

Abstract
Mitochondrial dysfunction is an underlying cause of ischemia-reperfusion injury. In particular, ischemic injury induces dramatic increases in mitochondrial permeability, thereby instigating a chain of events that leads to both apoptotic and necrotic cardiomyocyte death. The mitochondrial permeability transition (MPT) pore, a large, non-specific channel that spans the inner mitochondrial membrane, is known to mediate the lethal permeability changes that initiate mitochondrial-driven cardiomyocyte death. The purpose of this review is to focus on the role of the MPT pore in ischemia-reperfusion injury, the mechanisms involved, and, in particular, what we do and do not know regarding the pore's molecular composition.
Keywords: Ischemia-reperfusion, Mitochondrial permeability transition, Voltage-dependent anion channel, Adenine nucleotide translocase, Cyclophilin-D, Bcl-2 proteins
Introduction
It would be a gross understatement to say that mitochondrial research has undergone a considerable renaissance over the past decade or so. The discovery that mitochondria are not only the major suppliers of energy to a cell, but are also pivotal in the cell's decision to live or die has revolutionized many fields, especially that of cardiac biology. Indeed, it is now well recognized that mitochondrial dysfunction plays a crucial role in the pathogenesis of several cardiac diseases including ischemia-reperfusion injury. Moreover, the mitochondrial permeability transition (MPT) pore, a large, non-specific channel, is now known to mediate the lethal permeability changes that initiate mitochondrial-driven death in these diseases. In this review we will focus on the role of the MPT pore in ischemia-reperfusion injury, the mechanisms involved, and what we do and do not know regarding its molecular composition.
Mitochondrial death pathways
There are many components to the molecular machinery that mediates mitochondrial-dependent cell death, which due to space limitations we will only briefly summarize here. However, I would draw the reader's attention to a recent excellent review that comprehensively covers this subject [42]. The canonical mitochondrial death mechanism is the so-called “intrinsic” pathway that mediates apoptosis. Toxic stimuli such as oxidative stress induce translocation and integration of the pro-death members of the Bcl-2 family (e.g., Bax, Bak, Bid) into the outer mitochondrial membrane [17, 26, 42, 61, 64]. These proteins, by a mechanism that remains both elusive and controversial, permeabilize the outer membrane to an extent that allows the release of pro-apoptotic proteins from the inter-membrane space, most notably cytochrome c, Smac/DIABLO, htrA2/Omi protease, and endonuclease-G (endoG). Cytochrome c binds to the cytosolic protein apaf1 and the resultant “apoptosome” activates the caspase-9 and -3 protease system [17, 26, 42, 61, 64]. Smac/DIABLO and htrA2/Omi activate caspases by either sequestering or degrading caspase-inhibitory proteins, respectively. EndoG translocates to the nucleus and mediates DNA fragmentation. The coordinated action of these proteins subsequently results in the death of the cardiomyocyte by apoptosis.
Whereas the Bcl-2 proteins primarily induce cell death through permeabilization of the outer mitochondrial membrane, there is also a second mechanism that initiates cell death via permeabilization of the inner mitochondrial membrane. Noxious stimuli often lead to excessive production of reactive oxygen species (ROS), and Ca2+ overload of the mitochondrial matrix [20, 28, 41, 42, 45, 73]. These in turn cause the opening of a large, non-specific channel in the inner mitochondrial membrane. This phenomenon, termed the MPT, dissipates the proton electrochemical gradient (ΔΨm) that drives many mitochondrial functions, leading to ATP depletion, further ROS production, and ultimately swelling and rupture of the organelle [20, 28, 41, 45, 73]. Mitochondrial rupture can lead to the release of pro-apoptotic inter-membrane proteins described above, which would conceivably initiate the apoptotic program. However, if the stress is severe and/or prolonged, ATP (which is required for apoptosis to occur) will be depleted and the cell will instead die by necrosis. Indeed, the relative contribution of MPT to apoptosis versus necrosis is still the subject of debate.
Mitochondria in ischemia-reperfusion injury
In order to understand the regulation of the MPT during ischemia-reperfusion injury, it is perhaps best to begin with the ionic and metabolic alterations the mitochondria are subjected to during this stress. This is not intended to be an exhaustive list, but rather is focused on the main factors that ultimately contribute to the induction of MPT and cardiomyocyte death.
