

Abstract
Endothelial dysfunction is the earliest pathologic alteration in diabetic vascular injury and plays a critical role in the development of atherosclerosis. Plasma levels of adiponectin (APN), a novel vasculoprotective adipocytokine, are significantly reduced in diabetic patients, but its relationship with endothelial dysfunction remains unclear. The present study aims to determine whether APN deficiency may cause endothelial dysfunction and to investigate the involved mechanisms. Vascular rings were made from the aortic vessels of wild type (WT) or APN knockout (APN-/-) mice. Endothelial function, total NO production, eNOS expression/phosphorylation, superoxide production, and peroxynitrite formation were determined. ACh and acidified NaNO2 (endothelial dependent and independent vasodilators, respectively) caused similar concentration-dependent vasorelaxation in WT vascular rings. APN-/- rings had a normal response to acidified NaNO2, but a markedly reduced response to ACh (>50% reduction vs. WT, P<0.01). Both superoxide and peroxynitrite production were increased in APN-/- vessels (P<0.01 vs. WT). Pretreatment with superoxide scavenger Tiron significantly, but incompletely restored vascular vasodilatory response to ACh. In APN-/- vessels, eNOS expression was normal, but NO production and eNOS phosphorylation was significantly reduced (P<0.01 vs. WT). Treatment of APN-/- mice in vivo with the globular domain of adiponectin reduced aortic superoxide production, increased eNOS phosphorylation, and normalized vasodilatory response to ACh. Increased NO inactivation combined with decreased basal NO production contribute to endothelial dysfunction development when there is a paucity of APN production. Interventions directed towards increasing plasma APN levels may improve endothelial function, and reduce cardiovascular complications suffered by diabetic patients.
Keywords: Endothelial Dysfunction, Diabetes, Adipocytokine
1. Introduction
Cardiovascular disease accounts for an overwhelming proportion of the morbidity and mortality suffered by patients with obesity and type 2 diabetes mellitus[1]. Endothelial dysfunction characterized by a deficiency of nitric oxide (NO) production in response to normal secretion signals, is a characteristic abnormality observed in the diabetic vascular system, and is a critical component of atherosclerosis development[2]. Therefore, clarification of the mechanisms responsible for endothelial dysfunction in diabetic patients, and identification of therapeutic interventions that may improve endothelial function hold great potential in reducing cardiovascular complications and overall death in diabetic patients.
Adiponectin (APN) is a plasma protein secreted specifically from adipose tissue. It presents in multimeric complexes in the circulation of healthy human subjects at relatively high levels[3]. In contrast to the dramatic increase in plasma concentrations of several adipokines observed in visceral adiposity and diabetes, the plasma levels of adiponectin are markedly reduced in diabetic patients[4] as well as in patients with coronary artery disease[5]. Numerous epidemiological studies have shown that reduced adiponectin levels correlate with increased risk of cardiovascular disease in obesity and diabetes with hyperglycemia[6,7]. Moreover, several clinical observations have demonstrated that hypoadiponectinemia is associated with impaired endothelium-dependent vasodilation[8-11]. These clinical observations strongly suggest that reduced adiponectin may contribute to the development of vasculopathic states in diabetic patients. However, there exists no definitive evidence linking reduced adiponectin level with endothelial dysfunction pathogenesis, the critical first step towards cardiovascular complication development in diabetic patients.
Therefore, the aims of the present study were 1) to determine whether adiponectin deficiency may cause endothelial dysfunction in descending aortic vascular segments, the location where atherosclerosis frequently develops, and if so, 2) to dissect the involved mechanisms, and 3) to identify the effective interventions that can normalize endothelial function.
2. Materials and Methods
2.1. Determination of Endothelium-Dependent, Nitric Oxide-Mediated Vasorelaxation
Adult male adiponectin knockout mice (APN-/-) or their wild type littermates (WT) were used in all experiments in this study. The experiments were performed with adherence to NIH Guidelines on the Use of Laboratory Animals and were approved by the Thomas Jefferson University Committee on Animal Care. Mice were anesthetized with 3% isoflurane and the descending aortic segments were isolated. Vascular segments were placed into ice-cold Krebs-Henseleit (K-H) buffer consisting of (mM) NaCl 118, KCl 4.75, CaCl2.2H20 2.54, KH2PO4 1.19, MgSO4.7H20 1.19, NaHCO3 25, and glucose 10.0. Aortic segments were carefully cleaned of fat and loose connective tissue, and cut into 2-3 rings of 2-3 mm length. These rings were then mounted on stainless steel hooks, suspended in 37° C and aerated (95% O2 and 5% CO2) 5 ml K-H tissue baths, and connected to FORT-10 force transducers (WPI, Sarasota, FL) to record changes via a MacLab data acquisition system. The rings were then stretched to an optimum preload of 0.5 g of force (determined in preliminary experiments) and allowed to equilibrate for 60 min. During this period, the K-H buffer in the tissue bath was replaced every 15 min, and the tension of the vascular rings was adjusted until 0.5 g of preload was maintained.
After equilibration, the rings were first exposed to maximally effective concentration (100 nM) of U-46619 (9,11-epoxymethano-PGH2;, Biomol Research Laboratories, Plymouth Meeting, PA) to ensure stabilization of the vascular smooth muscle. The agonist was then washed out and the rings re-equilibrated. Twenty minutes after the initial washing, 50 nM of U-46619 was added to each ring bath to generate approximately 0.5 g of developed force. Once a stable contraction was obtained, acetylcholine (ACh), an agent that induces vasodilation via stimulation of endothelial NO production, was added to the bath in cumulative concentrations of 10-8-10-4 M. This was performed to determine endothelial function and agonist-stimulated NO production from the endothelium. After the cumulative response stabilized, the rings were washed and allowed to equilibrate to baseline. The procedure was then repeated with an endothelium-independent vasodilator, acidified NaNO2 (10-8-10-4 M), to determine smooth muscle function. NaNO2 was prepared by dissolving the compound in 0.1 N HCl and titrating it to pH 2.0. Titrating distilled water to pH 2.0 and adding aliquots to the bath did not produce any vasorelaxation.
