia were enlarged slightly following 5 minute MCAO (E) and sometimes much more so following 10 minute MCAO (F). The largest processes for many of the microglia (yellow arrows) were perpendicular to the surface of the cortex (F). G, H, and I. Microglia in the cortex (Iba-1 positive) around an infarct became activated and polarized (oriented perpendicular to the surface of the cortex) following 10 minute MCAO and 48h reperfusion (G), 10 minute MCAO and 72h reperfusion (H) and 120 minute MCAO and 24h reperfusion (I). A number of the microglia sometimes appeared to be aligned (H, green arrows; I, yellow arrows). Bipolar microglia had a shorter process facing the microinfarct and a longer process facing the cortex (G, white arrow). J. Hsp70 stained cells (J1) intermingled with Iba1 stained microglia (J2) following 120 minute MCAO and 24 h reperfusion. These Hsp70 stained cells (J1) were surrounded by enlarged, polarized, and Hsp70 negative microglia (J3, merged figure, yellow arrows). Bar=25μm.
Changes of microglial morphology were apparent in cortex following 5, 10 and 120-minutes of MCA ischemia. In sham animals, cortical microglial staining with Iba-1 showed regularly spaced lightly stained ramified microglia (Fig. 6D). Following 5 min MCA ischemia and 24h reperfusion, modest activation of some microglia occurred in the cortex (Fig. 6E) adjacent to (above) areas of striatal microinfarction. Following 10min MCA ischemia some of the Iba-1 stained microglia in cortex, located adjacent to the striatal infarctions, became hypertrophied at 24h (Fig. 6F). By 48h (Fig. 6G) and 72h of reperfusion (Fig. 6H) following 10 min MCA ischemia the microglia in some areas of cortex adjacent to the striatal infarctions were activated and polarized (rod-like), with the long axis of the cells oriented perpendicular to the cortex (Fig. 6G, H). Some polarized microglia could be classified as monopolar or bipolar cells. Monopolar microglia showed a long thin process directed towards the cortex following 10-minute MCA ischemia and 24h reperfusion (Fig. 6F, arrows). At 48h after ischemia, both monopolar and bipolar microglia were observed. Bipolar microglia were elongated and had a longer process facing toward the cortex and a shorter process facing toward the microinfarct (Fig. 6G, white arrow). Occasionally, the long and thin processes of two adjacent microglial cells appeared to be aligned (Fig. 6G, green arrow). At 72h after ischemia, the aligned processes were more obvious (Fig. 6H, green arrows) and occasionally three or more microglial cells appeared to be aligned particularly following 2h of ischemia (Fig. 6I).
Following 120 min MCA ischemia there were areas of cortical infarction 24h later. Around some of these cortical infarctions were Hsp70 stained microglia that had a “bushy” appearance (Fig. 3). Adjacent to these Hsp70 stained microglia (Fig. 6J1) were some Hsp70 negative microglia that had a “polarized” or rod-like appearance (Fig. 6J2, J3).
2.4 TUNEL and Cleaved, Activated Caspase-3 Stained Neurons
TUNEL stained cells were detected following 5 minutes, 10 minutes, and 2h of ischemia. Most of the Hsp70 positive cells following 10 minutes of ischemia at 24h (Fig. 7A1, arrows) and 48h (Fig. 7B1, arrows) were TUNEL negative (Fig. 7A2, B2) when co-labeling was performed (Fig. 7A3, B3). However, at 72h some Hsp70 positive cells (Fig. 7C1, arrows) were stained with TUNEL (Fig. 7C2) when co-labeling was performed (Fig. 7C3). Apoptosis was confirmed by double staining Hsp70 protein positive cells with an apoptosis marker, cleaved Caspase-3. Some Hsp70 positive cells (Fig. 7D1) were stained with cleaved Caspase-3 (Fig. 7D2) when double labeling was performed (Fig. 7D3) following 10 minutes of ischemia and 72h reperfusion (Fig. 7D) but not following 24 and 48h of reperfusion (not shown). Notably, some Hsp70 stained glial inclusions (Fig. 7E1, arrow) stained for cleaved, activated Caspase-3 (Fig. 7E2, E3) even though the Hsp70 stained cell body was devoid of activated caspase –3 (Fig. 7E3). The TUNEL and activated caspase-3 stained cells were adjacent to areas of infarction and within areas of Hsp70 immunostaining.
Fig. 7.