Adenine nucleotides
The basic definition of ischemia is the lack of oxygen supply to the affected area of the myocardium. As the electrons generated by the electron transfer chain in the mitochondria can no longer be transferred to molecular oxygen, the net result is a cessation of oxidative phosphorylation and inhibition of mitochondrial ATP synthesis [20, 28, 51, 73]. In addition, the inhibition of electron transfer prevents the pumping of H+ across the inner membrane, which is required to generate the ΔΨm. In an effort to maintain the ΔΨm the mitochondrion runs the F1F0 ATP synthase in reverse thereby hydrolyzing the remaining ATP [20, 51]. Thus adenine nucleotides are rapidly depleted after the onset of ischemia, with a concomitant rise in inorganic phosphate (Pi), both of which facilitate opening of the MPT pore (see below).
Mitochondrial Ca2+
As oxidative phosphorylation is inhibited, the ischemic cardiomyocyte must rely on anaerobic glycolysis for its ATP supply. This leads to a build up of lactate and therefore acidification of the cytosol. In an attempt to reestablish normal pH, the cell extrudes the H+ ions in exchange for Na+ via the sarcolemmal Na+/H+ exchanger (NHE) [20, 28, 51, 73]. In addition, Na+ ions also enter the cell through non-inactivating Na+ channels during ischemia [51]. The Na+ ions are in turn extruded in exchange for Ca2+ by the Na+/Ca2+ exchanger [20, 28, 51, 73]. Thus ischemia causes a slow elevation in cytosolic Ca2+ that is then accelerated upon reperfusion as extracellular H+ is washed out (thereby increasing the gradient for the NHE to work). One of the ways the cardiomyocyte deals with this lethal increase in Ca2+ is to take it up into the mitochondria via the mitochondrial Ca2+ uniporter, a protein that uses the negative ΔΨm to drive uptake of the positively charged Ca2+ ions into the matrix [51, 73]. If the elevations in mitochondrial Ca2+ become excessive they can trigger the MPT response [5, 7, 15, 19, 41, 42].
Mitochondrial ROS
It is paradoxical that the restoration of the oxygen supply to the ischemic region by reperfusion is most likely the biggest cause of myocyte death. The mitochondria damaged during ischemia are no longer able to efficiently transfer electrons thereby greatly increasing ROS generation from complexes-I and -III [20, 28, 51, 73]. Thus reperfusion is associated with a massive burst of mitochondrial-derived ROS. These ROS can interact with and damage a variety of mitochondrial proteins, including the components of the electron transfer chain, as well cause lipid peroxidation. In addition, like Ca2+, ROS are excellent inducers of the MPT response [2, 5, 41, 42, 52, 63].
Apoptotic Bcl-2 proteins
Ischemia-reperfusion also induces apoptotic cardiomyocyte death, although the incidence of this form of death is significantly lower than necrosis. In particular, activation of pro-death Bcl-2 proteins such as Bax, Bid, Puma, and BNIP3, and their translocation and integration into mitochondrial membranes has been reported in ischemically damaged myocytes and hearts [27, 29, 61, 67], and genetic deletion of Puma, Bax, or BNIP3 reduces infarct size [22, 32, 67]. Again, it appears that ischemia alone is not sufficient for Bcl-2 protein activation and that reperfusion is required, consistent with the fact that many of these proteins are redox-sensitive [55]. In addition to upregulating/activating the pro-death contingent of this protein family, there is a concomitant decrease in the anti-apoptotic family members such as Bcl-2 and Bcl-XL during ischemia-reperfusion injury [43, 48, 76]. In contrast, cardiac-specific transgenic overexpression of Bcl-2 is cardioprotective [14, 33]. The ability of the pro-death Bcl-2 proteins to induce MPT is a very controversial one, and we will come back to this issue later on in the review.
The MPT pore
The MPT phenomenon is mediated by the MPT pore, a non-specific channel thought to span the inner mitochondrial membrane. The pore itself is permeable to solutes up to 1.5 kDa [20, 27, 28, 41, 42, 73]. This causes equilibration of H+ across the inner membrane, which dissipates ΔΨm and inhibits ATP production. A concomitant influx of water causes swelling of the mitochondria, which stretches the membranes to the point where the outer membrane fails. The mitochondrial pore is redox, Ca2+, voltage, adenine nucleotide, Pi, and pH sensitive [20, 27, 28, 41, 42, 73]. Most importantly, increases in matrix Ca2+ and ROS induce pore opening, whereas adenine nucleotides inhibit the pore; indeed, as we have already discussed in the previous section, ischemia-reperfusion injury is associated with increases in MPT pore activators (Ca2+, ROS, Pi) and reductions in MPT pore inhibitors (ATP/ADP). The MPT pore is inhibited by low pH and is therefore believed to be quiescent during ischemia [21, 25, 28, 35]. However, the restoration of pH coupled with the rapid elevation in mitochondrial Ca2+ and ROS would lead to rapid opening of the pore upon reperfusion; a scenario that has been confirmed in isolated cardiomyocytes and whole heart preparations by a variety of techniques [21, 25, 35].
