

Summary
Cobra venom factor (CVF) is a functional analog of human complement component C3b, the active fragment of C3. Similar to C3b, in human and mammalian serum, CVF binds factor B, which is then cleaved by factor D giving rise to CVFBb complex that targets the same scissile bond in C3 as the authentic complement convertases, C4bC2a and C3bBb. Unlike the latter, CVFBb is a stable complex and an efficient C5-convertase. We solved the crystal structure of CVF, isolated from Naja naja kouthia venom, at 2.6 Å resolution. The CVF crystal structure, an intermediate between C3b and C3c, lacks the TED domain and has CUB domain in identical position to that seen in C3b. The similarly positioned CUB and slightly displaced C345c domains of CVF could play a vital role in the formation of C3-convertases by providing important primary binding sites for factor B.
Introduction
Cobra venom factor (CVF), a disulfide-linked three-chain 149 kDa nontoxic glycoprotein, is an activator of mammalian complement present in cobra venom. It is a functional homologue of complement component C3b, an active fragment of C3. Similar to C3b, CVF binds to serum factor B in the presence of Mg2+. The resulting complex has weak C3-convertase activity (Xu et al., 2001) but subsequent cleavage of factor B by factor D gives rise to CVFBb, an efficient C3-convertase targeting the same scissile bond of C3 as authentic complement convertases (Figure 1A). Although the closely related convertases C3bBb and CVFBb share identical restricted substrate specificity residing in their Bb subunit, they differ in other biochemical properties. Interesting differences include: 1) C3bBb is very short-lived with a half-life of 1.5 min at 37°C (Medicus et al., 1976; Pangburn and Müller-Eberhard, 1986), whereas CVFBb is rather stable with a half-life of 7 hours at room temperature and more than 20 days at 4°C (Vogel and Müller-Eberhard, 1982), though both C3bBb and CVFBb show spontaneous decay-dissociation into their two respective subunits, which abolishes their enzymatic activity. 2) Factor H disassembles C3bBb (Pangburn et al., 1977) and serves as cofactor for the subsequent proteolytic inactivation of C3b by factor I (Whaley and Ruddy, 1976), whereas CVFBb and CVF are completely resistant to factors H and I (Alper and Balavitch, 1976; Lachmann and Halbwachs, 1975). 3) C3bBb requires an additional C3b molecule to form an efficient C5-convertase (Daha et al., 1976; Vogt et al., 1978), whereas CVF, from certain cobra species like Naja naja kouthia forms the CVFBb complex that can efficiently cleave C5 (DiScipio et al., 1983; von Zabern et al., 1980).
Figure 1. Similarities and differences between CVF and C3b.
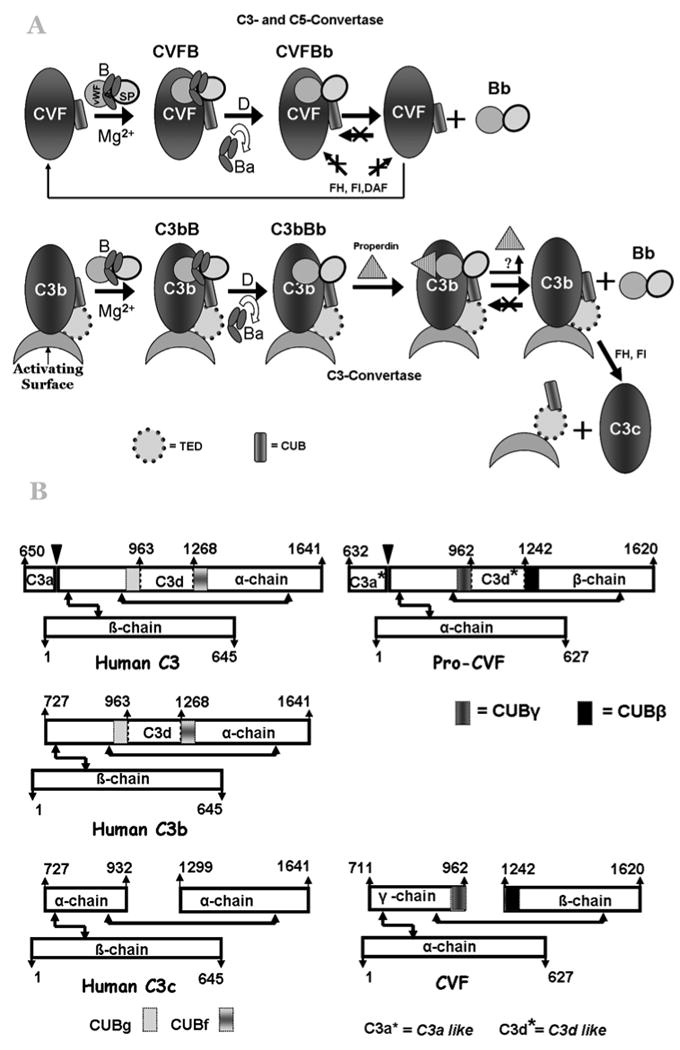
A) Cartoon representation of similarity and differences in the C3-convertase formation by CVF and C3b. Abbreviations used in the figure: CVF - Cobra Venom Factor; B - factor B; vWFA - von Willebrand factor type A domain; SP - serine protease domain; FH - factor H; FI - factor I; DAF – decay accelerating factor; Ba - N-terminal smaller fragment of B that consists of three complement control protein (CCP) modules; Bb - C-terminal fragment of B, consists of vWFA and SP domains; C3b - the active proteolytic product of C3; C3c - the inactive proteolytic product of C3, CUB - Complement C1r/C1s, Uegf, Bmp1 domain; TED - thioester-containing domain. B) Schematic representation of chain structures of Pro-CVF and CVF, and its homologous C3, C3b and C3c. Pro-C3 is processed into mature two-chain C3 by the removal of four arginine residues (646-649). The pro-CVF is processed into its mature three-chain CVF (α-chain, β-chain, and γ-chain) by proteolytic removal of C3a (632-710) and C3d (963-1241)-like domains and four arginine residues (628-631). The arrows with two heads indicate disulfide bridges.
Pro-C3 (1-1641) is processed into mature two-chain C3 (115 kDa α-chain (650-1641) and 70 kDa β-chain (1-645)) by the removal of four arginine residues (646-649), whereas pro-CVF (1-1620) is processed into its mature three-chain CVF (70 kDa α-chain (1-627), 48 kDa β-chain (1242 -1620), and 32 kDa γ-chain (711-962)) by proteolytic removal of C3a- (632-710) and C3d- (963-1241) like domains and four arginine residues (628-631) (Figure 1B). The pro-CVF contains 27 cysteine residues and their distribution is identical to those in the C3 (Fritzinger et al., 1994). However, there are differences in carbohydrate content and their sites. C3 has only 1.7% carbohydrate content (Tomana et al., 1985), whereas CVF has 7.4% (w/w) (Fritzinger et al., 1994; Gowda et al., 1992; Grier et al., 1987; Vogel and Müller-Eberhard, 1984). Studies have shown that the oligosaccharides of CVF are not required for its complement-activating function (Gowda et al., 2001; Gowda et al., 1994).