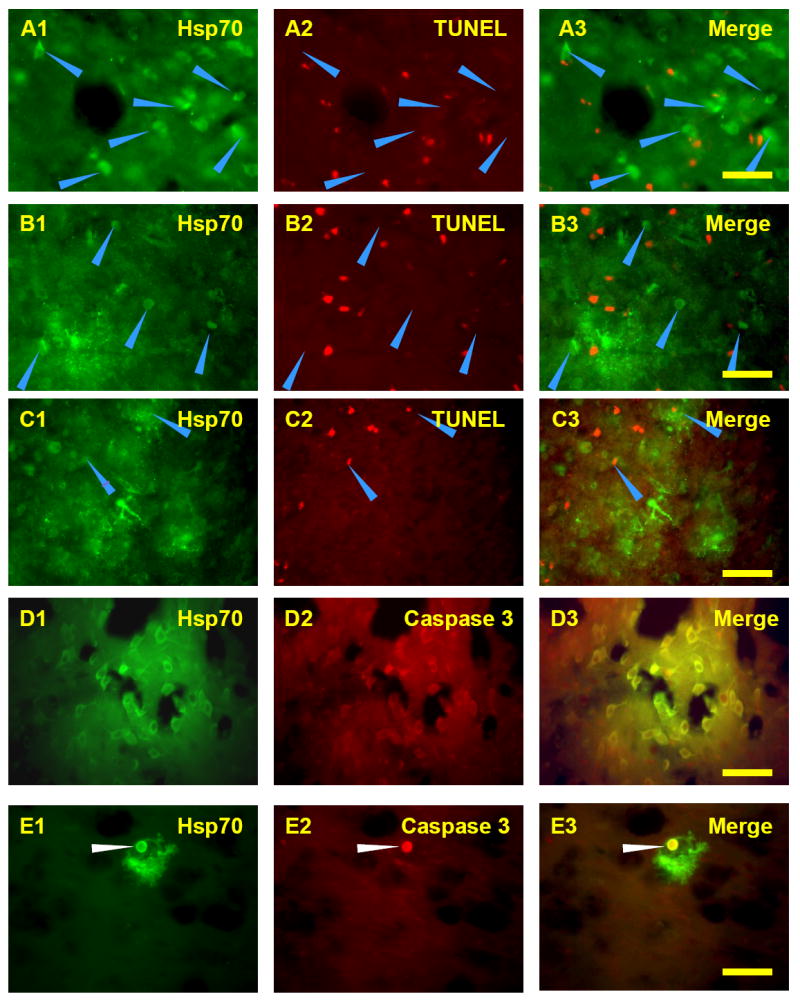
Double staining for Hsp70 and TUNEL or cleaved Caspase-3 following 10 minute MCAO and 24h reperfusion (A), 48h reperfusion (B), or 72h reperfusion (C, D, E). Note that most of the Hsp70 stained cells (A1, B1, arrows) at 24h and 48h were TUNEL negative (A2, A3, B2, B3, arrows). In contrast, some of the Hsp70 stained cells (C1, arrows) at 72h were TUNEL positive (C2, C3, arrows). Some Hsp70 positive cells (D1) and inclusions (E1, arrow) were colocalized with cleaved Caspase-3 (D2, D3, E2, E3) following 72h reperfusion. Bar=25μm.
3. DISCUSSION
This study demonstrates several significant changes in brain following 5 and 10 minutes of focal ischemia. Hsp70 protein is induced in neurons throughout the ischemic MCA territory in many but not all animals following 5 and 10 minutes of focal ischemia that simulates TIAs that occur in humans. Nine of thirty two animals with brief ischemia had microinfarcts detected by loss of GFAP stained astrocytes, loss of NeuN neurons and Nissl staining. Other novel findings in this study included infrequent Hsp70 protein positive inclusions in all glial subtypes; and, activated Hsp70 stained microglia and activated Hsp70 negative microglia.
3.1 A Molecular Penumbra Defined by Hsp70 Protein Induction
Our results are consistent with previous studies showing that 10 minutes of focal ischemia induced hsp70 mRNA throughout the MCA distribution (Soriano et al., 1995). The results also support suggestions that the region of Hsp70 protein induction following focal ischemia represents a molecular penumbra located outside of the infarction (Kinouchi et al., 1993a,b; Sharp et al., 1999, 2000). The possibility that Hsp70 protein mapped the salvageable penumbra has been supported by biochemical data that showed that infarction occurred in regions where blood flow decreases were associated with decreased protein synthesis and decreased ATP, whereas the penumbra was defined by decreased blood flow and decreased protein synthesis but preserved ATP (Hata et al., 2000). Hsp70 protein induction was found to be co-incident with the regions of decreased blood flow and decreased protein synthesis but retained ATP levels; hence, expression of Hsp70 mRNA combined with Hsp70 protein expression also defined the same penumbra (Hata et al., 2000; Kokubo et al., 2003).
Thus, based upon these previous studies we propose that the induction of Hsp70 protein in neurons throughout the MCA distribution following 5 and 10 minutes of focal ischemia represents the denatured protein penumbra (Hata et al., 2000; Sharp et al., 2000). The Hsp70 protein induction mainly in neurons in the MCA territory may occur because these are the cells that are most vulnerable to ischemia (Kinouchi et al., 1993a,b). The maximal induction of Hsp70 protein at 24h following ischemia is consistent with our previous studies (Gonzalez et al., 1989; Kinouchi et al., 1993a,b). The maximal induction of Hsp70 protein at 10 minutes of ischemia compared to 120 minutes of ischemia likely relates to the large hemispheric infarction following the 120 minutes of ischemia with little or no Hsp70 protein synthesis in the core of the infarction.
3.2 Hsp70 Protein, TUNEL, and cleaved Caspase-3 Positive Cells
Hsp70 protein protects against cerebral ischemia (Rajdev et al., 2000; Yenari et al., 2005; Giffard et al., 2004). Though this protection may occur in part by refolding proteins (Tsuchiya et al., 2003), there are indications that Hsp70 also protects brain cells by decreasing apoptosis (Sun et al., 2006) potentially by binding AIF (apoptosis inducible factor) and procaspase-9/APAF (Ran et al., 2004a; Matsumori et al., 2005), by decreasing necrosis (Giffard et al., 2004), by decreasing protein aggregation (Giffard et al., 2004), and by decreasing inflammation (Yenari et al., 2005; Ran et al., 2004b; Zheng et al., 2008). Whether Hsp70 protected cells in the present study was not tested though Hsp70 protein positive neurons at 24h and 48h were not TUNEL or activated caspase-3 positive. This is consistent with the idea that Hsp70 protein protected the cells in which it was expressed. However, a number of Hsp70 protein positive neurons at 72h reperfusion were TUNEL and activated caspase-3 stained. It is possible that the declining levels of Hsp70 protein at 72h reperfusion were not sufficient to protect these neurons at these times.