There is considerable evidence that inhibition of the MPT pore represents a powerful mechanism by which the heart can be protected against ischemia-reperfusion injury. Many studies have shown that direct pharmacological inhibition of the MPT pore with compounds such as cyclosporine-A blunts the loss of cardiac myocytes that underlies myocardial ischemia-reperfusion injury [5, 15, 21, 30, 52], as well as other cardiac pathologies [37, 50, 53, 54]. Moreover, the protective signaling cascades initiated by both pre- or post-conditioning appear to terminate, at least in part, in the inhibition of the MPT pore [4, 34, 46]. Needless to say, the MPT pore represents an obvious therapeutic target for inhibition of cardiomyocyte mortality and treatment of myocardial ischemia-reperfusion. Unfortunately, antagonizing the MPT pore in a clinical setting is currently hampered by the fact that the precise molecular architecture of the pore remains unknown; a problem we will address in the next section.
Molecular composition of the MPT pore
Based upon biochemical and pharmacological studies, the pore was proposed to consist of following [20, 28, 41, 42, 45, 73]: the voltage-dependent anion channel (VDAC) in the outer membrane, the adenine nucleotide translocase (ANT) in the inner membrane, plus cyclophilin-D (CypD) in the matrix (Fig. 1). VDAC, ANT, and CypD interact at membrane contact sites and reconstitution of this complex in vesicles yields a Ca2+-sensitive channel reminiscent of the MPT pore [16]. Moreover, pharmacological inhibitors of ANT and CypD, e.g., bongkrekic acid and cyclosporine-A, respectively, inhibit MPT and protect cardiac cells against oxidative stress, hypoxia, and ischemia-reperfusion [2, 5, 15, 21, 30, 35, 52]. However, recent genetic studies have seriously questioned the validity of this paradigm.
Fig. 1.
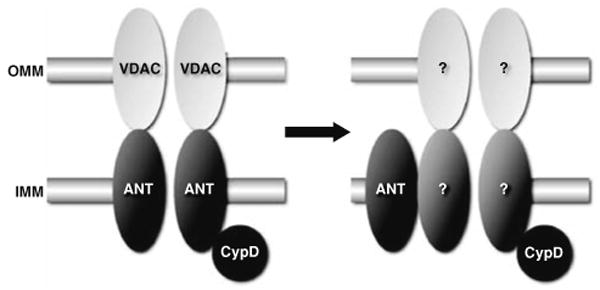
Molecular models for the mitochondrial permeability transition (MPT) pore. On the left is shown the original model for the MPT pore, consisting of the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane (OMM), the adenine nucleotide translocase (ANT) in the inner mitochondrial membrane (IMM), and cyclophilin-D (CypD) in the matrix. However, as shown on the right, if we apply recent findings in gene-targeted mice the model becomes very different. VDAC is no longer part of the model, ANT is now more of a regulatory protein, and only CypD remains as an established component. Consequently, the identity of the pore-forming protein(s) remains unknown
Voltage-dependent anion channel
The most abundant protein in the outer mitochondrial membrane, VDAC facilitates the efficient transport of ATP/ADP across the outer leaflet [11, 62]. The VDAC family consists of three gene products (VDAC1, 2, and 3) that exhibit a fairly high degree of structural and functional homology [11, 62]. Although VDAC has long been considered a key component of the MPT pore, almost to the point of dogma, the evidence supporting this paradigm has always been far from compelling. While putative VDAC inhibitors and VDAC “blocking” antibodies have been reported to prevent MPT in vitro [13, 77], the specificity of these agents is uncertain [40, 62]. Moreover, a mitochondrial pore-like channel has been reconstituted in proteoliposomes in the absence of VDAC [74]. From a genetic standpoint, Bernardi's group showed that mitochondria lacking VDAC1 still exhibited a normal cyclosporine-sensitive MPT response [40]. We have recently built upon this and shown that mitochondria and cells lacking all three VDAC isoforms still undergo MPT [6] and are still sensitive to ischemia-reperfusion injury, further demonstrating that VDAC is dispensable for MPT, and is not an essential component of the MPT pore.