2.2. Vascular Superoxide Production
Superoxide production was quantified by lucigenin-enhanced chemiluminescence as previously described[12] and expressed as relative light units (RLU) per second per mg protein (RLU/s/mg protein). In situ superoxide detection was performed with dihydroethidium staining as described previously.[13]
2.3. Determination of NO Production from Aortic Segments
To determine basal nitric oxide production by endothelium in situ, isolated aortic segments were placed in culture medium and incubated in a cell culture incubator (5% CO2 37°C). After 8 hours of incubation, NO and its oxidative metabolic products (NO2 and NO3), collectively known as NOx, were determined as described in our previous study.[14] In brief, vascular segments were homogenized and protein concentrations were determined by the BCA method (Pierce Chemical). 100 μl of culture medium was placed into each well of a 96-well plate and incubated with nitrate reductase and cofactors for 20 minutes at 37° C to reduce NO3 to NO2. Samples (50 μl) were then injected into a water-jacketed, oxygen-free purge vessel containing 5 ml of 20 mM potassium iodide in glacial acetic acid to reduce NO2 to NO. NO reacts with ozone in a reaction chamber and resultant chemiluminescence was detected with a nitric oxide analyzer (SIEVERS NOA 280I; Sievers, Boulder, CO). Signals from the detector were collected and analyzed using a PC-based data recording and processing system. Standard curves were generated using the area under curve after each injection of 50 μl samples containing nitrate reductase treated NaNO3 (0, 2.5, 5, 12.5, 25 and 50 μM). To determine the nitric oxide content of the culture medium, calculations of the slope of the regression analysis using the linear formula y=a +bx were performed. The amount of NOx released was expressed in nmol/mg protein.
2.4. Quantitation of Tissue Nitrotyrosine Content
Nitrotyrosine content, a footprint of in vivo peroxynitrite (ONOO-) formation, was determined utilizing an ELISA method described in our recent publication.[15] In brief, aortic segments were homogenized in lysis buffer and centrifuged for 10 min at 12,500 g at 4° C. The supernatants were collected and protein concentrations were determined. A nitrated protein solution was prepared and diluted for use as a standard. These standard samples, along with tissue samples from aortic segments, were applied to disposable sterile ELISA plates and incubated overnight with primary antibody against nitrotyrosine (Transduction Laboratories, San Jose, CA). After extensive wash and incubation with the peroxidase-conjugated secondary antibody, the peroxidase reaction product was generated using OPD solution. The optical density was measured at 430 nm with a SpectraMax-Plus microplate spectrophotometer. The amount of nitrotyrosine content in tissue samples was calculated using standard curves generated from nitrated BSA containing known amounts of nitrotyrosine, and expressed as pmol/mg protein.
2.5. Immunoblotting
Proteins from tissue homogenate were separated on SDS-PAGE gels, transferred to nitrocellulose membranes, and Western blotted with monoclonal antibody against eNOS, polyclonal antibody against Ser1177 phosphorylated eNOS or monoclonal antibody against gp91phox (Cell Signaling, Danvers, MA). Nitrocellulose membranes were then incubated with horseradish peroxide-conjugated anti-mouse or anti-rabbit IgG antibodies (1:2,000, Cell Signaling, Danvers, MA, USA) for 1 h. The blot was developed using SuperSignal Reagent (Pierce) and visualized with a Kodak Image Station 400. The blot densities were analyzed with Kodak 1D software.
2.6. In vivo Treatment of APN-/- Mice with Globular Domain of Adiponectin
Adiponectin knockout mice were treated with vehicle (PBS, n=6), apocynin (NADPH oxidase inhibitor, 15 mg/kg/day × 3 days, n=8)[16] or recombinant human globular domain of adiponectin (gAd, 2 μg/g/day × 3 days, n=8)[13]. Two hours after the last treatment, animals were sacrificed, and blood samples and aortic vessels were extracted. Endogenous plasma adiponectin level was determined with mouse adiponectin ELISA kit and exogenous gAd was determined with human adiponectin ELISA kit (Phoenix Pharmaceuticals, Inc., Belmont, CA) per manufacturer's instructions. Endothelial function, superoxide production, nitrotyrosine formation, eNOS expression and eNOS phosphorylation were determined as described above.
2.7. Statistical Analysis
All values in the text, table, and figures are presented as means ± SEM of n independent experiments. Data (except Western blot density) were subjected to t test (two groups) or ANOVA (Three or more groups) followed by Bonferoni correction for post hoc t-test. Western blot densities were analyzed with the Kruskal-Wallis test followed by Dunn's post test. Probabilities of 0.05 or less were considered to be statistically significant.
3. Results
3.1. The anatomical and metabolic parameters of the APN-/- and WT littermates
APN-/- mice and their WT littermates at age 8 to 9 weeks were used in this study. All mice developed normally and there was no significant difference in their body weights between the two groups (APN-/-: 23.8±1.5 g, WT: 22.9±1.7 g, P>0.5). Plasma TNFa (APN-/-: 1.36±0.18 pg/ml, WT: 1.09±0.21 pg/ml, P>0.05), fasting glucose (APN-/-: 108±14 mg/dl, WT: 102±11 mg/dl, P>0.05) and insulin levels (APN-/-: 1.45 ng/ml, WT: 1.21 ng/ml, P>0.05) were all slightly increased in APN-/- mice. However, the differences were not statistically significant. Plasma adiponectin was not detectable in APN-/- mice but vascular expression of adiponectin receptor 1 and receptor 2 was normal in these animals (data not shown).