It has been suggested that the CVF structure may resemble the inactive C3c fragment of C3 (Vogel et al., 1984). However, as expected from the functional homology, CVF and C3b molecules exhibit several similarities including amino acid compositions, isoelectric points, circular dichroism spectra and secondary structures, electron microscopic ultrastructures, immunological cross-reactivity in the polypeptide and carbohydrate moieties and amino-terminal sequences (Vogel et al., 1984). On the other hand, structural differences must exist between these two molecules dictating different properties of their respective convertases. Similar to C3, Pro-CVF also contains a thioester site in its C3d-like region (Cys971 and Glu974), but this region is removed during the processing toward CVF. Hence, the predicted C3c-like structure and observed C3b-like function of CVF combined with a C3c-like resistance to inactivation by the control proteins factors H and I need to be reconciled.
Formation of stable C3/C5 -convertase by CVF leads to severe depletion of serum complement activity in both in vitro and in vivo. This property has been exploited in numerous animal studies to investigate the role of complement in host defense, immune response and pathogenesis of inflammatory diseases. CVF has also been used for targeted complement activation by coupling it to monoclonal antibodies or other targeting moieties with specificity for targeted cells, such as cancer cells (Grier et al., 1987; Vogel and Müller-Eberhard, 1981). CVF has thus also become a gold standard for assessing the inhibitory effect of experimental anti-complementary agents (Hughes et al., 1992; Morgan and Harris, 2003; Morganroth et al., 1989; Morganroth et al., 1986).
The three-dimensional structure of several complement proteins have been determined, particularly those of the closely related human C3 (Janssen et al., 2005), C3b (Janssen et al., 2006; Wiesmann et al., 2006), C3c (Janssen et al., 2005), C5 (Fredslund et al., 2008), bovine C3 (Fredslund et al., 2006), and insect thioester protein (TEP) (Baxter et al., 2007). However, the crystal structure of CVF, a protein that has been used extensively in complement research both in vitro and in vivo experiments, has not been reported. Here we present the crystal structure of de-glycosylated CVF (dCVF) and we discuss its structural and functional similarities and differences from C3b. In contrast to C3b crystal structure (Janssen et al., 2006), the TED (thioester-containing domain) domain is missing in the present dCVF crystal structure, and its CUB (complement C1r/C1s, Uegf, Bmp1) domain is positioned in identical position and the flexible C345c domain is seen shifted towards the CUB domain.
Rseults and Discussion
CVF Crystal Structure
The crystal structure of CVF was determined by molecular replacement methods and refined using 2.6 Å resolution diffraction data. The crystal structure of C3c (Janssen et al., 2005) and its individual domains were used as a starting search models (see methods for details). The three chains of CVF together form eleven domains (Figure 2A & 2B) with overall dimensions of 150 Å × 76 Å × 66 Å and the domain arrangement is similar to C3b (Janssen et al., 2006). In contrast to C3b, which displays one domain made of two chains, two CVF domains are formed by two chains. One domain is composed of regions from the α- and γ- chains, while the other is comprised of γ- and β-chain regions.
Figure 2. The crystal structure of CVF.
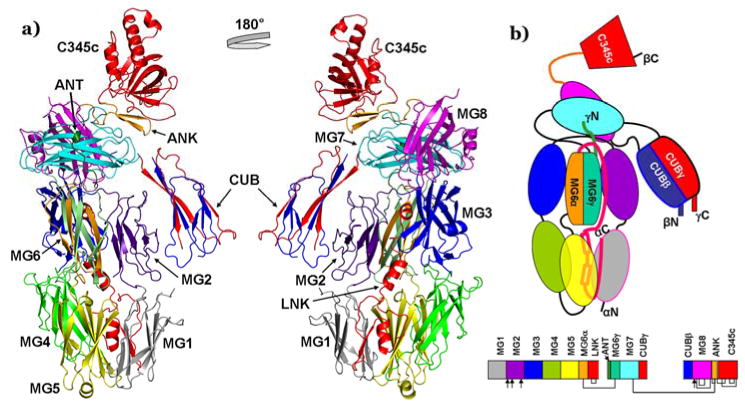
A) The ribbon diagram of overall CVF crystal structure (left) and with its 180° rotated view (right). The α-chain forms MG1 (grey), MG2 (indigo), MG3 (blue), MG4 (green), MG5 (yellow), MG6α (orange), and LNK (red) domains. The γ-chain forms α'NT (dark green), MG6γ (light green), MG7 (cyan), and CUBγ (red) domains. The β-chain forms CUBβ (blue), MG8 (magenta), ANK (orange), and C345c (red) domains. B) Schematic representation of CVF domains (colored as in the ribbon diagram of crystal structure 2A). The chain ends are labeled. The connected lines and arrows indicate disulfide bridges and glycosylation sites, respectively. The secondary structure details and domain boundaries are presented in supplementary Figure 1.
The core of the CVF structure is formed by eight macroglobulin (MG1-8) domains. MG1-5 domains are formed by the N-terminal residues of the α-chain, while MG7 and MG8 domains are formed by residues from the γ-chain and β-chain, respectively. The MG6 domain is formed by residues from both the α-chain (MG6α) and γ-chain (MG6γ). The six MG domains (MG1-MG6) form the so-called key β-ring, the body of the structure. The α-chain is linked to the γ-chain via an inter-chain disulfide bridge (α522C- γ779C) near the MG5-MG6 interface. The α'NT-like segment is formed by the N-terminal residues of the γ-chain and a linker (LNK) region formed by residues of the α-chain traverses the β-ring. The LNK region consists of three helices in an extended configuration and one β-strand that align with the first strand of MG1. The C-terminal end of LNK is stabilized by a disulfide bond (587C-622C) at the end of the second helix. The CUB domain formed by residues from both the γ-chain (CUBγ) and β-chain (CUBβ) is inserted between the MG7 and MG8 domains. Finally, the C-terminal C345c domain formed by residues of the β-chain, containing six intra chain disulfide bridges is covalently linked to MG8 via an anchor (ANK) region and to MG7 by an inter-chain disulphide bond (γ835C-β1470C). The ten domains of CVF (MG1-8, LNK, C345c) are arranged similar to C3c, while the CUB domain is placed like an extended arm and is located in a way similar to that seen in the C3b structure (Janssen et al., 2006), even though the thioester-containing domain (TED) is absent. In summary, the core of CVF is similar to C3b and C3c, but the CUB and C345c domains of CVF, which are implicated in ligand binding, differ in composition and location, as the latter is rotated toward the former domain by about 38° when compared to C3b and C3c (Figure 3). In all the published structures of human C3, C3b, C3c, C5, bovine C3, and TEP the MG1-6 (β-ring core) domains and LNK form a stable platform, although subtle differences exist among them; for example the β-ring core displays a more open arrangement in the TEP structure (Baxter et al., 2007). Superposition of the β-ring MG1-6 domains and LNK of CVF on related C3c and C3b structures yields an rmsd of 1.6 Å for 598 common Cα atoms. Difference electron density map (>7σ) revealed a calcium binding site with six ligands at the center of the key β-ring near MG5-MG6 interface. The coordination for this calcium ion comes from MG5 (side chains of Asp517, Asp520, and main chain carbonyl oxygen atoms of Val518 and Pro494), LNK region (side chain of Glu581 through a water molecule) and one weakly occupied water molecule. The average bond length of the calcium ion to its ligand O atoms is 2.35 Å.