Adenine nucleotide translocase
The ANT family mediates the exchange of ATP and ADP across the inner mitochondrial membrane. Two homologous isoforms, ANT1 and ANT2 are present in the mouse, with a third isoform, ANT3, found in humans [24]. The ANT1 isoform is found primarily in striated muscle, with ANT2 being more ubiquitously expressed. A third mouse isoform (ANT4) has been recently reported but appears to be restricted to testicular germ cells [12]. Unlike VDAC, the evidence supporting a role for ANT in MPT has, until recently, been far more convincing. For example, pharmacological manipulation of ANT with atractyloside or bongkrekic acid influences MPT [2, 9, 18, 31]. ANT1, but not ANT2, interacts with CypD at contact sites between the inner and outer mitochondrial membranes, where the pore is believed to localize [69], and reconstitution of ANT plus CypD can yield an MPT-like pore in proteoliposomes [74]. Furthermore, expression of ANT1 can elicit mitochondrial-dependent death in non-cardiac cells [8, 75]. However, although ANT1 is primarily restricted to cardiac and skeletal muscle, MPT occurs in all major organs suggesting that ANT1 may not in fact be a bona fide component of the MPT pore. Indeed, as with VDAC, the genetic data has not corroborated the biochemical findings. We have found that overexpression of ANT1 in myocytes, although cytotoxic, appears to act through mechanisms independent of MPT (unpublished data) whereas specific expression of ANT1 in the rat heart was actually cardioprotective in a model of cardiomyopathy [70]. Genetic deletion of both ANT isoforms in mice does not appreciably alter MPT thus raising doubts as to ANT's identity as a necessary component of the MPT pore [38]. Instead, this study indicated that ANT may act more as a peripheral regulatory protein that confers sensitivity of the MPT pore to adenine nucleotides and ANT ligands. One major caveat with this study is that these are not healthy mice and there appear to be wholesale changes in the mitochondrial protein profile that could of compensated for the lack of ANT at some level, or sensitized the cells to death through another mechanism.
Cyclophilin-D
The last piece of the original MPT paradigm is CypD. This mitochondrial matrix protein is a member of the large family of peptidylprolyl isomerases, which catalyze the rotation of proline peptide bonds, thereby inducing a conformational change in the target protein [71]. Although the physiological role of CypD remains unknown, its pathological role as a component of the MPT pore has been widely accepted. Inhibition of CypD's isomerase activity by cyclosporine-A or its non-immunosuppressive analogues inhibits MPT and cell death in numerous cell culture systems. Moreover, such CypD-targeting pharmacological agents protect the heart against the noxious effects of ischemia-reperfusion injury [5, 15, 21, 30, 52]. Unlike VDAC and ANT, there is only one known gene for CypD (the Ppif gene), and, several groups, including our own, have independently generated Ppif-null mice [5, 7, 52, 63]. All four studies demonstrated that CypD-deficient mitochondria and cells were resistant to Ca2+ and oxidative stress-induced MPT and cell death. Furthermore, we, and others, have shown that CypD-null mice are significantly more resistant to myocardial ischemia-reperfusion injury than their wild type counterparts [5, 46, 52], thereby confirming the previous pharmacological studies. Perhaps most excitingly, recent data in humans undergoing angioplasty has suggested that inhibition of CypD, and therefore the MPT pore, could be of benefit in the clinical setting of ischemia-reperfusion [58]. Thus, of the three components of the original model for the MPT pore, only CypD has survived the genetic testing somewhat intact (Fig. 1).
Mitochondrial phosphate carrier
Given that ANT and VDAC do not appear to be the critical channel-forming units of the mitochondrial pore, we must look elsewhere for alternatives. The most likely candidates are inner membrane exchangers/ion channels that have the capacity to form a pore-like structure (as was originally thought with ANT). Using a proteomic approach, Halestrap's group has recently identified the mitochondrial phosphate carrier (PiC) as a CypD-interacting protein [44, 45]. They then went on to demonstrate that MPT-inducing agents enhanced the PiC–CypD interaction, whereas MPT-blocking compounds reduced it. Most importantly, agents that were able to inhibit mitochondrial Pi transport activity also blocked MPT in isolated mitochondria. Together, these data suggest that the PiC may indeed be a component of the MPT pore. Consistent with this, using a non-biased screen for death-inducing proteins, Alcalá and colleagues reported that overexpression of the PiC induced apoptosis in non-cardiac cells [1]. Obviously the question still remains as to whether the PiC is in fact the pore-forming channel of the MPT pore or whether it represents yet another regulatory protein such as CypD or ANT. Moreover, the agents used to block PiC activity can affect other mitochondrial transporters thus raising the spectre of specificity. Genetic analyses using RNAi or gene-targeting technologies to deplete the PiC are clearly needed before a role for PiC in MPT can be conclusively established.