3.2. Adiponectin Deficiency Caused Severe Endothelial Dysfunction
In aortic rings isolated from wild type littermate controls, the addition of ACh (an endothelium-dependent vasodilator) induced a concentration-dependent vasorelaxation which is comparable to that caused by acidified NaNO2 (an endothelium-independent vasodilator). At the maximal concentration studied (100 μM), ACh and NaNO2 caused 77.9±3.9 and 79.8±3.8% vasorelaxation in these vascular segments (P>0.5), indicating that endothelial function is intact in our small vascular ring preparations. However, aortic vascular rings isolated from APN-/- mice demonstrated a severely impaired vasorelaxation response to ACh, while maintaining a normal dilation response to acidified NaNO2. Specifically, addition of 100 μM ACh caused only 34.7±4.5% relaxation (P<0.01 vs. WT), whereas addition of acidified NaNO2 of the same concentration caused 71.9±4.0% relaxation (P>0.05 vs. WT) (Figure 1). These results demonstrated that a severe endothelial dysfunction occurs in adiponectin deficient animals.
Figure 1.
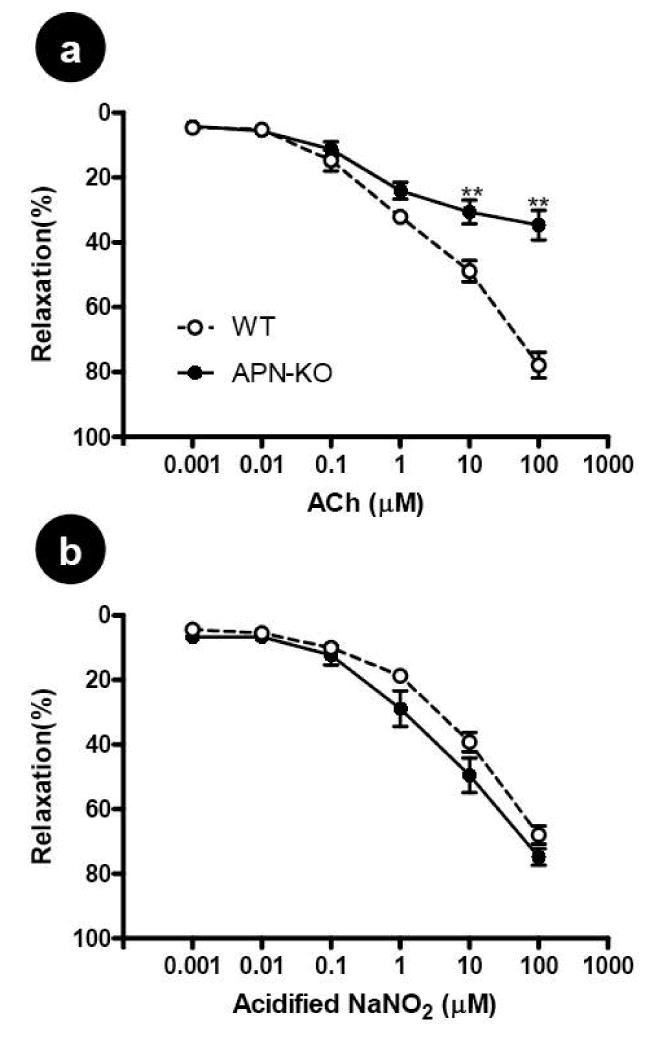
Concentration-dependent vasorelaxation of aortic vascular rings to ACh, an endothelium-dependent vasodilator (A) and to acidified NaNO2, an endothelium-independent vasodilator (B). Each mouse aorta generated 2-3 rings. n=15-17 rings/group. WT=Wild type littermates, APN-KO=adiponectin knockout mice. **P<0.01 vs. WT.
3.3. Increased Superoxide Production and Nitric Oxide Inactivation as a Cause of Endothelial Dysfunction in Adiponectin Deficiency Mice
Experimental and clinical studies have demonstrated that increased superoxide production and its resultant superoxide/nitric oxide bi-radical reaction is the primary cause for endothelial dysfunction in pathological conditions such as hypertension and hyperlipidemia. To determine whether endothelial dysfunction observed in adiponectin deficient animals is related to increased superoxide production and nitric oxide inactivation, three series of experiments were performed. In the first experiment series, in situ superoxide generation was detected by dihydroethidium staining and superoxide production was quantified by lucigenin-enhanced chemiluminescence. As illustrated in Figure 2, a weak dihydroethidium staining was observed in aortic rings isolated from wild type animals, indicating that there was a basal superoxide production in the vascular segment. However, dihydroethidium staining was significantly intensified in aortic rings isolated from adiponectin knockout animals throughout the vascular structure, indicating increased vascular superoxide production in the absence of adiponectin (Figure 2a). The quantitative assay utilizing lucigenin-enhanced chemiluminescence demonstrated that adiponectin deficiency caused a greater than 1.7-fold increase in superoxide generation (Figure 2b). Considerable evidence exists that NADPH oxidase is the major resource for increased vascular superoxide production in many diseases. As illustrated in Figure 2c and 2d, expression of gp91phox, the essential membrane component of NADPH oxidase, is significantly increased in vascular tissue samples from APN-/- mice.
Figure 2.
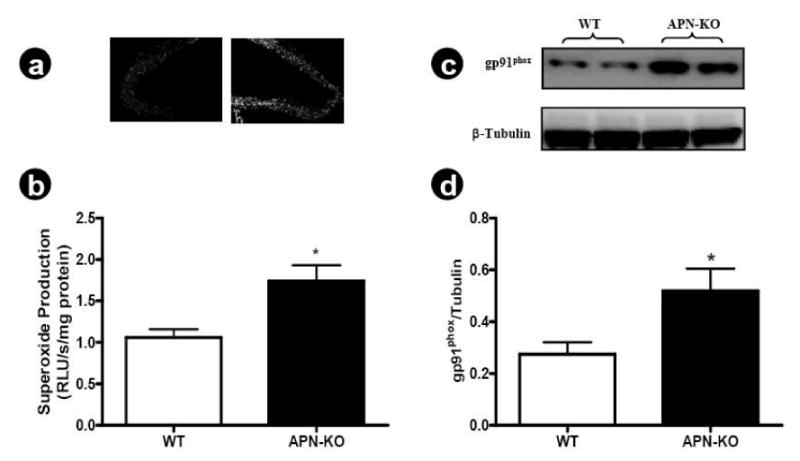
(A) Representative photos of in situ superoxide detection with dihydroethidium staining, n=6 mice/group; (B) Quantitative assay of superoxide production determined by lucigenin-enhanced chemiluminescence, n=8 mice/group; (C) Representative Western blots; (D) Western blot density analysis results from 6 animals. *P<0.05 vs. WT.