Figure 3. Comparison of CVF with C3c and C3b structures.
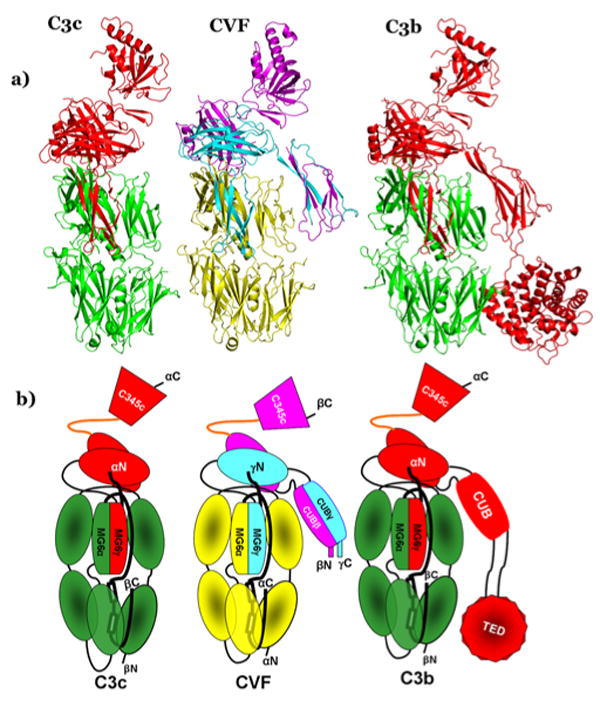
A) Ribbon diagrams of C3c (left), CVF (middle) and C3b (right). C3c and C3b are made of two chains, β-chain (green), and α-chain (red): whereas, CVF is composed of three chains α (yellow), β (magenta), and γ (cyan). B) Schematic depiction of domain organizations of C3c, CVF and C3b (colored according to chains as in the ribbon diagram 3A).
Comparison with C3b, and C3c
Since CVF is functionally similar to C3b and suggested to have C3c-like structure, we restrict comparisons of CVF to these proteins. The structures of human C3, Bovine C3, C5 and TEP have suggested that all members of the α2M super family have similar architecture and their comparison with active C3b and inactive C3c has been discussed previously. The CVF structure displays all C3b domains except the TED domain (Figure 3B). The eight homologous (MG1-8) domains that represent the core of CVF align with the corresponding domains of C3b quite well (see supplementary Table 1). For most of them there are small variations in orientation, translation and rms deviation (Figure 3A and 3B). The MG1 domain of CVF displays disordered EF loop similar to that of C3b MG1, but has density for additional 3 residues in the C-terminal end of the loop. The MG2 domain of CVF has a disordered BC loop, though this loop is surrounded by MG1, MG5, MG6 and LNK domains where its C-terminal part is exposed to the surface. However, the corresponding loop of C3b is ordered. The MG3 domain of CVF is very similar to that of C3b and density is seen for the EF loop in CVF MG3, unlike its absence in the C3b structure. The CC' loop of the CVF MG4 domain deviates toward the core of the domain compared to C3b. The FG loop of the MG5 domain is 5 residues shorter than the C3b MG5, where it is extended toward the MG4-MG5 linker.
The MG6 domain is made up of two chains in both CVF and C3b. The MG6 of CVF (Figure 4A) is formed by the α- and γ- chain s, whereas in C3b, it is formed by the α- and β- chains. The similarly positioned MG7 domain in C3b and CVF is connected to the anchor (ANK) region via a disulfide bridge (γ835C-β1470C) in CVF and (β851C-β1491C) in C3b. The MG8 domain of CVF is formed by the β-chain similar to C3b. A structurally variable segment that is not part of the MG core fold is inserted between βC and βC' of the MG fold and has been observed in the MG8 domain of the C3b and C3c structures with conformation β-α-α motif. This segment has also been observed in the structure of the isolated RBD region of α2M and is considered important for the α2M receptor binding (Jenner et al., 1998). This segment in C3b is thought to be involved in binding of properdin that stabilizes the C3bBb, C3-convertase complex. Notice that a β-α-β motif is observed for this segment in the C3 structure to which properdin is not binding. In CVF, the conformation of this segment is a β-α motif followed by an extended loop and a β-sheet of two short anti-parallel β-strands. The LNK and ANK regions of CVF are similarly positioned to those in C3b and C3c. The ANK region of CVF connects the C345c domain to the core of the molecule via a peptide bond to MG8 and via a disulfide bond (835C-1470C) to MG7 as in C3b. This disulfide bond is an inter-chain bond in CVF linking β- and γ- chains. The ANK region of CVF has an additional intra-chain disulfide bond (1463C-1468C) as observed in C3b. The region undergoes a drastic conformational change during the conversion of C3 to C3b from an α-helix to a β-hairpin, apparently not restricted by the internal disulfide bond (Janssen et al., 2006; Janssen et al., 2005).
Figure 4. Topology diagrams of intertwined domains of CVF and C3b.