Pro-apoptotic Bcl-2 proteins
Pro-apoptotic proteins such as Bax or Bak do not appear to constitute the MPT pore per se [19, 72]. However, a considerable area of controversy that deserves to be addressed here is whether such proteins can bind to and regulate the MPT pore. This in of itself is part of the bigger controversy surrounding the mechanisms by which pro-death Bcl-2 proteins induce outer mitochondrial membrane permeabilization and the release of apoptogenic factors. There are currently three main models (Fig. 2): (1) pro-death Bcl-2 members simply form their own protein-permeant pore; (2) pro-death Bcl-2 proteins interact with VDAC to form a protein-permeant channel that specifically permeabilizes the outer membrane; and (3) pro-death Bcl-2 proteins bind to and evoke opening of the MPT pore [26, 42, 64].
Fig. 2.
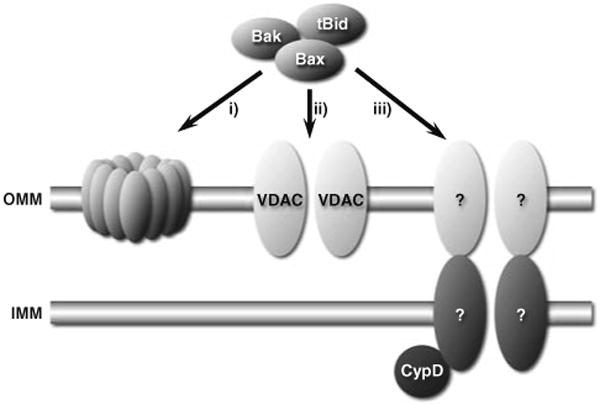
The three main models for how pro-death Bcl-2 proteins induce apoptogen release from mitochondria: (1) pro-death members simply form their own protein-permeant pore; (2) pro-death Bcl-2 proteins interact with VDAC to form a protein-permeant channel that specifically permeabilizes the outer membrane; or (3) pro-death Bcl-2 proteins bind to and evoke opening of the MPT pore. Although there is considerable evidence supporting and refuting, respectively, the first two models, the role of the MPT pore in this process remains controversial
There is a substantial amount of data demonstrating that hetero- or homo-oligermization of Bax and/or Bak with other pro-death proteins is sufficient to form a channel in the outer membrane that is large enough to allow protein efflux [3, 39, 49]. Moreover, despite pro-death proteins being reported to interact with VDAC to form cytochrome c release channels [65, 66], other groups have failed to recapitulate these studies [59, 60], and we have demonstrated that pro-death Bcl-2 proteins can still induce cytochrome c release and apoptosis in mitochondria and cells devoid of VDAC [6].
Up to this point things seem fairly clear-cut. Unfortunately, it starts to get messy when we look at the role of the MPT pore in this process. Kroemer's group has published several studies indicating that pro-death Bcl-2 proteins can directly interact with ANT to elicit MPT and cytochrome c release [10, 47], and others have shown that Bax-induced cell death can be blocked by cyclosporine-A [56, 57]. However, others have shown that Bax-induced cytochrome c efflux can occur independent of any changes in inner membrane permeability and is cyclosporine-insensitive [23, 36, 68]. Such discrepancies maybe dose-related, such that MPT only occurs at high concentrations of the toxic proteins. Perhaps more telling is that in the CypD-null mitochondria, which are more resistant to MPT, Bax and Bid proteins were still able to elicit cytochrome c release to the same extent as normal mitochondria and CypD-null cells were just as sensitive to apoptotic stimuli as wild type cells [5, 52, 63]. These findings suggest that the MPT pore is not essential for the action of cytotoxic Bcl-2 proteins. However, these data do not rule out the possibility that proteins such as Bax and Bak can open the MPT pore through a CypD-independent mechanism, and, unfortunately, a more definitive answer to this question will not be forthcoming until we identify the other components of the MPT pore.