In the second experiment series, tissue nitrotyrosine level was determined. Superoxide reacts with NO at a near-diffusion limited rate to form a cytotoxic species, peroxynitrite (ONOO-). Due to its short half life, direct determination of tissue ONOO- is difficult and nitrotyrosine content has been used and accepted as an in vivo foot-print of peroxynitrite formation. As illustrated in Figure 3, nitrotyrosine content was significantly increased in aortic segments isolated from adiponectin knockout mice.
Figure 3.
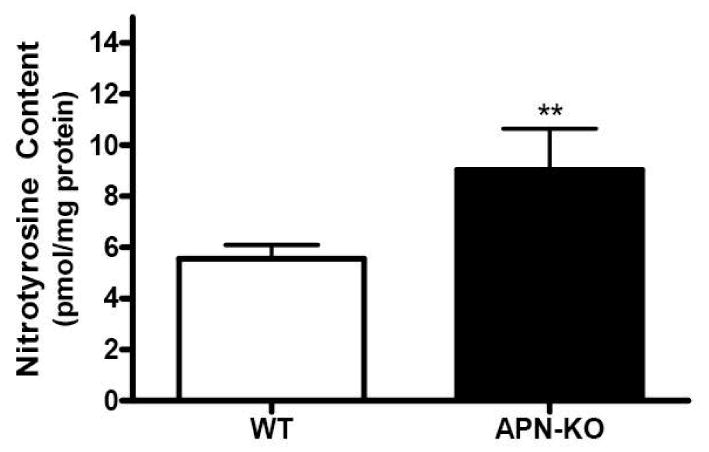
Aortic nitrotyrosine content determined by ELISA. n=10 mice/group. **P<0.01 vs. WT.
The data from the two experiment series described above demonstrate that vessels deficient of adiponectin have significantly increased superoxide generation and nitric oxide inactivation, suggesting that increased superoxide production might be responsible for endothelial dysfunction observed in these adiponectin knockout animals. To obtain more evidence to support this hypothesis, the third experiment series was performed. Aortic segments isolated from APN-/- mice were suspended in the tissue bath as described above. The rings were pre-incubated with vehicle, Tiron (4,5-dihydroxy-1,3-benzene-disulphonic acid, 10 mM, a cell permeable superoxide scavenger) or diphenyleneiodonium (DPI, 2.5 μM)[17] for 10 min before the vasorelaxation to ACh was determined. As summarized in Figure 4, pre-incubation of vascular rings from APN-/- mice with Tiron or DPI significantly improved their vasorelaxation response to ACh (59±2.3% and 62.5+2.7% vs. 37±4.2% in vehicle group, P<0.05). These results further indicate that increased superoxide production in adiponectin deficient animals is partially responsible for endothelial dysfunction in these animals. Moreover, there was no difference in the maximal vasorelaxation response to ACh between Tiron and DPI treated vascular segments. Together with those data presented in figure 2 showing that NADPH oxidase expression is significantly increased in vascular segments isolated from adiponectin knockout mice, our results strongly indicate that increased NADPH oxidase expression is largely responsible for increased vascular superoxide production in the adiponectin deficiency animals.
Figure 4.
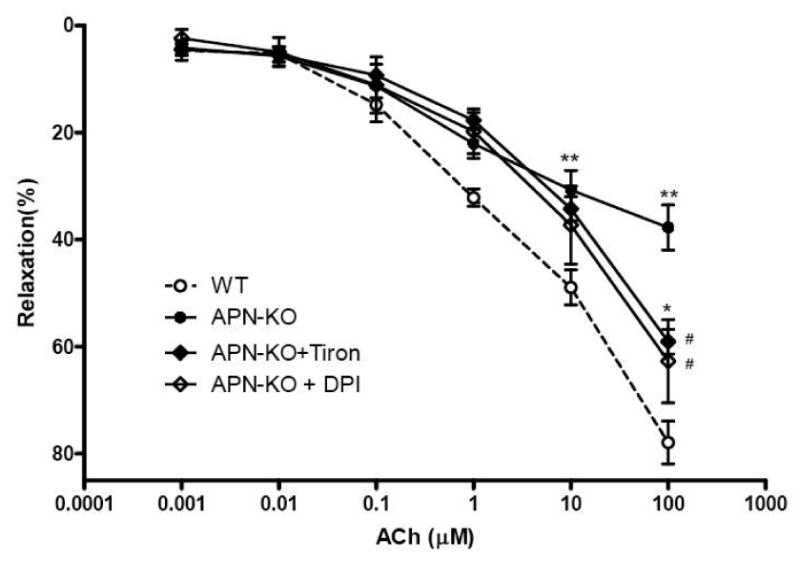
Effect of acute treatment of aortic rings from adiponectin knockout mice with Tiron, a cell permeable superoxide scavenger (APN-KO+Tiron), or DPI, an NADPH oxidase inhibitor (APN-KO+DPI), on vasorelaxation to ACh, an endothelium-dependent vasodilator. Each mouse aorta generated 2-3 rings. n=15-17 rings/group. *P<0.05, **P<0.01 vs. WT; #P<0.05 vs. APN-KO.
3.4. Basal NO Production was Significantly Decreased in Vascular Segments from Adiponectin Deficiency Animals
The data obtained from the three experiment series above indicate that increased superoxide production contribute to endothelial dysfunction development in adiponectin deficient animals. However, the fact that pre-incubation with very high concentration Tiron fails to completely normalize endothelial vasorelaxation to ACh suggests that other mechanisms exist (in addition to increased superoxide production) as contributing pathogenic processes to endothelial dysfunction in adiponectin deficient animals. To directly address this issue, NO production under basal condition was determined. Aortic segments were isolated from either wild type or adiponectin knockout animals and placed in cell culture medium. After 8 hours of incubation in a tissue culture incubator, total NO accumulation in culture medium was determined. As summarized in Figure 5, total NO concentration was significantly reduced in vascular segments isolated from adiponectin knockout mice, indicating that the NO producing system is impaired in adiponectin knockout mice at basal conditions.