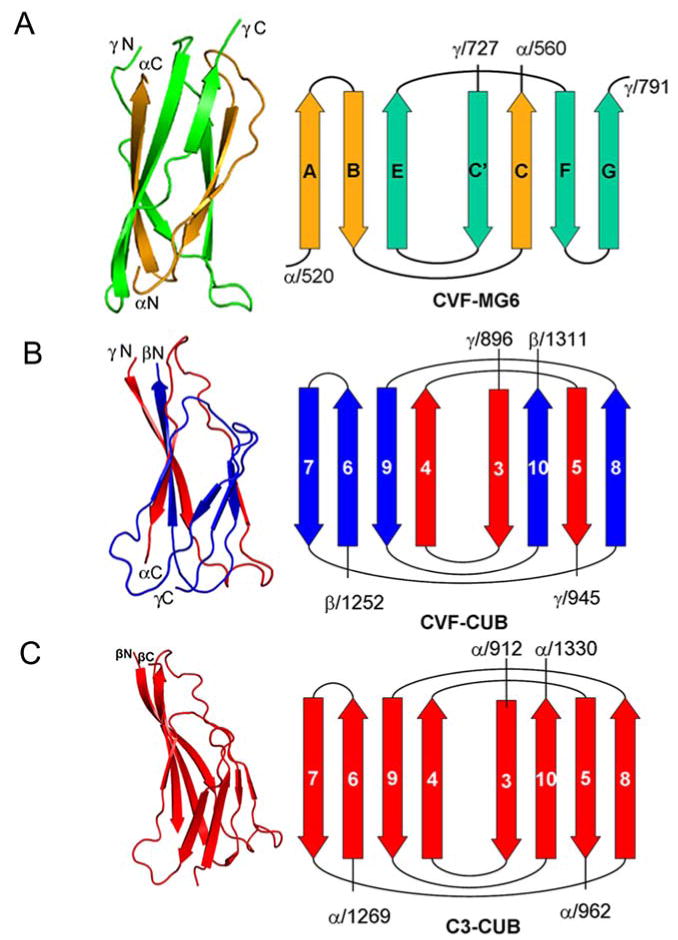
A) CVF MG6 is made of the α- (orange), and γ-chains (green). The terminals of the domains are labeled. B) CVF CUB is composed of the γ- (red), and β-chains (blue). C) C3b CUB domain is composed of two distinct parts of α-chain.
CUB domain
The CUB domain and its conformation deserve special attention. The CUB domain in CVF is formed by the segments from γ- (896-945) and β- (1252-1311) chains (Figure 4B), and in C3b, it is formed by two distinct parts (913-963; 1270-1329) of the α-chain (Janssen et al., 2006)(Figure 4C). The C-terminal segment after MG7 domain in γ-chain and the N-terminal segment preceding MG8 domain of β-chain constitute the CUB domain in CVF, whereas in C3b the CUB domain, with TED domain inserted between its β5-β6-loop connects MG7 to MG8 of α-chain. There is a glycosylation site at position 917 on the CUB domain of C3b, which is absent in CVF. The electron density is clear for the core β-strands of the CVF CUB domain, although the connecting loops have weak density. Its conformation and position relative to the remaining domains are similar to those of the CUB domain of C3b (Janssen et al., 2006), except that the CVF CUB domain is translated by 3 Å with 15.5° rotation compared to the C3b CUB domain. Maybe because of its solvent accessibility and freedom from packing constraints, the CUB domain in CVF exhibits higher temperature factors with poorer electron density, especially for many side chains compared to all other domains. However, disordered regions are often functional and may play a role in the assembly of macromolecular arrays (Dyson and Wright, 2005).
C345c domain
The C345c domain at the C-terminal end is common to the complement proteins C3, C4 and C5, but absent from other members of the α2M family. The C345c domain of CVF is formed by the C-terminal region of the β-chain; it is connected to MG8 covalently via the ANK region and to the MG7 domain via a disulfide bridge as in C3b. Superposition of the C345c domain structure on that of C3b and C3c yields rmsd of 1.0 Å for 129 common Cα atoms. This domain is translated by 5.2 Å and 7.7 Å and rotated by 38.5° and 36.9° toward the CUB domain, compared to C3b and C3c, respectively. The electron density for this domain is relatively weak compared to other domains but better defined than the CUB domain. This may be partly due to weak crystal lattice contacts and relative mobility that is also observed in the C3c, C3b, C3, and C5 structures.
Convertase formation
The catalytic activity of the C3-convertases C3bBb and CVFBb is mainly due to the proteolytic apparatus provided by the serine protease factor B, the primary ligand for CVF. Many studies have investigated the factor B interaction sites on C3 and C3b, but not on CVF. Therefore, the binding site for factor B on CVF has to be defined in comparison to C3b. Multiple C3b sites have been proposed for factor B binding. Residues 730DEDIIAEENT739 at the α'NT region of C3b that include four acidic residues (730DE and 736EE) have been indicated as the major binding site by peptide binding and mutagenesis studies (Becherer et al., 1992; Clemenza and Isenman, 2000; Taniguchi-Sidle and Isenman, 1994). Analogously, it has also been proposed that the NH2 terminus of the C4 α'-chain is a binding site for C2 (Pan et al., 2000). In Bovine and human C3, the α'NT region is buried but becomes exposed on the surface of C3b (Janssen et al., 2006; Wiesmann et al., 2006). In the CVF structure, the structurally equivalent region 714EDGFIADSDI723 is found at the N-terminal end of the γ-chain occupying the same position as the α'NT region of C3b and C3c (Figure 5). Thus, two clusters of acidic residues on C3b are also present in CVF at the N-terminal end of the γ-chain (714DE and 720DSD). Additionally, CVF has one more acidic acid cluster at the very beginning of the γ-chain (711DDN), while the equivalent region of C3b is 727SNL. The above two clusters of acidic residues present in C3b are also present in C3c. However, C3c does not bind factor B, suggesting that additional sites are needed for factor B binding. The efficient binding of factor B by CVF, which is structurally similar to C3c, except for the presence of the CUB domain, suggests a role for CUB in factor B binding.
Figure 5. Proposed factor B binding sites.
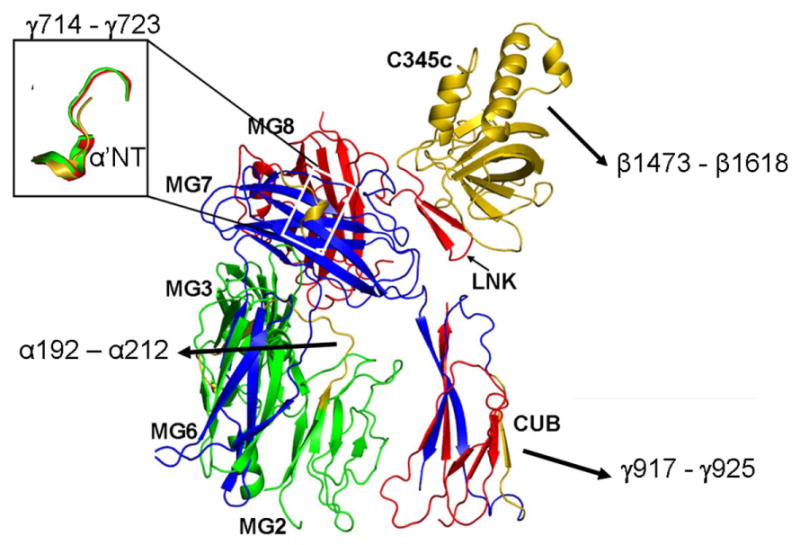
These sites are indicated (in gold) on the structure of CVF (only the top half of the molecule is shown). The small panel depict the CVF site α'NT (gold) in comparison with the corresponding sites of C3b (green) and C3c (red).