Where do we go from here?
Undoubtedly, the MPT pore is one of the most promising clinical targets for the treatment of myocardial infarction and other cardiac diseases. Indeed, the clinical cyclosporine-A study by Ovize's group [58], suggests that we are tantalizingly close to being able to move from the bench into the clinic. However, this will remain a pipe dream until we solve the jigsaw puzzle that is the protein makeup of the pore. By using what we do know to fish for what we do not, we will hopefully be able to find new pieces of the puzzle and fit them together. As in Halestrap's PiC study, the use of known components of the MPT pore (e.g., CypD) as “bait” coupled with the increased efficiency and availability of proteomic technologies should help accelerate the process. Similarly, genetic screens in yeast and Drosophila can add another powerful dimension to the search. Alternatively, protein or drug library screens may yield candidates in a more unbiased manner. Together, these technologies should provide us with novel contenders that can then be tested from the molecular and biochemical levels all the way through to whole animals for their role in MPT and cardiomyocyte death.
Acknowledgments
We apologize that due to space constraints we have not been able to cite every important study pertaining to this subject. Work in the author's laboratory is supported by grants from the National Institutes of Health (HL094404 and HL092327) and by an American Heart Association Scientist Development Grant (0635134N).
References
- 1.Alcalá S, Klee M, Fernández J, Fleischer A, Pimentel-Muioñs FX. A high-throughput screening for mammalian cell death effectors identifies the mitochondrial phosphate carrier as a regulator of cytochrome c release. Oncogene. 2008;27:44–54. doi: 10.1038/sj.onc.1210600. [DOI] [PubMed] [Google Scholar]
- 2.Akao M, O'Rourke B, Kusuoka H, Teshima Y, Jones SP, Marbán E. Differential actions of cardioprotective agents on the mitochondrial death pathway. Circ Res. 2003;92:195–202. doi: 10.1161/01.res.0000051862.16691.f9. [DOI] [PubMed] [Google Scholar]
- 3.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000;345:271–278. [PMC free article] [PubMed] [Google Scholar]
- 4.Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M. Postconditioning inhibits mitochondrial permeability transition. Circulation. 2005;111:194–197. doi: 10.1161/01.CIR.0000151290.04952.3B. [DOI] [PubMed] [Google Scholar]
- 5.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 6.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 8.Bauer MK, Schubert A, Rocks O, Grimm S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol. 1999;147:1493–1502. doi: 10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belzacq AS, Vieira HL, Kroemer G, Brenner C. The adenine nucleotide translocator in apoptosis. Biochimie. 2002;84:167–176. doi: 10.1016/s0300-9084(02)01366-4. [DOI] [PubMed] [Google Scholar]
- 10.Belzacq AS, Vieira HL, Verrier F, Vandecasteele G, Cohen I, Prevost MC, Larquet E, Pariselli F, Petit PX, Kahn A, Rizzuto R, Brenner C, Kroemer G. Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Res. 2003;63:541–546. [PubMed] [Google Scholar]
- 11.Blachly-Dyson E, Forte M. VDAC channels. IUBMB Life. 2001;52:113–118. doi: 10.1080/15216540152845902. [DOI] [PubMed] [Google Scholar]
- 12.Brower JV, Rodic N, Seki T, Jorgensen M, Fliess N, Yachnis AT, McCarrey JR, Oh SP, Terada N. Evolutionarily conserved mammalian adenine nucleotide translocase 4 is essential for spermatogenesis. J Biol Chem. 2007;282:29658–29666. doi: 10.1074/jbc.M704386200. [DOI] [PubMed] [Google Scholar]
- 13.Cesura AM, Pinard E, Schubenel R, Goetschy V, Friedlein A, Langen H, Polcic P, Forte MA, Bernardi P, Kemp JA. The voltage-dependent anion channel is the target for a new class of inhibitors of the mitochondrial permeability transition pore. J Biol Chem. 2003;278:49812–49818. doi: 10.1074/jbc.M304748200. [DOI] [PubMed] [Google Scholar]
- 14.Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol. 2001;280:H2313–H2320. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 15.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–34799. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 16.Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 17.Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–970. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 18.de Macedo DV, Nepomuceno ME, Pereira-da-Silva L. Involvement of the ADP/ATP carrier in permeabilization processes of the inner mitochondrial membrane. Eur J Biochem. 1993;215:595–600. doi: 10.1111/j.1432-1033.1993.tb18070.x. [DOI] [PubMed] [Google Scholar]