Figure 5.
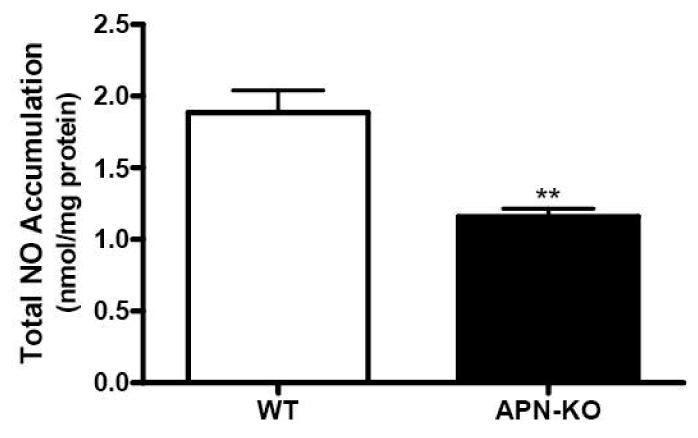
Nitric oxide concentration in culture medium after 8 hours incubation of aortic segments from WT or adiponectin knockout mice. Each mouse aorta generated 1 aortic segment. N=8 mice/group. **P<0.01 vs. WT.
3.5. Expression of eNOS was Normal but its Phosphorylation was Significantly Reduced in Aortic Tissue from Adiponectin Knockout Animals
Endothelial nitric oxide synthase (eNOS) is responsible for physiologic vascular tissue NO production. Our experimental results presented in Figure 5 thus suggest that either eNOS expression or its phosphorylation is impaired in APN-/- aortic vessel. As summarized in Figure 6, there was no difference in total eNOS expression between aortic segments isolated from WT and APN-/- mice, indicating that eNOS expression was normal in these animals. However, compared to WT mice samples, eNOS phosphorylation was significantly reduced in aortic tissue from adiponectin knockout mice.
Figure 6.
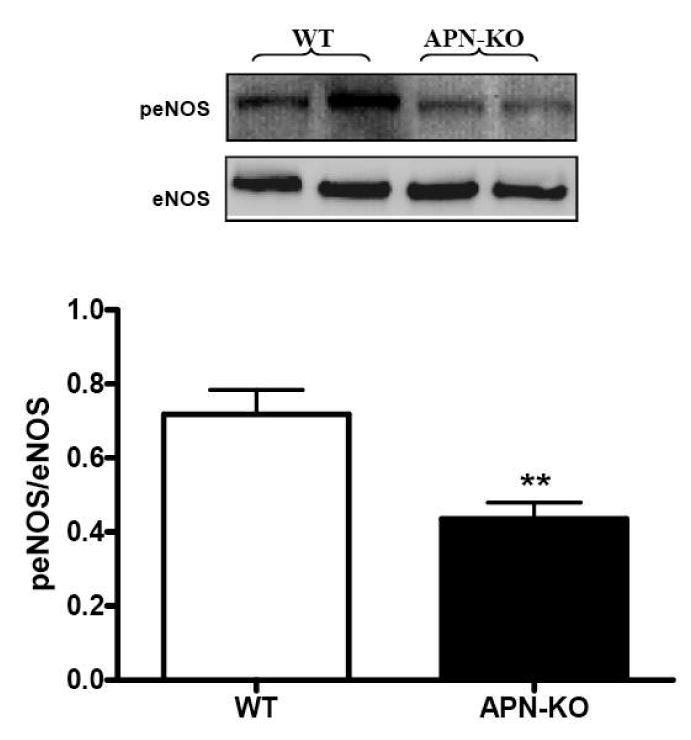
Expression of eNOS and peNOS in aortic tissues obtained from WT or adiponectin knockout mice. Top: representative Western blots. Bottom: Summary of density analysis expressed as arbitrary unit. n= 8 mice/group
3.6. Treatment of APN-/- Animals with Adiponectin Reduced Superoxide Production and Peroxynitrite Formation, Increased eNOS Phosphorylation and Normalized the Vasorelaxation Response to ACh
The above presented data suggest that the mechanisms responsible for endothelial dysfunction observed in adiponectin knockout mice are multifactorial. Increased NO inactivation (due to increased superoxide production) and decreased NO production (due to reduced eNOS phosphorylation) both play a significant role. In a final attempt to dissect the mechanisms responsible for endothelial dysfunction in adiponectin knockout animals, 22 adiponectin knockout mice were treated with vehicle, recombinant human gAd, or apocynin (NADPH oxidase inhibitor) for 3 days. As expected, neither mouse nor human adiponectin was detected in APN-/- mice treated with vehicle or apocynin. Intraperitoneally administered human gAd was partially absorbed and human adiponectin was detected in APN-/- mice treated with gAd (21.8±1.68 ng/ml). In vivo treatment with apocynin significantly reduced superoxide production (Figure 7A), decreased nitrotyrosine formation (Figure 7B), and partially restored endothelium dependent vasorelaxation (Figure 7D) without altering eNOS phosphorylation (Figure 7C). Most importantly, replenishment with exogenous gAd suppressed aortic superoxide production (Figure 7A), blocked peroxynitrite formation (Figure 7B), increased eNOS phosphorylation (Figure 7C) and completely restored vasodilatory response to ACh (Figure 7D).
Figure 7.