O'Keefe et al. (1988) identified another possible site for factor B binding when they characterized the C3o fragment produced by cleavage of C3 by a cobra venom protease (O'Keefe et al., 1988). C3o resembles C3c, but unlike C3c it can form a C3-convertase with factor B. N-terminal sequencing and molecular weight comparisons of C3c and C3o suggested that residues 933EGVQKEDIPPA943 at the N-terminus of C3dg might be responsible for the difference in activity. In the C3b structure, the above said region forms β4 strand and the flanking loops of the CUB domain. The structurally corresponding region 917GGTQLEVIKARK927, more polar and charged, in the CVF (Figure 5) is located at the C-terminal end of the γ-chain on the β4 strand of the CUBg half of CUB. The CUB domain is extended downwards from the head during C3b formation (Janssen et al., 2006) and in CVF it is located in a similar position, except for the small deviation discussed above, suggesting similar binding sites for factor B.
It has been suggested that residues 1496-1641 at the C-terminal of the C345c domain are involved in the factor B binding (Kolln et al., 2005). The suggestion was based on experiments replacing various segments of C345c with the corresponding regions of CVF, which resulted in enhanced lifetime of the C3bBb convertase complex. However, the C345c domain of CVF is translated and rotated compared to C3b and C3c as mentioned above (Figure 3). Hence, in addition to the specific residue differences between C3b and CVF in the C345c domain, its relative position to the CUB domain may also have an affect on the stability of the C3-convertase complex.
In C3b and CVF, the three sites, α'NT, CUB and C345c are located on the same side of molecule and near to each other, like corners of a triangle. In CVF, the α'NT and C345c are separated by 40 Å while CUB is placed 60 Å away from each of the other two domains. Factor B consists of 739 amino acids (MW 90kDa) that form three modules. The N-terminal smaller module Ba, which is removed during convertase formation, consists of three complement control protein (CCP1-3) domains (Figure 1A). The C-terminal major fragment Bb consists of a von Willebrand factor A (vWFA) domain and a serine protease (SP) domain (Figure 1A). Proposed C3b binding sites are found on all three CCPs of Ba (Hourcade et al., 1995), the MIDAS of vWFA domain (Hinshelwood et al., 1999; Hourcade et al., 1999; Sánchez-Corral et al., 1990) and the SP domain (Lambris and Müller-Eberhard, 1984). Analogously, multiple C4b-binding sites have also been proposed on C2 (Laich and Sim, 2001), located on all three CCP domains (Xu and Volanakis, 1997), and the MIDAS motif of the vWFA domain (Horiuchi et al., 1991). Various studies have shown an acidic residue of the ligand that completes the coordination sphere of the divalent ion bound at the MIDAS (Emsley et al., 2000; Shimaoka et al., 2003), and it is expected that such a residue to be located in the α'NT. The conformational changes induced in the vWFA and SP domains of C3b- or CVF-bound factor B upon removal of Ba are irreversible. Once dissociated, Bb can no longer associate with C3b. The distance (∼44 Å in FB and ∼55 Å in Bb) between the MIDAS and the positively charged L2-loop of the SP domain of Bb corresponds to the above sites on CVF. Therefore, two of the above sites of CVF could be the binding sites for flexible SP and vWFA domain while the third site could be the binding site for CCP modules during the initial convertase formation.
Two additional sites for factor B binding on C3b have been proposed. The first, analogous to a C2-binding site on C4 (Inal and Schifferli, 2002; Oh et al., 2003), is formed by the βG strand of MG2, βA strand of MG3 and the MG2-MG3 linker loop. The corresponding residues 192-212 in CVF (Figure 5) and C3b (200-220) are on the buried side of the CUB domain and relatively inaccessible. Conformational changes in the β-ring may make this site accessible. Second site that has been implicated is present on the C3d domain of C3b (Koistinen et al., 1989; Lambris et al., 1988); however CVF does not carry a C3d-like domain, thus excluding this possibility.
Regulation of Convertase
Several regulatory proteins (factor H, complement receptor 1 (CR1), decay-accelerating factor (DAF), membrane co-factor protein (MCP), factor I) prevent assembly and dissociate the C3bBb complex and/or act as co-factors for proteolysis of C3b by factor I. Fishelson (Fishelson, 1991) suggested that the 744EFPESWLWNVE754 region at the α'NT of C3b, contributes to factor H and CR1 binding. Two acidic residues, 744E and 747E in that segment located in the MG6 domain were found important for factor H and CR1 binding. In CVF, the corresponding segment 728DFPKSWLWLTK738 has one acidic residue 728D while the second one is replaced by a lysine 731K, which possibly explains the inability of CVF to bind factor H and CR1. Moreover, residues 1187-1249 of the C3b α-chain provide at least one additional site for factor H binding (Janssen et al., 2006; Jokiranta et al., 2000; Koistinen et al., 1989; Lambris et al., 1988). This segment forms helices α9-α11 and neighboring loops in the TED domain. Since the TED domain is not present in the CVF, factor H loses an additional potential binding site. Consequently, the decay-acceleration of CVFBb complex by factor H and CR1 is prevented contributing to its stability in serum.
Properdin, a 45 kDa protein binds to the C3-convertase C3bBb significantly extending its half-life. A binding site for properdin has been proposed to be at residues 1402-1435 in the C-terminal of the C3 α-chain, which forms the structurally variable MG8 β-α-β motif converted to a β-α-α motif in C3b and C3c. The corresponding site in the CVF is at residues 1381–1414 of the β-chain. This loop is exposed as in C3b and retains its hydrophobic nature, but deviates in secondary structure as the segment displays a loop element with a short β-sheet after the β-α motif.
Conclusion
It appears that the CUB domain, which in CVF occupies the same position as in C3b, probably plays a vital role in the formation of C3- convertases CVFBb and C3bBb by providing an important primary binding site for factor B. The C345c domain exhibits considerable differences in its position and orientation between CVF and C3b, suggesting a possible role for the higher stability of the CVFBb complex. However, the position of C345c domain seems to be influenced by molecular packing in many of the similar protein structures. No significant differences are observed among the CVF, C3b, and C3c structures in the position and composition of the α'NT and the same hold true for the MG2-MG3 interface; hence these regions are expected to play a minor role in the formation and stability of the convertases. The segment 730DEDIIAEENT739 in the α'NT may provide a binding site but that may not be enough for factor B and Bb binding, since C3c, which does not bind factor B, also has the α'NT region at the same position. Moreover, C3o lacks residues 727-736 of the α'NT but supports C3-convertase formation, perhaps because it retains residues 933-942 of the CUB domain. In both CVF and C3b crystal structures these residues are present on the CUB domain surface at the C-terminal end of a β-strand that is physically closer to C345c domain. If this stretch of residues has a role in factor B binding (O'Keefe et al., 1988), the charged nature of this β-strand in CVF in contrast to the hydrophobic environment present in C3b (Janssen et al., 2006), may suggest some bearing on the binding and affinity of CVF towards Bb. The predicted factor B binding site (Fishelson, 1991) in the C3d domain could be an additional binding site but probably is not essential because of its absence in CVF. Factor B may interact with α'NT, CUB and C345c domains through multiple binding sites, and at least two of them, one in Ba and another in the middle vWFA domain, have been identified. Resistance of CVF to regulatory proteins is apparently due to the absence of the C3d domain, which provides binding site(s) for them. In conclusion the presence of the CUB domain and the absence of the TED domain in CVF, when compared to C3c and C3b, reveals some possible reasons why the CVFBb complex is more stable and escapes from the regulatory proteins during complement activation in plasma.