3.3 Microinfarctions Following Brief Focal Ischemia
Our data are the first to show that durations of experimental focal ischemia equivalent to the durations of symptoms observed in classical TIAs were associated with cell death in brain. The data show that 5 or 10 minutes of brief focal ischemia followed by reperfusion caused microinfarctions in brain of 28% of the animals. These small infarcts could be caused by embolization produced at the time the suture occluded the MCA. These regions could also represent areas of poor collaterals and watershed ischemia and infarction.
Previous clinical studies show that as many as a third of classical TIA patients eventually go on to have ischemic strokes (Rothwell et al., 2006). A number of these classical TIA patients may already have had microinfarcts before they have a clinically diagnosed stroke since as many as 25-40% of TIA patients have DWI abnormalities on MRI brain scans (Nagura et al., 2003; Rothwell et al., 2006). TIAs are under recognized, under reported and under treated (Albers et al., 2002). With the recognition of DWI positive TIA patients, a new TIA definition has been proposed: “a TIA is a brief episode of neurologic dysfunction caused by focal brain or retinal ischemia, with clinical symptoms typically lasting less than one hour, and without evidence of acute infarction” (Albers et al., 2002). Thus, in animals and as likely in humans, it is probably important to distinguish subjects with brief focal ischemia with and without any infarction.
It is notable that the 5 and 10 minutes of focal ischemia used here produce microinfarctions in some animals, but do not produce any detectable cell death in most animals. This contrasts to 5 and 10 minute periods of global ischemia that do not produce infarction but instead produce very reliable selective neuronal cell death of the CA1 pyramidal and dentate hilar neurons of hippocampus (Vass et al., 1988; Ito et al., 1975; Kirino, 1982; Pulsinelli et al., 1982; Vass et al., 1988; Gaspary et al., 1995; Harukuni and Bhardwaj, 2006; Tanaka et al., 2004). Though large increases of lactate are thought to contribute to infarction following focal ischemia, further study is needed.
3.4 Hsp70 Stained Intracellular Inclusions in Glia
An unexpected, previously unreported finding in brain ischemia was the presence of Hsp70 stained inclusions in some scattered glia around microinfarcts, including microglia, astrocytes and oligodendrocytes. We postulate these inclusions represent denatured protein aggregates associated with accumulation of Hsp70 protein in intracellular organelles like lysosomes or vacuoles. Though 5 and 10 minute occlusions did not produce these inclusions with 24h of reperfusion, longer durations of reperfusion and the longer duration 2 hour MCA occlusions were associated with these inclusions. This suggests that the formation of the Hsp70 stained glial inclusions may depend on both the severity and duration of cerebral ischemia.
Some of the Hsp70 stained inclusions also stained for activated, cleaved caspase-3. To explain this, we postulate that the encapsulated denatured proteins induce Hsp70 protein and other chaperones that then target the denatured proteins either for refolding or for proteolysis. If the proteins are targeted for proteolysis, this could occur via activated caspase-3, the proteasome and other pathways. The source of the denatured proteins in the glial inclusions could be denatured proteins from within the cells where the inclusions form, or could be by phagocytosis of extracellular denatured proteins from other injured cells. Notably, Hsp70 positive inclusions were observed in glia and not neurons, perhaps suggesting different mechanisms of sequestration of denatured proteins in glia versus neurons.
Similar inclusions, often associated with heat shock proteins, have been described in various degenerative neurological diseases including synucleinopathies (Parkinsons) and tauopathies, some motor neuron diseases, and in some triplet repeat diseases (Cummings et al., 2001; Dou et al., 2003; Shorter et al., 2004; Giffard et al., 2004; Chai et al., 1999; Satyal et al., 2000; Thomas et al., 2006; Koyama et al., 2006; Klucken et al., 2004). These inclusions have been interpreted to be denatured protein aggregates that induce heat shock proteins, and likely represent a response of brain cells to segregate or compartmentalize cytotoxic denatured proteins (Hinault et al., 2006; Sherman and Goldberg, 2001).
3.5 Two kinds of activated microglia - Hsp70 positive and negative
Another novel finding of this study relates to the two kinds of activated microglia following focal ischemia: activated Hsp70 positive microglia and activated Hsp70 negative microglia.
Based on developmental and pathophysiological studies, several types of brain microglia have been distinguished on morphological grounds. Ramified microglia, considered to be the typical resting microglia, are characterized by small, round or oval cell bodies, a large nucleus, a small volume of perinuclear cytoplasm, and highly branched, thin processes (Soltys et al., 2001). Activated microglia may demonstrate several morphologies including hypertrophic (Ziaja et al., 1999), bushy (Ziaja et al., 1999) or rod cell-shaped or polarized (Wierzba-Bobrowicz, et al., 2002; Marín-Teva et al., 1998; Soltys et al., 2001; Ziaja et al., 1999; Gehrmann et al., 1995a,b; Davis et al., 1994; Kreutzberg, 1996). In the present study, all these groups of activated microglia were noted.
Hsp70 stained microglia associated with microinfarcts and at the margins of large infarctions had a “bushy” appearance and corresponded to a number of descriptions of “activated” microglia following ischemia and other types of injury (Streit, 2000; Zheng and Yenari, 2004; Nakamura, 2002). Activated Hsp70 stained “bushy” microglia at the margins of infarcts are well described (Soriano et al., 1994). In this study we also describe activated Hsp70 negative microglia, usually somewhat further from the infarction, with a “polarized” or rod-like morphology.