- 19.De Marchi U, Campello S, Szabò I, Tombola F, Martinou JC, Zoratti M. Bax does not directly participate in the Ca2+-induced permeability transition of isolated mitochondria. J Biol Chem. 2004;279:37415–37422. doi: 10.1074/jbc.M314093200. [DOI] [PubMed] [Google Scholar]
- 20.Di Lisa F, Canton M, Menabò R, Kaludercic N, Bernardi P. Mitochondria and cardioprotection. Heart Fail Rev. 2007;12:249–260. doi: 10.1007/s10741-007-9028-z. [DOI] [PubMed] [Google Scholar]
- 21.Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 22.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- 24.Fiore C, Trézéguet V, Le Saux A, Roux P, Schwimmer C, Dianoux AC, Noel F, Lauquin GJ, Brandolin G, Vignais PV. The mitochondrial ADP/ATP carrier: structural, physiological and pathological aspects. Biochimie. 1998;80:137–150. doi: 10.1016/s0300-9084(98)80020-5. [DOI] [PubMed] [Google Scholar]
- 25.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 27.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77:334–343. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 28.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 29.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 30.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60:617–625. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 31.Haworth RA, Hunter DR. Control of the mitochondrial permeability transition pore by high-affinity ADP binding at the ADP/ATP translocase in permeabilized mitochondria. J Bioenerg Biomembr. 2000;32:91–96. doi: 10.1023/a:1005568630151. [DOI] [PubMed] [Google Scholar]
- 32.Hochhauser E, Kivity S, Offen D, Maulik N, Otani H, Barhum Y, Pannet H, Shneyvays V, Shainberg A, Goldshtaub V, Tobar A, Vidne BA. Bax ablation protects against myocardial ischemia-reperfusion injury in transgenic mice. Am J Physiol. 2003;284:H2351–H2359. doi: 10.1152/ajpheart.00783.2002. [DOI] [PubMed] [Google Scholar]
- 33.Imahashi K, Schneider MD, Steenbergen C, Murphy E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ Res. 2004;95:734–741. doi: 10.1161/01.RES.0000143898.67182.4c. [DOI] [PubMed] [Google Scholar]
- 34.Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549:513–524. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jürgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerkela R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB, Force T. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12:908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 38.Kokoszka J, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 40.Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1−/− mitochondria. Biochim Biophys Acta. 2006;1757:590–595. doi: 10.1016/j.bbabio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Kroemer G. The mitochondrial permeability transition pore complex as a pharmacological target. An introduction. Curr Med Chem. 2003;10:1469–1472. doi: 10.2174/0929867033457232. [DOI] [PubMed] [Google Scholar]
- 42.Kroemer G, Galluzzi L, Brenner C. Mitochondrial permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 43.Lazou A, Iliodromitis EK, Cieslak D, Voskarides K, Mousikos S, Bofilis E, Kremastinos DT. Ischemic but not mechanical preconditioning attenuates ischemia/reperfusion induced myocardial apoptosis in anaesthetized rabbits: the role of Bcl-2 family proteins and ERK1/2. Apoptosis. 2006;11:2195–2204. doi: 10.1007/s10495-006-0292-5. [DOI] [PubMed] [Google Scholar]
- 44.Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leung AW, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 46.Lim SY, Davidson SM, Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res. 2007;75:530–535. doi: 10.1016/j.cardiores.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 48.Maulik N, Engelman RM, Rousou JA, Flack JE, 3rd, Deaton D, Das DK. Ischemic preconditioning reduces apoptosis by upregulating anti-death gene Bcl-2. Circulation. 1999;100:II369–II375. doi: 10.1161/01.cir.100.suppl_2.ii-369. [DOI] [PubMed] [Google Scholar]
- 49.Mikhailov V, Mikhailova M, Degenhardt K, Venkatachalam MA, White E, Saikumar P. Association of Bax and Bak homo-oligomers in mitochondria Bax requirement for Bak reorganization and cytochrome c release. J Biol Chem. 2003;278:5367–5376. doi: 10.1074/jbc.M203392200. [DOI] [PubMed] [Google Scholar]