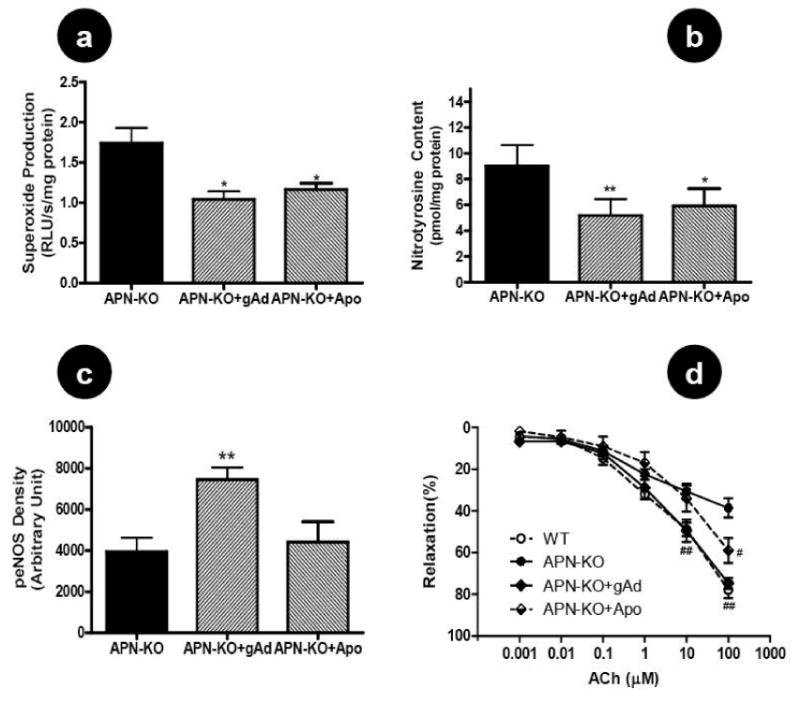
Effect of in vivo treatment of adiponectin knockout mice with gAd (global adiponectin domain) or apocynin (NADPH oxidase inhibitor) on aortic superoxide production (A), nitrotyrosine content (B), eNOS phosphorylation (C), and endothelium-dependent vasorelaxation to ACh (D). Each mouse aorta generated 2-3 rings. n=12-13 rings/group. #P<0.05, ##P<0.01 vs. APN-KO.
4. Discussion
Several important observations were made in the current study. First, we have provided the first direct evidence that adiponectin deficiency caused severe endothelial dysfunction in aortic vasculature. Endothelial cells produce an array of bioactive molecules that play critical roles in general homeostasis. Among the endothelium-derived bioactive molecules, NO is the most potent vasculoprotective molecule. Besides its vasodilatory action, NO blocks platelet activation/adhesion, inhibits leukocyte/endothelial interaction, promotes endothelial cell regeneration, inhibits smooth muscle cell proliferation, and reduces lipid oxidation[18]. Under pathologic conditions, when NO production is impaired in response to agonist stimulation or hyperemia (endothelial dysfunction), platelets are activated, leukocyte/endothelial interactions are promoted, endothelial apoptosis and smooth muscle cell proliferation are increased, lipid oxidation is stimulated, and atherosclerosis is developed[19].
Substantial animal experimental evidence, as well as clinical observations, have demonstrated that atherosclerosis, the hallmark of macroangiopathy, is markedly accelerated in diabetes[20, 21]. Although it is well-documented that severe endothelial dysfunction in diabetic patients contributes to the development of atherosclerosis[22], the mechanisms underlying endothelial dysfunction in diabetic patients remain incompletely understood and effective treatment is currently limited. Studies in human subjects and animal models have demonstrated an association between circulating adiponectin levels and endothelial function. Specifically, forearm blood flow in human subjects during reactive hyperemia is highly negatively correlated with adiponectin plasma concentrations, indicating that adiponectin is closely associated with endothelium-dependent vasodilation[8-11]. Since plasma adiponectin levels are severely reduced in patients with type 2 diabetes[4], these clinical observations strongly suggest that reduced circulating adiponectin may contribute to the development of vasculopathic state in diabetic patients. Moreover, adiponectin knockout mice show striking altered vascular histology, including severe neointimal thickening and increased proliferation of vascular smooth muscle cells in mechanically injured arteries[23].
In the current study, we have demonstrated that bioactive NO was markedly reduced in aortic rings isolated from adiponectin knockout mice. This result provided direct evidence supporting the notion that reduced adiponectin levels in diabetics is a major etiologic factor for endothelial dysfunction and accelerated atherosclerosis suffered by these patients. Moreover, reduced ambient bioactive NO (as demonstrated in our study) also constitutes a likely mechanism for accelerated pathologic remodeling following mechanical vascular injury in adiponectin knockout mice.[23] Previous studies have demonstrated that adiponectin directly stimulates NO production by increasing eNOS phosphorylation[24]. Moreover, adiponectin has been shown to be a potent anti-inflammatory molecule that inhibits leukocyte/endothelial interaction. Therefore, endothelial dysfunction observed in the current study may result from either direct or indirect effects of adiponectin deficiency.
Second, we have demonstrated that increased nitric oxide inactivation (due to increased superoxide production) as well as reduced nitric oxide production (due to decreased eNOS phosphorylation) both contribute to the pathogenesis of endothelial dysfunction in adiponectin deficient animals. Substantial evidence exists that the mechanisms of endothelial dysfunction are multifactorial- including deficiencies of arginine supply, alteration of signaling mechanisms, alterations of NO synthase expression or one of the cofactors involved in NO synthase activation, and increased destruction of NO[25]. Our preliminary experimental results demonstrated that plasma and tissue arginine levels, co-factors required for eNOS activation, and endothelial morphology are normal in adiponectin knockout mice (data not shown), suggesting that endothelial dysfunction in adiponectin deficient animals cannot be attributed to these factors. However, we have now demonstrated the significant increased production of vascular superoxide and peroxynitrite (the toxic product of superoxide and NO) in adiponectin knockout animals. Moreover, in vitro acute incubation of aortic vascular rings from adiponectin knockout animals with Tiron, a superoxide scavenger, significantly improved vascular vasodilatory response to ACh. Although it has been previously reported that adiponectin possesses strong anti-oxidant effects in vivo and in vitro[13, 26, 27], the current study provided the first direct evidence that increased inactivation of NO by superoxide is one of the major mechanisms responsible for endothelial dysfunction in pathologic conditions where adiponectin production is reduced, such as type 2 diabetes.