The functions of the activated Hsp70 positive and activated Hsp70 negative microglia are uncertain. Activated microglia have been suggested to provide a “molecular” index of the “penumbra” (Lehrmann et al., 1997; Gehrmann et al., 1995a,b). Polarization of microglia has been postulated to be a sign of migration (Marín-Teva et al., 1998). The short thick processes are often at the leading edge whereas the long thin processed are often at the trailing edge of the cell (Marín-Teva et al., 1998). Therefore, the activated, Hsp70 negative polarized microglia may represent activated microglia that migrated in response to the infarctions. Activated Hsp70 stained “bushy” microglia might have phagocytosed dying or dead cells leading to the presence of denatured proteins within the microglia that induce the Hsp70 protein. The Hsp70 stained microglia could have also sustained ischemic injury which denatured proteins within the cell and induced Hsp70 protein. These Hsp70 stained microglia might undergo apoptosis themselves (Gehrmann et al., 1995b). Alternatively, the presence of Hsp70 protein in some activated microglia might suppress inflammation by inhibiting the activation of the inflammatory transcription factor, nuclear factor-κB (NF-κB) (Ran et al., 2004b; Zheng, et al., 2008).
We have previously described dividing microglia in regions of ischemic brain where there was no detectable cell injury or cell death (Liu et al., 2001). It would be interesting to determine if BrdU labeled dividing microglia are associated with activated Hsp70 positive microglia and or the activated Hsp70 negative microglia or both since selective ablation of dividing microglia worsens focal ischemic injury in brain (Lalancette-Hebert et al., 2007).
3.6 Technical Considerations
The variable Hsp70 induction following 5-10 minute focal ischemia could relate to different degrees of ischemia from subject to subject that could be better controlled in the future by monitoring cerebral blood flow. It could also relate to somewhat different susceptibility to ischemic injury from animal to animal. In addition, the suture itself and various differences in the tips of each suture might contribute to the variability of the microinfarcts. Though the markers for the individual cell types are probably fairly specific there are exceptions. For example, the microglial markers used here (OX42/CD11 and Iba-1) can also stain neutrophils in the core of an infarction (Matsumoto et al., 2007). However the stained neutrophils are small and are morphologically quite different from the process bearing microglial cells stained in this study. ED1 likely stains macrophages and microglia and is not specific for either one.
4. Experimental Procedures
4.1 Animals
Sixty-six male Sprague-Dawley rats weighing 280-350 g (Charles River Labs, USA) were used in this study. Eighteen were used for Western blot analysis, and forty-eight were used for all other analyses. The University of California Animal Care Committee at Davis approved the animal protocol in accordance with NIH guidelines.
4.2 Focal ischemia
Brief focal cerebral ischemia was produced by occluding the middle cerebral artery (MCA) using the intraluminal suture technique (Longa et al., 1989; States et al., 1996). Briefly, rats were anesthetized with 3% isoflurane and maintained on 1.5% isoflurane. The right common carotid artery was exposed via a ventral midline incision. To occlude the MCA, a 3-0 monofilament nylon suture with the tip rounded by heat was inserted into the external carotid artery (ECA) and advanced into the internal carotid artery approximately 20-23 mm beyond the carotid bifurcation until mild resistance was felt. Rectal temperature was maintained at 36.5°C to 37.5°C with a heating blanket throughout the procedure.
Rats subjected to 5 minutes of focal ischemia were allowed to survive 24h (n=11). Rats subjected to 10 minutes of focal ischemia were allowed to survive 24h (n=11), 48h (n=11) and 72h (n=11). Rats with 2h MCA occlusions were allowed to survive for 24h (n=11). Sham-operated rats were subjected to the identical surgical protocol except that no suture was inserted into ECA and allowed to survive for 24h (n=11). Three animals from each group were used for Western blot analysis and eight animals from each group were used for all other analyses.
4.3 Tissue preparation
After a 24h, 48h or 72h period of reperfusion, forty eight rats were anesthetized with isoflurane and perfused transcardially with 0.9% saline, followed by 4% paraformaldehyde in 0.01 M phosphate buffered saline (PBS, pH 7.4). The brains were rapidly removed and immersed in a fixative solution containing 4% paraformaldehyde in PBS. After 6 hours of post fixation, the brains were placed in a 30% sucrose solution until they sank. Coronal sections 50μm thick were cut in a cryostat (-20°C) and collected at 600 μm intervals through the entire MCA distribution.
4.4 Immunohistochemistry
Brain sections were treated with 3% H2O2 in PBS for 20 minutes to quench endogenous peroxidase activity. After blocking nonspecific sites with a buffer containing 2% horse serum, 0.2% Triton X-100, and 0.1% bovine serum albumin (BSA) in PBS, the sections were incubated with a mouse monoclonal antibody against Hsp70 (1:200 dilution, BioVision, USA) overnight at 4°C. Secondary antibody was a biotinylated horse anti-mouse IgG (1:200 dilution, Vector Labs, USA). The antibody complex was detected using ABC reagent and a substrate solution of H2O2 and diaminobenzidine (DAB) according to the manufacturer’s instructions (Vector Labs). Primary antibody was omitted to assess nonspecific staining.