- 50.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 53.Nakayama N, Chen X, Baines CP, Klevitsky R, Zhang H, Jaleel N, Chua BHL, Zhang X, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oliveira PJ, Seica R, Coxito PM, Rolo AP, Palmeira CM, Santos MS, Moreno AJ. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003;554:511–514. doi: 10.1016/s0014-5793(03)01233-x. [DOI] [PubMed] [Google Scholar]
- 55.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 56.Pastorino JG, Chen ST, Tafani M, Snyder JW, Farber JL. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J Biol Chem. 1998;273:7770–7775. doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- 57.Pastorino JG, Tafani M, Rothman RJ, Marcinkeviciute A, Hoek JB, Farber JL. Functional consequences of the sustained or transient activation by Bax of the mitochondrial permeability transition pore. J Biol Chem. 1999;274:31734–31739. doi: 10.1074/jbc.274.44.31734. [DOI] [PubMed] [Google Scholar]
- 58.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 59.Polcic P, Forte M. Response of yeast to the regulated expression of proteins in the Bcl-2 family. Biochem J. 2003;374:393–402. doi: 10.1042/BJ20030690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Priault M, Chaudhuri B, Clow A, Camougrand N, Manon S. Investigation of bax-induced release of cytochrome c from yeast mitochondria permeability of mitochondrial membranes, role of VDAC and ATP requirement. Eur J Biochem. 1999;260:684–691. doi: 10.1046/j.1432-1327.1999.00198.x. [DOI] [PubMed] [Google Scholar]
- 61.Regula KM, Kirshenbaum LA. Apoptosis of ventricular myocytes: a means to an end. J Mol Cell Cardiol. 2005;38:3–13. doi: 10.1016/j.yjmcc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 62.Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr. 2005;37:129–142. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- 63.Schinzel A, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharpe JC, Arnoult D, Youle RJ. Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta. 2004;1644:107–113. doi: 10.1016/j.bbamcr.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 65.Shimizu S, Shinohara Y, Tsujimoto Y. Bax and Bcl-xL independently regulate apoptotic changes of yeast mitochondria that require VDAC but not adenine nucleotide translocator. Oncogene. 2000;19:4309–4318. doi: 10.1038/sj.onc.1203788. [DOI] [PubMed] [Google Scholar]
- 66.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 67.Toth A, Jeffers JR, Nickson P, Min JY, Morgan JP, Zambetti GP, Erhardt P. Targeted deletion of Puma attenuates cardiomyocyte death and improves cardiac function during ischemia-reperfusion. Am J Physiol. 2006;291:H52–H60. doi: 10.1152/ajpheart.01046.2005. [DOI] [PubMed] [Google Scholar]
- 68.von Ahsen O, Renken C, Perkins G, Kluck RM, Bossy-Wetzel E, Newmeyer DD. Preservation of mitochondrial structure and function after Bid- or Bax-mediated cytochrome c release. J Cell Biol. 2000;150:1027–1036. doi: 10.1083/jcb.150.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vyssokikh MY, Katz A, Rueck A, Wuensch C, Dorner A, Zorov DB, Brdiczka D. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem J. 2001;358:349–358. doi: 10.1042/0264-6021:3580349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walther T, Tschöpe C, Sterner-Kock A, Westermann D, Heringer-Walther S, Riad A, Nikolic A, Wang Y, Ebermann L, Siems WE, Bader M, Shakibaei M, Schultheiss HP, Dörner A. Accelerated mitochondrial adenosine diphosphate/adenosine triphosphate transport improves hypertension-induced heart disease. Circulation. 2007;115:333–344. doi: 10.1161/CIRCULATIONAHA.106.643296. [DOI] [PubMed] [Google Scholar]
- 71.Wang P, Heitman J. The cyclophilins. Genome Biol. 2005;6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 73.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 74.Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zamora M, Granell M, Mampel T, Vinas O. Adenine nucleotide translocase 3 (ANT3) overexpression induces apoptosis in cultured cells. FEBS Lett. 2004;563:155–160. doi: 10.1016/S0014-5793(04)00293-5. [DOI] [PubMed] [Google Scholar]
- 76.Zhao ZQ, Nakamura M, Wang NP, Wilcox JN, Shearer S, Ronson RS, Guyton RA, Vinten-Johansen J. Reperfusion induces myocardial apoptotic cell death. Cardiovasc Res. 2000;45:651–660. doi: 10.1016/s0008-6363(99)00354-5. [DOI] [PubMed] [Google Scholar]
- 77.Zheng Y, Shi Y, Tian C, Jiang C, Jin H, Chen J, Almasan A, Tang H, Chen Q. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene. 2004;23:1239–1247. doi: 10.1038/sj.onc.1207205. [DOI] [PMC free article] [PubMed] [Google Scholar]