Previous experimental studies have demonstrated that under pathologic conditions such as hypercholesterolemia, endothelial NO production is increased rather than decreased. Increased NO inactivation is therefore considered to be the single most crucial mechanism responsible for reduced NO activity and endothelial dysfunction in these diseases[28-30]. Our current study demonstrated that in vitro incubation of aortic segments from adiponectin knockout mice with high concentrations of a superoxide scavenger or in vivo treatment with optimal dose NADPH oxidase inhibitor[16] significantly improved ACh-induced vasorelaxation. However, a significant portion of endothelial dysfunction was not restored despite the anti-oxidant treatments. These results indicate that, unlike other pathologic conditions, endothelial dysfunction in adiponectin deficiency is multifactorial. Our data demonstrated significantly reduced basal NO production in adiponectin knockout mice aortic segments, indicating the diminished capacity of NOS to generate NO in the adiponectin deficiency state. Our additional experimental results revealed normal vascular eNOS expression in adiponectin knockout mice, but significantly reduced eNOS phosphorylation, a critical post-translational modification that increases eNOS activity. Although it has been reported previously that adiponectin phosphorylates eNOS through the AMPK/Akt signaling system[24, 31], our study provided the first evidence that links reduced eNOS phosphorylation with endothelial dysfunction in adiponectin deficiency.
Finally, we have demonstrated that the pathologic alterations observed in adiponectin deficiency are completely rescued by administration of recombinant human gAd. These results not only provided further evidence that reduced adiponectin levels as seen in diabetes plays a causative role in the pathogenesis of vascular injury in these patients, but also demonstrated that gAd is a potent therapeutic molecule with pharmacologic potential in the treatment of patients with vascular injury and reduced adiponectin production. These results are consistent with the published findings of other investigators showing that adiponectin treatment attenuates neointimal proliferation in mechanically-injured arteries of adiponectin knockout mice,[23, 32] decreases atherosclerotic lesion formation in ApoE-deficient mice[33, 34] and ameliorates hypertension in obese mice[35].
Factors associated with endothelial dysfunction in diabetes are complex and include activation of protein kinase C, overexpression of growth factors and/or cytokines, and oxidative stress[2]. Several therapeutic interventions, including insulin sensitizers, ACE inhibitors, hypolipidemic therapy, arginine supplementation and antioxidants, have been tested in clinical trials directed at improving endothelial function in diabetic patients[2]. Unfortunately, the outcome of these treatments on endothelial function is inconsistent or even controversial, and no definitive conclusion regarding efficacy has been made for any of previously proposed treatments[2]. Our study provides further evidence to the beneficial actions of adiponectin, specifically its ability to increase NO production by eNOS phosphorylation and decreasing NO inactivation by blocking superoxide production. Given that adiponectin is a potent vasculoprotective molecule with multiple arms of action, and that its levels are significantly reduced in diabetic patients, it is conceivable that adiponectin deficiency might be a novel target of attack in the prevention of vascular injury in the diabetic population.
Acknowledgments
Funding: This research was supported by the following grants: NIH 2R01HL-63828, American Diabetes Association Research Award 7-08-RA-98, American Heart Association Grant-in-Aid 0855554D, Commonwealth of Pennsylvania-Department of Health (to X.L.M.), American Diabetes Association 7-06-JF59 (to L.T.), and Emergency Medicine Foundation Career Development Grant (to W.L.).
Footnotes
Conflict of interest: none declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Buse JB, Ginsberg HN, Bakris GL, Clark NG, Costa F, Eckel R, et al. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation. 2007;115(1):114–126. doi: 10.1161/CIRCULATIONAHA.106.179294. [DOI] [PubMed] [Google Scholar]
- 2.Calles-Escandon J, Cipolla M. Diabetes and Endothelial Dysfunction: A Clinical Perspective. Endocr Rev. 2001;22(1):36–52. doi: 10.1210/edrv.22.1.0417. [DOI] [PubMed] [Google Scholar]
- 3.Kadowaki T, Yamauchi T. Adiponectin and Adiponectin Receptors. Endocr Rev. 2005;26(3):439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- 4.Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20(6):1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 5.Koenig W, Khuseyinova N, Baumert J, Meisinger C, Lowel H. Serum concentrations of adiponectin and risk of type 2 diabetes mellitus and coronary heart disease in apparently healthy middle-aged men: results from the 18-year follow-up of a large cohort from southern Germany. J Am Coll Cardiol. 2006;48(7):1369–1377. doi: 10.1016/j.jacc.2006.06.053. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein BJ, Scalia R. Adipokines and vascular disease in diabetes. Curr Diab Rep. 2007;7(1):25–33. doi: 10.1007/s11892-007-0006-6. [DOI] [PubMed] [Google Scholar]
- 7.Zhu W, Cheng KK, Vanhoutte PM, Lam KS, Xu A. Vascular effects of adiponectin: molecular mechanisms and potential therapeutic intervention. Clin Sci (Lond) 2008;114(5):361–374. doi: 10.1042/CS20070347. [DOI] [PubMed] [Google Scholar]
- 8.Ouchi N, Ohishi M, Kihara S, Funahashi T, Nakamura T, Nagaretani H, et al. Association of Hypoadiponectinemia With Impaired Vasoreactivity. Hypertension. 2003;42(3):231–234. doi: 10.1161/01.HYP.0000083488.67550.B8. [DOI] [PubMed] [Google Scholar]