4.5 Immunofluorescence
To perform double label staining, sections were incubated with a first set of primary and secondary antibodies followed by a second set of primary and secondary antibodies. The first sets of primary antibodies were directed against NeuN (neuronal nuclei), GFAP (astrocytes), ED1 (macrophage), OX42 (microglia), Iba-1 (microglia), RIP (oligodendrocyte), or active Caspase 3 (apoptosis). NeuN, GFAP, RIP (Chemicon, USA), ED1, OX42 (Serotec, UK), Iba-1 (Wako, Japan), and activated, cleaved Caspase 3 (Abcam, USA) were diluted at 1:1000, 1:1000, 1:20,000, 1:100, 1:200, 1:1000, and 1:100 respectively. The second primary antibody was a rabbit anti-Hsp70 (1:2000 dilution, Stressgen, Canada) or a mouse anti Hsp70 (1:2000 dilution, BioVision, USA) depending on the species of the first set of primary antibodies. Goat anti-mouse or goat anti-rabbit Alexa Fluor® 488 or 594 conjugated antibodies (Invitrogen, USA) were used for secondary antibodies depending on the species of the primary antibody.
Hsp70 antibody was incubated with sections at 4°C overnight. All other antibodies were incubated at room temperature (RT) for 2h. Slides were cover slipped with a medium containing 4,6-diamidino-2-phenylindole (DAPI, Vector Labs). Microscopy was conducted at excitation/emmision wavelengths of 493/520 (for green fluorochrome Alexa 488) or 590/619 (for red fluorochrome Alexa 594) nm or 358/463 nm (for blue fluorochrome DAPI).
4.6 TUNEL
A modification of the TUNEL technique described by Chen et al. (Chen et al., 1997) was used. Brain sections were treated with 1% Triton X-100 for 30 minutes and pre-incubated in equilibration buffer containing 0.1 M potassium cacodylate, 2 mM CoCl2, and 0.2 mM DTT (Invitrogen) for 10 minutes at room temperature. They were then incubated in the TUNEL reaction mixture containing 30 μM biotin-aha-dUTP (Invitrogen), 300 U/ml recombinant terminal deoxynucleotidyl transferase (rTdT, Invitrogen), 0.1 M potassium cacodylate, 2 mM CoCl2, and 0.2 mM DTT for 2 hours at 37°C. The biotinylated dUTP 3’-OH DNA end-label was detected using the biotin-streptavidin-fluorescence (1:200 dilution, Invitrogen) method. Sections immunostained for Hsp70 were double stained with TUNEL. Non-specific labeling was assessed by omitting rTdT.
4.7 Infarction
Brain sections were stained with cresyl violet. Any large or small unstained, contiguous area was considered an infarction. Infarction was confirmed microscopically by atrophic changes in both neurons and glial cells or absence of both neurons and glial cells. Infarction was also confirmed by the absence of NeuN stained neuronal nuclei and absence of GFAP stained astrocytes in the same region of abnormal Nissl staining. Loss of GFAP staining was the most sensitive marker for microinfarcts and showed a larger extent of cell damage than the other two markers.
4.8 Western Blots Analysis
Eighteen animals with various durations of ischemia (sham, 5min, 10 min, and 2h) and various durations of reperfusion (0h, 24h, 48h, and 72h) were euthanized. The cortex and basal ganglia in the entire ischemic hemisphere were dissected and frozen at - 70°C. Frozen tissues were homogenized in ice-cold immunoprecipitation buffer containing a complete protease inhibitor mixture (Sigma). The homogenates were centrifuged at 14,000g for 30 min at 4°C, and the pellet was discarded. The protein in the supernatant was loaded (50 μg each) onto lanes and separated on 12.5% SDS-polyacrylamide gels and transferred to nitrocellulose. The membranes were probed overnight at 4°C with anti-Hsp70 (1:4000 dilution, Stressgen) or anti-β-Actin (1:10,000 dilution, Santa Cruz) antibodies. Primary antibody was detected using horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Bio-Rad). The signal was detected using the ECL chemiluminescent detection system (PIERS Inc). Blots were imaged on the Fluorchem 8900 system (Alpha Innotech), and the intensity of the bands was quantified with NIH Image J software. β-Actin was used as a loading control. Band intensity was expressed relative to the intensity of the band in the control samples.
4.9 Statistics
Data are given as Mean ± standard error of the mean. One-way ANOVA was performed with a Student-Neuman-Keuls post hoc test. A p value of less than 0.05 was considered statistically significant.
Acknowledgments
We thank Dr. Bradley Ander, Dr. Nikita Dirugen, Dr. Zena Vexler, Dr. Jialing Liu and Dr. Midori Yenari for providing training and assistance for these studies.