Experimental Procedures
Protein purification
CVF was isolated from lyophilized Naja naja kouthia cobra venom using published methods (Vogel and Müller-Eberhard, 1984; Sharma et al., 2001). Briefly, 200 mg of cobra venom was dissolved in 1 ml of 50 mM Tris-HCL, 100 mM NaCl pH 7.5 and mixed at room temperature for 10 minutes. The sample was then centrifuged at 18 k for 10 min and the cleared supernatant was applied to a Sephacryl S-200 (16/60) column (Pharmacia) in the same buffer. The second elution peak containing the CVF was collected and dialyzed overnight against 50 mM Tris-HCL pH 8.5. This was subsequently applied to a DEAE Bioscale column (Bio Rad) and eluted with a linear gradient to 1 M NaCl (Sharma et al., 2001). Fractions containing CVF were combined for deglycosylation. The deglycosylation was carried out using PNGase F (New England BioLabs Inc.) according to the manufactures instructions. The deglycosylated CVF (dCVF) was further purified by gel-filtration chromatography and dialyzed against 25 mM phosphate buffer pH 6.5 and concentrated to 7 mg/ml. Purity of dCVF was examined by SDS-PAGE analysis. Activity was assayed and confirmed by hemolytic assay (Vogel and Müller-Eberhard, 1984).
Crystallization and data collection
Rod shaped dCVF crystals were obtained through sequential seeding in hanging drops equilibrated against 1 ml reservoir solution containing 13% PEG 3350, 10 mM MES pH 6.8, 5 mM CaCl2, and 200 mM NH4Cl at 20°C. Crystals were flash-cooled in liquid nitrogen using 30% Ethylene Glycol plus the well solution containing 15% PEG 3350 as a cryoprotectant. The dCVF crystals diffracted to 2.6 Å on the SER-CAT BM beam line at APS (Advanced Photon Source, Chicago). However, crystals were sensitive and suffered from radiation damage and anisotropic decay. Hence, multiple crystals were used for data collection. Processed and scaled with HKL2000 (Otwinowski et al., 2003), diffraction data from three crystals were scaled to obtain a single and suitable data set for the structure determination (see Table 1 for statistics). Data set is complete to 2.8 Å resolution and completeness dropped to 75% in the last resolution shell. Crystals exhibited space group P21212 with one molecule per asymmetric unit.
Table 1. Data collection statistics of dCVF native crystala.
| Space group | P21212 |
| a (Å) | 134.14 |
| b (Å) | 151.14 |
| c (Å) | 76.79 |
| Resolution (Å) | 50.0-2.6 (2.69-2.60) |
| Rmergeb (%) | 0.08 (0.31) |
| Unique reflections | 43934 |
| Completeness (%) | 92.8 (76.6) |
| Redundancy | 7.1 (2.1) |
| I/σI | 21.7 (2.0) |
Values in the parentheses refer to the outermost resolution shell.
Rmerge = Σhkl Σj | Ij − <I> | / Σhkl Σj Ij, where <I> is the mean intensity of j observations from a reflection hkl and its symmetry equivalents.
Structure determination
The structure was determined by molecular replacement methods with PHASER (McCoy et al., 2005; Read, 2001; Storoni et al., 2004), using individual domains of C3c as starting search models. The key MG1-MG6 β-ring was first located and the remaining domains of C3c, i.e., MG7, MG8 and C345C were added one after another. After placing all other domains by PHASER, search and building of the CUB domain was carried out with the help of CUB domain model of C3b and sequential difference electron density maps. Electron density for the CUB domain was weak initially but improved gradually over multiple cycles of model building done with the help of difference and omit maps. Rigid body refinement was done using CNS (Brunger et al., 1998) and model fitting with COOT (Emsley and Cowtan, 2004) and final refinement using REFMAC (1994). After the final cycle of refinement, the model had Rwork value of 24.7% and Rfree of 29.7% (see Table 2 for statistics and supplementary Figure 2 for the quality of the electron density maps).
Table 2. Refinement statistics for dCVF structure.
| Resolution range (Å) | 50 - 2.6 |
| Rwork/Rfree (%)c | 24.7/29.7 |
| Rmsd for bonds (Å) | 0.011 |
| Rmsd for angles (°) | 1.323 |
| No. of Non-Hydrogen atoms | 9158 |
| No. of water molecules | 80 |
| No. of metal ions (Calcium) | 1 |
| Wilson B-factor (Å2) | 68.7 |
| Average B-factor (Å2) | |
| Overall | 57.4 |
| Protein atoms | 57.6 |
| Solvent atoms | 43.0 |
| Unique reflections | 41713d |
| Ramachandran plot (favored/allowed/disallowed) (%) |
85.7/14.1/0.2 |
R-work = Σhkl | |Fobs| − |Fcalc| | / Σhkl |Fobs|, where Fo and Fc are measured and calculated structure factors respectively. Rfree was calculated using 5% of data that were omitted from refinement.
the count excludes 5% test data (2221 reflections) set used in the refinement.
Supplementary Material
Acknowledgments
The project is supported by NIH grant AI1064815 to S.V.L.N.