- 9.Shimabukuro M, Higa N, Asahi T, Oshiro Y, Takasu N, Tagawa T, et al. Hypoadiponectinemia Is Closely Linked to Endothelial Dysfunction in Man. J Clin Endocrinol Metab. 2003;88(7):3236–3240. doi: 10.1210/jc.2002-021883. [DOI] [PubMed] [Google Scholar]
- 10.Tan KCB, Xu A, Chow WS, Lam MCW, Ai VHG, Tam SCF, et al. Hypoadiponectinemia Is Associated with Impaired Endothelium-Dependent Vasodilation. J Clin Endocrinol Metab. 2004;89(2):765–769. doi: 10.1210/jc.2003-031012. [DOI] [PubMed] [Google Scholar]
- 11.Maruyoshi H, Kojima S, Otsuka F, Funahashi T, Kaikita K, Sugiyama S, et al. Hypoadiponectinemia is associated with coronary artery spasm in men. Circ J. 2005;69(9):1154–1156. doi: 10.1253/circj.69.1154. [DOI] [PubMed] [Google Scholar]
- 12.Lund DD, Faraci FM, Miller FJ, Jr, Heistad DD. Gene transfer of endothelial nitric oxide synthase improves relaxation of carotid arteries from diabetic rabbits. Circulation. 2000;101(9):1027–1033. doi: 10.1161/01.cir.101.9.1027. [DOI] [PubMed] [Google Scholar]
- 13.Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, et al. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115(11):1408–1416. doi: 10.1161/CIRCULATIONAHA.106.666941. [DOI] [PubMed] [Google Scholar]
- 14.Liang F, Gao E, Tao L, Liu H, Qu Y, Christopher TA, et al. Critical timing of L-arginine treatment in post-ischemic myocardial apoptosis-role of NOS isoforms. Cardiovasc Res. 2004;62(3):568–577. doi: 10.1016/j.cardiores.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 15.Liu HR, Tao L, Gao E, Lopez BL, Christopher TA, Willette RN, et al. Anti-apoptotic effects of rosiglitazone in hypercholesterolemic rabbits subjected to myocardial ischemia and reperfusion. Cardiovasc Res. 2004;62(1):135–144. doi: 10.1016/j.cardiores.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 16.Cotter MA, Cameron NE. Effect of the NAD(P)H oxidase inhibitor, apocynin, on peripheral nerve perfusion and function in diabetic rats. Life Sci. 2003;73(14):1813–1824. doi: 10.1016/s0024-3205(03)00508-3. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka K, Honda M, Takabatake T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J Am Coll Cardiol. 2001;37(2):676–685. doi: 10.1016/s0735-1097(00)01123-2. [DOI] [PubMed] [Google Scholar]
- 18.Lamas S, Lowenstein CJ, Michel T. Nitric oxide signaling comes of age: 20 years and thriving. Cardiovasc Res. 2007;75(2):207–209. doi: 10.1016/j.cardiores.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 19.Arnal JF, Dinh-Xuan AT, Pueyo M, Darblade B, Rami J. Endothelium-derived nitric oxide and vascular physiology and pathology. Cell Mol Life Sci. 1999;55(89):1078–1087. doi: 10.1007/s000180050358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laight DW, Carrier MJ, Anggard EE. Antioxidants, diabetes and endothelial dysfunction. Cardiovasc Res. 2000;47(3):457–464. doi: 10.1016/s0008-6363(00)00054-7. [DOI] [PubMed] [Google Scholar]
- 21.Goldberg IJ. Why does diabetes increase atherosclerosis? I don't know! Journal of Clinical Investigation. 2004;114(5):613–615. doi: 10.1172/JCI22826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Busse R, Fleming I. Endothelial dysfunction in atherosclerosis. J Vasc Res. 1996;33:181–194. doi: 10.1159/000159147. [DOI] [PubMed] [Google Scholar]
- 23.Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277(29):25863–25866. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- 24.Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem. 2003;278(45):45021–45026. doi: 10.1074/jbc.M307878200. [DOI] [PubMed] [Google Scholar]
- 25.Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture) Am J Physiol Heart Circ Physiol. 2006;291(3):H985–1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- 26.Li R, Wang WQ, Zhang H, Yang X, Fan Q, Christopher TA, et al. Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/iNOS activity. Am J Physiol Endocrinol Metab. 2007;293(6):E1703–E1708. doi: 10.1152/ajpendo.00462.2007. [DOI] [PubMed] [Google Scholar]
- 27.Motoshima H, Wu X, Mahadev K, Goldstein BJ. Adiponectin suppresses proliferation and superoxide generation and enhances eNOS activity in endothelial cells treated with oxidized LDL. Biochem Biophys Res Commun. 2004;315(2):264–271. doi: 10.1016/j.bbrc.2004.01.049. [DOI] [PubMed] [Google Scholar]
- 28.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 29.Doughan AK, Harrison DG, Dikalov SI. Molecular Mechanisms of Angiotensin II-Mediated Mitochondrial Dysfunction: Linking Mitochondrial Oxidative Damage and Vascular Endothelial Dysfunction. Circ Res. 2008;102(4):488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 30.Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. Journal of Clinical Investigation. 1993;91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonon AT, Widegren U, Bulhak A, Salehzadeh F, Persson J, Sjoquist PO, et al. Adiponectin protects against myocardial ischaemia-reperfusion injury via AMP-activated protein kinase, Akt, and nitric oxide. Cardiovasc Res. 2008;78(1):116–122. doi: 10.1093/cvr/cvn017. [DOI] [PubMed] [Google Scholar]
- 32.Matsuda M, Shimomura I, Sata M, Arita Y, Nishida M, Maeda N, et al. Role of Adiponectin in Preventing Vascular Stenosis. The missing link of adipo-vascular axis. J Biol Chem. 2002;277(40):37487–37491. doi: 10.1074/jbc.M206083200. [DOI] [PubMed] [Google Scholar]
- 33.Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106(22):2767–2770. doi: 10.1161/01.cir.0000042707.50032.19. [DOI] [PubMed] [Google Scholar]
- 34.Yamauchi T, Kamon J, Waki H, Imai Y, Shimozawa N, Hioki K, et al. Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem. 2003;278(4):2461–2468. doi: 10.1074/jbc.M209033200. [DOI] [PubMed] [Google Scholar]
- 35.Ohashi K, Kihara S, Ouchi N, Kumada M, Fujita K, Hiuge A, et al. Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension. 2006;47(6):1108–1116. doi: 10.1161/01.HYP.0000222368.43759.a1. [DOI] [PubMed] [Google Scholar]