Sources of Support: This work was supported by NIH/NINDS grants to FRS (NS028167, NS043252 and NS054652).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albers GW, Caplan LR, Easton JD, Fayad PB, Mohr JP, Saver JL, Sherman DG. Transient Ischemic Attack -- Proposal for a New Definition. N Engl J Med. 2002;347:1713–1716. doi: 10.1056/NEJMsb020987. [DOI] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Bonini NM, Paulson HL. Analysis of the role of heat shock protein Hsp molecular chaperones in polyglutamine disease. J Neurosci. 1999;19:10338–10347. doi: 10.1523/JNEUROSCI.19-23-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Graham SH, Zhu RL, Simon RP. Stress proteins and tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:566–577. doi: 10.1097/00004647-199607000-00006. [DOI] [PubMed] [Google Scholar]
- Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP. Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem. 1997;69:232–245. doi: 10.1046/j.1471-4159.1997.69010232.x. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- Currie RW, Ellison JA, White RF, Feuerstein GZ, Wang X, Barone FC. Benign focal ischemic preconditioning induces neuronal Hsp70 and prolonged astrogliosis with expression of Hsp27. Brain Res. 2000;863:169–181. doi: 10.1016/s0006-8993(00)02133-8. [DOI] [PubMed] [Google Scholar]
- Davis EJ, Foster TD, Thomas WE. Cellular forms and functions of brain microglia. Brain Res Bull. 1994;34:73–87. doi: 10.1016/0361-9230(94)90189-9. [DOI] [PubMed] [Google Scholar]
- Davis DP, Robertson T, Imbesi SG. Diffusion-weighted magnetic resonance imaging versus computed tomography in the diagnosis of acute ischemic stroke. J Emerg Med. 2006;31:269–77. doi: 10.1016/j.jemermed.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Dhodda VK, Sailor KA, Bowen KK, Vemuganti R. Putative endogenous mediators of preconditioning-induced ischemic tolerance in rat brain identified by genomic and proteomic analysis. J Neurochem. 2004;89:73–89. doi: 10.1111/j.1471-4159.2004.02316.x. [DOI] [PubMed] [Google Scholar]
- Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspary H, Graham SH, Sagar SM, Sharp FR. HSP70 heat shock protein induction following global ischemia in the rat. Brain Res Mol Brain Res. 1995;34:327–332. doi: 10.1016/0169-328x(95)00195-x. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Rev. 1995a;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Banati RB. Microglial turnover in the injured CNS: activated microglia undergo delayed DNA fragmentation following peripheral nerve injury. J Neuropathol Exp Neurol. 1995b;54:680–688. doi: 10.1097/00005072-199509000-00010. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Xu L, Zhao H, Carrico W, Ouyang Y, Qiao Y, Sapolsky R, Steinberg G, Hu B, Yenari MA. Chaperones, protein aggregation, and brain protection from hypoxic/ischemic injury. J Exp Biol. 2004;207:3213–3220. doi: 10.1242/jeb.01034. [DOI] [PubMed] [Google Scholar]
- Gonzalez MF, Shiraishi K, Hisanaga K, Sagar SM, Mandabach M, Sharp FR. Heat shock proteins as markers of neural injury. Brain Research. 1989;6:93–100. doi: 10.1016/0169-328x(89)90033-8. [DOI] [PubMed] [Google Scholar]
- Harukuni I, Bhardwaj A. Mechanisms of brain injury after global cerebral ischemia. Neurol Clin. 2006;24:1–21. doi: 10.1016/j.ncl.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Hata R, Maeda K, Hermann D, Mies G, Hossmann KA. Evolution of brain infarction after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2000;20:937–946. doi: 10.1097/00004647-200006000-00006. [DOI] [PubMed] [Google Scholar]
- Hinault MP, Ben-Zivi A, Goloubinoff P. Chaperones and proteases: cellular fold-controlling factors of proteins in neurodegenerative diseases and aging. J Mol Neurosci. 2006;30(3):249–265. doi: 10.1385/JMN:30:3:249. [DOI] [PubMed] [Google Scholar]
- Ito U, Spatz M, Walker JT, Jr, Klatzo I. Experimental cerebral ischemia in mongolian gerbils. I. Light microscopic observations. Acta Neuropathol Berl. 1975;32:209–223. doi: 10.1007/BF00696570. [DOI] [PubMed] [Google Scholar]
- Johnston SC. Clinical practice. Transient ischemic attack. N Engl J Med. 2002;347:1687–1692. doi: 10.1056/NEJMcp020891. [DOI] [PubMed] [Google Scholar]
- Johnston SC, Fayad PB, Gorelick PB, Hanley DF, Shwayder P, van Husen D, Weiskopf T. Prevalence and knowledge of transient ischemic attack among US adults. Neurology. 2003;60:1429–1434. doi: 10.1212/01.wnl.0000063309.41867.0f. [DOI] [PubMed] [Google Scholar]
- Kinouchi H, Sharp FR, Hill MP, Koistinaho J, Sagar SM, Chan PH. Induction of 70-kDa heat shock protein and hsp70 mRNA following transient focal cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1993a;13:105–115. doi: 10.1038/jcbfm.1993.13. [DOI] [PubMed] [Google Scholar]
- Kinouchi H, Sharp FR, Koistinaho J, Hicks K, Kamii H, Chan PH. Induction of heat shock hsp70 mRNA and HSP70 kDa protein in neurons in the ‘penumbra’ following focal cerebral ischemia in the rat. Brain Res. 1993b;619:334–338. doi: 10.1016/0006-8993(93)91630-b. [DOI] [PubMed] [Google Scholar]
- Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biol Chem. 2004;279:25497–25502. doi: 10.1074/jbc.M400255200. [DOI] [PubMed] [Google Scholar]
- Kokubo Y, Liu J, Rajdev S, Kayama T, Sharp FR, Weinstein PR. Differential cerebral protein synthesis and heat shock protein 70 expression in the core and penumbra of rat brain after transient focal ischemia. Neurosurgery. 2003;53:186–190. doi: 10.1227/01.neu.0000069023.01440.d6. [DOI] [PubMed] [Google Scholar]