Footnotes
Accession Number: Atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank under the ID code 3FRP.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Alper CA, Balavitch D. Cobra venom factor: evidence for its being altered cobra C3 (the third component of complement) Science. 1976;191:1275–1276. doi: 10.1126/science.56780. [DOI] [PubMed] [Google Scholar]
- Baxter RH, Chang CI, Chelliah Y, Blandin S, Levashina EA, Deisenhofer J. Structural basis for conserved complement factor-like function in the antimalarial protein TEP1. Proc Natl Acad Sci USA. 2007;104:11615–11620. doi: 10.1073/pnas.0704967104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherer JD, Alsenz J, Esparza I, Hack CE, Lambris JD. Segment spanning residues 727-768 of the complement C3 sequence contains a neoantigenic site and accommodates the binding of CR1, factor H, and factor B. Biochemistry. 1992;31:1787–1794. doi: 10.1021/bi00121a029. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Clemenza L, Isenman DE. Structure-guided identification of C3d residues essential for its binding to complement receptor 2 (CD21) J Immunol. 2000;165:3839–3848. doi: 10.4049/jimmunol.165.7.3839. [DOI] [PubMed] [Google Scholar]
- Daha MR, Fearon DT, Austen KF. C3 requirements for formation of alternative pathway C5 convertase. J Immunol. 1976;117:630–634. [PubMed] [Google Scholar]
- DiScipio RG, Smith CA, Müller-Eberhard HJ, Hugli TE. The activation of human complement component C5 by a fluid phase C5 convertase. J Biol Chem. 1983;258:10629–10636. [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Emsley J, Knight CG, Farndale RW, Barnes MJ, Liddington RC. Structural basis of collagen recognition by integrin α2β1. Cell. 2000;101:47–56. doi: 10.1016/S0092-8674(00)80622-4. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fishelson Z. Complement C3: a molecular mosaic of binding sites. Mol Immunol. 1991;28:545–552. doi: 10.1016/0161-5890(91)90169-k. [DOI] [PubMed] [Google Scholar]
- Fredslund F, Jenner L, Husted LB, Nyborg J, Andersen GR, Sottrup-Jensen L. The structure of bovine complement component 3 reveals the basis for thioester function. J Mol Biol. 2006;361:115–127. doi: 10.1016/j.jmb.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Fredslund F, Laursen NS, Roversi P, Jenner L, Oliveira CL, Pedersen JS, Nunn MA, Lea SM, Discipio R, Sottrup-Jensen L, Andersen GR. Structure of and influence of a tick complement inhibitor on human complement component 5. Nat Immunol. 2008;9:753–760. doi: 10.1038/ni.1625. [DOI] [PubMed] [Google Scholar]
- Fritzinger DC, Bredehorst R, Vogel CW. Molecular cloning and derived primary structure of cobra venom factor. Proc Natl Acad Sci USA. 1994;91:12775–12779. doi: 10.1073/pnas.91.26.12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda DC, Glushka J, Halbeek H, Thotakura RN, Bredehorst R, Vogel CW. N-linked oligosaccharides of cobra venom factor contain novel α (1-3)galactosylated Lex structures. Glycobiology. 2001;11:195–208. doi: 10.1093/glycob/11.3.195. [DOI] [PubMed] [Google Scholar]
- Gowda DC, Petrella EC, Raj TT, Bredehorst R, Vogel CW. Immunoreactivity and function of oligosaccharides in cobra venom factor. J Immunol. 1994;152:2977–2986. [PubMed] [Google Scholar]
- Gowda DC, Schultz M, Bredehorst R, Vogel CW. Structure of the major oligosaccharide of cobra venom factor. Mol Immunol. 1992;29:335–342. doi: 10.1016/0161-5890(92)90020-x. [DOI] [PubMed] [Google Scholar]
- Grier AH, Schultz M, Vogel CW. Cobra venom factor and human C3 share carbohydrate antigenic determinants. J Immunol. 1987;139:1245–1252. [PubMed] [Google Scholar]
- Hinshelwood J, Spencer DI, Edwards YJ, Perkins SJ. Identification of the C3b binding site in a recombinant vWF-A domain of complement factor B by surface-enhanced laser desorption-ionisation affinity mass spectrometry and homology modelling: implications for the activity of factor B. J Mol Biol. 1999;294:587–599. doi: 10.1006/jmbi.1999.3223. [DOI] [PubMed] [Google Scholar]
- Horiuchi T, Macon KJ, Engler JA, Volanakis JE. Site-directed mutagenesis of the region around Cys-241 of complement component C2. Evidence for a C4b binding site. J Immunol. 1991;147:584–589. [PubMed] [Google Scholar]
- Hourcade DE, Mitchell LM, Oglesby TJ. Mutations of the type A domain of complement factor B that promote high-affinity C3b-binding. J Immunol. 1999;162:2906–2911. [PubMed] [Google Scholar]
- Hourcade DE, Wagner LM, Oglesby TJ. Analysis of the short consensus repeats of human complement factor B by site-directed mutagenesis. J Biol Chem. 1995;270:19716–19722. doi: 10.1074/jbc.270.34.19716. [DOI] [PubMed] [Google Scholar]
- Hughes TR, Piddlesden SJ, Williams JD, Harrison RA, Morgan BP. Isolation and characterization of a membrane protein from rat erythrocytes which inhibits lysis by the membrane attack complex of rat complement. Biochem J. 1992;284(Pt 1):169–176. doi: 10.1042/bj2840169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inal JM, Schifferli JA. Complement C2 receptor inhibitor trispanning and the β-chain of C4 share a binding site for complement C2. J Immunol. 2002;168:5213–5221. doi: 10.4049/jimmunol.168.10.5213. [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Christodoulidou A, McCarthy A, Lambris JD, Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature. 2006;444:213–216. doi: 10.1038/nature05172. [DOI] [PubMed] [Google Scholar]
- Janssen BJ, Huizinga EG, Raaijmakers HC, Roos A, Daha MR, Nilsson-Ekdahl K, Nilsson B, Gros P. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature. 2005;437:505–511. doi: 10.1038/nature04005. [DOI] [PubMed] [Google Scholar]
- Jenner L, Husted L, Thirup S, Sottrup-Jensen L, Nyborg J. Crystal structure of the receptor-binding domain of α2-macroglobulin. Structure. 1998;6:595–604. doi: 10.1016/s0969-2126(98)00061-6. [DOI] [PubMed] [Google Scholar]
- Jokiranta TS, Hellwage J, Koistinen V, Zipfel PF, Meri S. Each of the three binding sites on complement factor H interacts with a distinct site on C3b. J Biol Chem. 2000;275:27657–27662. doi: 10.1074/jbc.M002903200. [DOI] [PubMed] [Google Scholar]
- Koistinen V, Wessberg S, Leikola J. Common binding region of complement factors B, H and CR1 on C3b revealed by monoclonal anti-C3d. Complement Inflamm. 1989;6:270–280. doi: 10.1159/000463102. [DOI] [PubMed] [Google Scholar]
- Kolln J, Bredehorst R, Spillner E. Engineering of human complement component C3 for catalytic inhibition of complement. Immunol Lett. 2005;98:49–56. doi: 10.1016/j.imlet.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Lachmann PJ, Halbwachs L. The influence of C3b inactivator (KAF) concentration on the ability of serum to support complement activation. Clin Exp Immunol. 1975;21:109–114. [PMC free article] [PubMed] [Google Scholar]