- Koyama S, Arawaka S, Chang-Hong R, Wada M, Kawanami T, Kurita K, Kato M, Nagai M, Aoki M, Itoyama Y, Sobue G, Chan PH, Kato T. Alteration of familial ALS-linked mutant SOD1 solubility with disease progression: its modulation by the proteasome and Hsp70. Biochem Biophys Res Commun. 2006;343:719–730. doi: 10.1016/j.bbrc.2006.02.170. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrmann E, Christensen T, Zimmer J, Diemer NH, Finsen B. Microglial and macrophage reactions mark progressive changes and define the penumbra in the rat neocortex and striatum after transient middle cerebral artery occlusion. J Comp Neurol. 1997;386:461–476. [PubMed] [Google Scholar]
- Li F, Liu KF, Silva MD, Omae T, Sotak CH, Fenstermacher JD, Fisher M, Hsu CY, Lin W. Transient and permanent resolution of ischemic lesions on diffusion-weighted imaging after brief periods of focal ischemia in rats. Stroke. 2000;31:956–964. doi: 10.1161/01.str.31.4.946. [DOI] [PubMed] [Google Scholar]
- Li W, Luo Y, Zhang F, Signore AP, Gobbel GT, Simon RP, Chen J. Ischemic preconditioning in the rat brain enhances the repair of endogenous oxidative DNA damage by activating the base-excision repair pathway. J Cereb Blood Flow Metab. 2006;26:181–198. doi: 10.1038/sj.jcbfm.9600180. [DOI] [PubMed] [Google Scholar]
- Liu J, Bartels M, Lu A, Sharp FR. Microglia/macrophages proliferate in striatum and neocortex but not in hippocampus after brief global ischemia that produces ischemic tolerance in gerbil brain. J Cereb Blood Flow Metab. 2001;21:361–373. doi: 10.1097/00004647-200104000-00005. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Marín-Teva JL, Almendros A, Calvente R, Cuadros MA, Navascués J. Tangential migration of ameboid microglia in the developing quail retina: mechanism of migration and migratory behavior. Glia. 1998;22:31–52. doi: 10.1002/(sici)1098-1136(199801)22:1<31::aid-glia4>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Matsumori Y, Hong SM, Aoyama K, Fan Y, Kayama T, Sheldon RA, Vexler ZS, Ferriero DM, Weinstein PR, Liu J. Hsp70 over expression sequesters AIF and reduces neonatal hypoxic/ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:899–910. doi: 10.1038/sj.jcbfm.9600080. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, Ii C, Takahashi H, Imai Y, Tanaka J. Antibodies to CD11b, CD68, and lectin label neutrophils rather than microglia in traumatic and ischemic brain lesions. J Neurosci Res. 2007;85:994–1009. doi: 10.1002/jnr.21198. [DOI] [PubMed] [Google Scholar]
- Moseley ME, Cohen Y, Mintorovitch J, Chileuitt L, Shimizu H, Kucharczyk J, Wendland MF, Weinstein PR. Early detection of regional cerebral ischemia in cats: comparison of diffusion- and T2-weighted MRI and spectroscopy. Magn Reson Med. 1990;14:330–346. doi: 10.1002/mrm.1910140218. [DOI] [PubMed] [Google Scholar]
- Nagura J, Suzuki K, Johnston SC, Nagata K, Kuriyama N, Ozasa K, Watanabe Y, Nakajima K. Diffusion-weighted MRI in evaluation of transient ischemic attack. J Stroke Cerebrovasc Dis. 2003;12:137–142. doi: 10.1016/S1052-3057(03)00040-5. [DOI] [PubMed] [Google Scholar]
- Nakamura Y. Regulating factors for microglial activation. Biol Pharm Bull. 2002;25:945–953. doi: 10.1248/bpb.25.945. [DOI] [PubMed] [Google Scholar]
- Naylor M, Bowen KK, Sailor KA, Dempsey RJ, Vemuganti R. Preconditioning-induced ischemic tolerance stimulates growth factor expression and neurogenesis in adult rat hippocampus. Neurochem Int. 2005;47:565–572. doi: 10.1016/j.neuint.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Pessin MS, Duncan GW, Mohr JP, Poskanzer DC. Clinical and angiographic features of carotid transient ischemic attacks. N Engl J Med. 1977;296:358–362. doi: 10.1056/NEJM197702172960703. [DOI] [PubMed] [Google Scholar]
- Puisieux F, Deplanque D, Bulckaen H, Maboudou P, Gele P, Lhermitte M, Lebuffe G, Bordet R. Brain ischemic preconditioning is abolished by antioxidant drugs but does not up-regulate superoxide dismutase and glutathione peroxidase. Brain Res. 2004;1027:30–37. doi: 10.1016/j.brainres.2004.08.067. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Rajdev S, Hara K, Kokubo Y, Mestril R, Dillmann W, Weinstein PR, Sharp FR. Mice over expressing rat heat shock protein 70 are protected against cerebral infarction. Ann Neurol. 2000;47:782–791. [PubMed] [Google Scholar]
- Ran R, Zhou G, Lu A, Zhang L, Tang Y, Rigby AC, Sharp FR. Hsp70 mutant proteins modulate additional apoptotic pathways and improve cell survival. Cell Stress Chaperones. 2004a;9:229–242. doi: 10.1379/CSC-19R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran R, Lu A, Zhang L, Tang Y, Zhu H, Feng Y, Han C, Zhou G, Rigby AC, Sharp FR. Hsp70 promotes TNF-mediated apoptosis by binding IKK gamma and impairing NF-kappa B survival signaling. Genes Dev. 2004b;18(12):1466–1481. doi: 10.1101/gad.1188204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PM, Buchan A, Johnston SC. Recent advances in management of transient ischaemic attacks and minor ischaemic strokes. Lancet Neurol. 2006;5:323–331. doi: 10.1016/S1474-4422(06)70408-2. [DOI] [PubMed] [Google Scholar]
- Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2000;97:5750–5755. doi: 10.1073/pnas.100107297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR, Lu A, Tang Y, Millhorn DE. Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:1011–1032. doi: 10.1097/00004647-200007000-00001. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Massa SM, Swanson RA. Heat-shock protein protection. Trends Neurosci. 1999;22:97–99. doi: 10.1016/s0166-2236(98)01392-7. [DOI] [PubMed] [Google Scholar]
- Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29(1):15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Nagayama T, Jin KL, Zhu L, Loeffert JE, Watkins SC, Graham SH, Simon RP. bcl-2 Antisense treatment prevents induction of tolerance to focal ischemia in the rat brain. J Cereb Blood Flow Metab. 2001;21:233–243. doi: 10.1097/00004647-200103000-00007. [DOI] [PubMed] [Google Scholar]
- Shorter J, Lindquist S, Cashikar AG, Schirmer EC, Hattendorf DA, Glover JR, Ramakrishnan MS, Ware DM, Lindquist SL, Queitsch C, Kowal AS, et al. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- Soltys Z, Ziaja M, Pawlinski R, Setkowicz Z, Janeczko K. Morphology of reactive microglia in the injured cerebral cortex. Fractal analysis and complementary quantitative methods. J Neurosci Res. 2001;63:90–97. doi: 10.1002/1097-4547(20010101)63:1<90::AID-JNR11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Sommer C. Ischemic preconditioning: postischemic structural changes in the brain. J Neurophathol Exp Neurol. 2008;67:85–92. doi: 10.1097/nen.0b013e3181630ba6. [DOI] [PubMed] [Google Scholar]
- Soriano MA, Planas AM, Rodriguez-Farre E, Ferrer I. Early 72-kDa heat shock protein induction in microglial cells following focal ischemia in the rat brain. Neurosci Lett. 1994;182:205–207. doi: 10.1016/0304-3940(94)90798-6. [DOI] [PubMed] [Google Scholar]
- Soriano MA, Ferrer I, Rodriguez-Farre E, Planas AM. Expression of c-fos and inducible hsp-70 mRNA following a transient episode of focal ischemia that had non-lethal effects on the rat brain. Brain Res. 1995;670:317–320. doi: 10.1016/0006-8993(94)01352-i. [DOI] [PubMed] [Google Scholar]
- States B, Honkaniemi J, Weinstein P, Sharp FR. DNA fragmentation and HSP70 protein induction in hippocampus and cortex occur in separate neurons following permanent middle cerebral artery occlusion. Journal of Cerebral Blood Flow and Metabolism. 1996;16(6):1165–1175. doi: 10.1097/00004647-199611000-00011. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglial response to brain injury: a brief synopsis. Toxicol Pathol. 2000;28:28–30. doi: 10.1177/019262330002800104. [DOI] [PubMed] [Google Scholar]
- Sun Y, Ouyang YB, Xu L, Chow AM, Anderson R, Hecker JG, Giffard RG. The carboxyl-terminal domain of inducible Hsp70 protects from ischemic injury in vivo and in vitro. J Cereb Blood Flow Metab. 2006;26:937–950. doi: 10.1038/sj.jcbfm.9600246. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, Bennett MV, Zukin RS. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci. 2004;24:2750–2759. doi: 10.1523/JNEUROSCI.5475-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Harrell JM, Morishima Y, Peng HM, Pratt WB, Lieberman AP. Pharmacologic and genetic inhibition of hsp90-dependent trafficking reduces aggregation and promotes degradation of the expanded glutamine androgen receptor without stress protein induction. Hum Mol Genet. 2006;15:1876–1883. doi: 10.1093/hmg/ddl110. [DOI] [PubMed] [Google Scholar]
- Tong DC, Albers GW. Diffusion and perfusion magnetic resonance imaging for the evaluation of acute stroke: potential use in guiding thrombolytic therapy. Curr Opin Neurol. 2000;13:45–50. doi: 10.1097/00019052-200002000-00009. [DOI] [PubMed] [Google Scholar]
- Tsuchiya D, Hong S, Matsumori Y, Kayama T, Swanson RA, Dillman WH, Liu J, Panter SS, Weinstein PR. Over expression of rat heat shock protein 70 reduces neuronal injury after transient focal ischemia, transient global ischemia, or kainic acid-induced seizures. Neurosurgery. 2003;53:1179–1187. doi: 10.1227/01.neu.0000090341.38659.cf. [DOI] [PubMed] [Google Scholar]
- Vass K, Welch WJ, Nowak TS., Jr Localization of 70-kDa stress protein induction in gerbil brain after ischemia. Acta Neuropathology. 1988;77:128–135. doi: 10.1007/BF00687422. [DOI] [PubMed] [Google Scholar]
- Wierzba-Bobrowicz T, Gwiazda E, Kosno-Kruszewska E, Lewandowska E, Lechowicz W, Bertrand E, Szpak GM, Schmidt-Sidor B. Morphological analysis of active microglia--rod and ramified microglia in human brains affected by some neurological diseases (SSPE, Alzheimer’s disease and Wilson’s disease) Folia Neuropathol. 2002;40:125–31. [PubMed] [Google Scholar]
- Yenari MA, Liu J, Zheng Z, Vexler ZS, Lee JE, Giffard RG. Antiapoptotic and anti-inflammatory mechanisms of heat-shock protein protection. Ann N Y Acad Sci. 2005;1053:74–83. doi: 10.1196/annals.1344.007. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Yenari MA. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res. 2004;26:884–892. doi: 10.1179/016164104X2357. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Kim JY, Ma H, Lee JE, Yenari MA. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab. 2008;28:53–63. doi: 10.1038/sj.jcbfm.9600502. [DOI] [PubMed] [Google Scholar]
- Ziaja M, Janeczko K. Spatiotemporal patterns of microglial proliferation in rat brain injured at the postmitotic stage of postnatal development. J Neurosci Res. 1999;58:379–386. [PubMed] [Google Scholar]