- Laich A, Sim RB. Complement C4bC2 complex formation: an investigation by surface plasmon resonance. Biochim Biophys Acta. 2001;1544:96–112. doi: 10.1016/s0167-4838(00)00208-9. [DOI] [PubMed] [Google Scholar]
- Lambris JD, Avila D, Becherer JD, Müller-Eberhard HJ. A discontinuous factor H binding site in the third component of complement as delineated by synthetic peptides. J Biol Chem. 1988;263:12147–12150. [PubMed] [Google Scholar]
- Lambris JD, Müller-Eberhard HJ. Isolation and characterization of a 33,000-dalton fragment of complement Factor B with catalytic and C3b binding activity. J Biol Chem. 1984;259:12685–12690. [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- Medicus RG, Gotze O, Müller-Eberhard HJ. Alternative pathway of complement: recruitment of precursor properdin by the labile C3/C5 convertase and the potentiation of the pathway. J Exp Med. 1976;144:1076–1093. doi: 10.1084/jem.144.4.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BP, Harris CL. Complement therapeutics; history and current progress. Mol Immunol. 2003;40:159–170. doi: 10.1016/s0161-5890(03)00111-1. [DOI] [PubMed] [Google Scholar]
- Morganroth ML, Schoeneich SO, Till GO, Pickett W, Ward PA. Lung injury caused by cobra venom factor is reduced in rats raised on an essential fatty acid-deficient diet. Am J Physiol. 1989;257:H1192–1199. doi: 10.1152/ajpheart.1989.257.4.H1192. [DOI] [PubMed] [Google Scholar]
- Morganroth ML, Till GO, Kunkel RG, Ward PA. Complement and neutrophil-mediated injury of perfused rat lungs. Lab Invest. 1986;54:507–514. [PubMed] [Google Scholar]
- O'Keefe MC, Caporale LH, Vogel CW. A novel cleavage product of human complement component C3 with structural and functional properties of cobra venom factor. J Biol Chem. 1988;263:12690–12697. [PubMed] [Google Scholar]
- Oh KS, Kweon MH, Rhee KH, Ho Lee K, Sung HC. Inhibition of complement activation by recombinant Sh-CRIT-ed1 analogues. Immunology. 2003;110:73–79. doi: 10.1046/j.1365-2567.2003.01706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Borek D, Majewski W, Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- Pan Q, Ebanks RO, Isenman DE. Two clusters of acidic amino acids near the NH2 terminus of complement component C4 α'-chain are important for C2 binding. J Immunol. 2000;165:2518–2527. doi: 10.4049/jimmunol.165.5.2518. [DOI] [PubMed] [Google Scholar]
- Pangburn MK, Müller-Eberhard HJ. The C3 convertase of the alternative pathway of human complement. Enzymic properties of the bimolecular proteinase. Biochem J. 1986;235:723–730. doi: 10.1042/bj2350723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein β1H for cleavage of C3b and C4b in solution. J Exp Med. 1977;146:257–270. doi: 10.1084/jem.146.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57:1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- Sánchez-Corral P, Anton LC, Alcolea JM, Marques G, Sánchez A, Vivanco F. Proteolytic activity of the different fragments of factor B on the third component of complement (C3). Involvement of the N-terminal domain of Bb in magnesium binding. Mol Immunol. 1990;27:891–900. doi: 10.1016/0161-5890(90)90156-t. [DOI] [PubMed] [Google Scholar]
- Sharma S, Jabeen T, Singh RK, Bredhorst R, Vogel CW, Betzel C, Singh TP. Structural studies on the cobra venom factor: isolation, purification, crystallization and preliminary crystallographic analysis. Acta Crystallogr D Biol Crystallogr. 2001;57:596–598. doi: 10.1107/s0907444901001342. [DOI] [PubMed] [Google Scholar]
- Shimaoka M, Salas A, Yang W, Weitz-Schmidt G, Springer TA. Small molecule integrin antagonists that bind to the β2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity. 2003;19:391–402. doi: 10.1016/s1074-7613(03)00238-3. [DOI] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Taniguchi-Sidle A, Isenman DE. Interactions of human complement component C3 with factor B and with complement receptors type 1 (CR1, CD35) and type 3 (CR3, CD11b/CD18) involve an acidic sequence at the N-terminus of C3 α'-chain. J Immunol. 1994;153:5285–5302. [PubMed] [Google Scholar]
- Tomana M, Niemann M, Garner C, Volanakis JE. Carbohydrate composition of the second, third and fifth components and factors B and D of human complement. Mol Immunol. 1985;22:107–111. doi: 10.1016/s0161-5890(85)80004-3. [DOI] [PubMed] [Google Scholar]
- Vogel CW, Müller-Eberhard HJ. Induction of immune cytolysis: tumor-cell killing by complement is initiated by covalent complex of monoclonal antibody and stable C3/C5 convertase. Proc Natl Acad Sci USA. 1981;78:7707–7711. doi: 10.1073/pnas.78.12.7707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CW, Müller-Eberhard HJ. The cobra venom factor-dependent C3 convertase of human complement. A kinetic and thermodynamic analysis of a protease acting on its natural high molecular weight substrate. J Biol Chem. 1982;257:8292–8299. [PubMed] [Google Scholar]
- Vogel CW, Müller-Eberhard HJ. Cobra venom factor: improved method for purification and biochemical characterization. J Immunol Methods. 1984;73:203–220. doi: 10.1016/0022-1759(84)90045-0. [DOI] [PubMed] [Google Scholar]
- Vogel CW, Smith CA, Müller-Eberhard HJ. Cobra venom factor: structural homology with the third component of human complement. J Immunol. 1984;133:3235–3241. [PubMed] [Google Scholar]
- Vogt W, Schmidt G, Von Buttlar B, Dieminger L. A new function of the activated third component of complement: binding to C5, an essential step for C5 activation. Immunology. 1978;34:29–40. [PMC free article] [PubMed] [Google Scholar]
- von Zabern I, Hinsch B, Przyklenk H, Schmidt G, Vogt W. Comparison of Naja n. naja and Naja h. haje cobra-venom factors: correlation between binding affinity for the fifth component of complement and mediation of its cleavage. Immunobiology. 1980;157:499–514. doi: 10.1016/s0171-2985(80)80018-0. [DOI] [PubMed] [Google Scholar]
- Whaley K, Ruddy S. Modulation of the alternative complement pathways by β1H globulin. J Exp Med. 1976;144:1147–1163. doi: 10.1084/jem.144.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmann C, Katschke KJ, Yin J, Helmy KY, Steffek M, Fairbrother WJ, McCallum SA, Embuscado L, DeForge L, Hass PE, van Lookeren Campagne M. Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature. 2006;444:217–220. doi: 10.1038/nature05263. [DOI] [PubMed] [Google Scholar]
- Xu Y, Volanakis JE. Contribution of the complement control protein modules of C2 in C4b binding assessed by analysis of C2/factor B chimeras. J Immunol. 1997;158:5958–5965. [PubMed] [Google Scholar]
- Xu Y, Narayana SV, Volanakis JE. Structural biology of the alternative pathway convertase. Immunol Rev. 2001;180:123–135. doi: 10.1034/j.1600-065x.2001.1800111.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.