

Abstract
It has been suggested that patients with nonalcoholic steatohepatitis (NASH) have a high risk for liver cancer. However, it is unknown whether high-fat diet induced NASH promotes hepatocarcinogenesis. In the present study, Sprague-Dawley rats were injected with a low dose of hepatic carcinogen diethylnitrosamine (DEN) and then fed either Lieber-DeCarli control diet (CD) or high-fat diet (HFD) for 6 weeks. Liver histology and the hepatic placental form of glutathione S-transferase (P-GST) positive foci were examined. Expression levels of proliferating cell nuclear antigen (PCNA), cyclinD1, phosphorylated MAPK including extracellular signal-regulated kinase (ERK) and p38, as well as TNF-α and NF-κB were measured in the liver. Induction of lipid peroxidation end products (malondialdehyde plus 4-hydroxynonenal) in liver and apoptotic hepatocytes were also assessed. Results showed that HFD-fed rats developed significantly higher incidence and multiplicity of P-GST positive foci along with more fat accumulation, infiltration of inflammatory cells and higher lipid peroxidation in the liver, as compared with rats fed the CD. This high prevalence of hepatic lesions in the liver was accompanied by greater PCNA expression and cyclinD1 protein concentration but little change in hepatocyte apoptosis. HFD feeding elevated hepatic phosphorylated ERK but reduced phosphorylated p38 as compared with the CD feeding. In addition, a significantly higher expression of TNF-α mRNA and nuclear NF-κB p65 protein were observed in HFD group than those in CD group. These data clearly demonstrate that NASH induced by high-fat diet promoted DEN-initiated early hepatocarcinogenesis, which was associated with elevated TNF-α/NF-κB signaling and MAPK related hepatocyte proliferation.
Keywords: nonalcoholic steatohepatitis, high-fat, diethylnitrosamine, preneoplastic foci
Nonalcoholic steatohepatitis (NASH) is a chronic progressive liver disease which is mainly characterized by the concurrence of fat accumulation and infiltration of abundant inflammatory cells in the liver. NASH patients are reported to possess a high risk for progression to cirrhosis 1, the most common risk factor for hepatocellular carcinoma 2. Moreover, an increasing number of clinical cases report that some patients with NASH can develop liver cancer 3. The increasing prevalence of NASH worldwide 4 and its close association with obesity and type II diabetes 5, 6 raise the necessity to investigate whether NASH and its related micro environmental changes in the liver carry a substantial risk for early development of liver cancer. However, experimental data on whether the pathophysiological features and mechanisms involved in NASH formation possess carcinogenic potential are lacking.
In the “2nd hit” model for NASH pathogenesis, increased oxidative stress was proposed to be one of the major driving forces for NASH development in the context of steatosis 7. At the cellular level, oxidant injury can elicit a wide spectrum of cellular responses ranging from proliferation to growth arrest that usually reflect the balance between multiple intracellular stresses-induced signaling pathways activated in response to an oxidant insult such as mitogen-activated protein kinase (MAPK). MAPK is an essential component of intracellular signal transduction and constitutes mainly extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38. Activation of the ERK pathway is frequently linked to cell proliferation and may be a key signaling pathway involved in the regulation of G1 phase progression in proliferating hepatocytes 8. The association between sustained JNK activation and increased rates of cell apoptosis is well established 9. Although many studies have shown that p38 is correlated with oxidative stress-induced apoptosis, an increasing amount of evidence from the past few years indicates a tumor suppressing effect by its activation 10. However, the MAPK responses and their potential effects on cell growth in NASH or NASH related carcinogenesis have not been investigated.
Aside from oxidative stress, the causal relationship between inflammation and tumorigenesis has also been implicated in a variety of chronic inflammatory diseases such as Helicobacter pylori-induced gastritis for gastric cancer, and inflammatory bowel disease (ulcerative colitis and Crohn’s disease) for colorectal cancer 11. Viral hepatitis such as hepatitis B virus causes a general inflammatory response in the liver that is a strong risk factor for cirrhosis and hepatocellular carcinoma development 12. One of the key molecules mediating the inflammatory processes is the pro-inflammatory cytokine tumor necrosis factor-alpha (TNF-α). TNF-α is a vital cytokine involved in inflammation and immunity, and is reported to be significantly increased in NASH patients 13. A previous study demonstrated reduced rates of preneoplastic lesions and liver tumor formation in TNF-α receptor (TNFR) knock-out mice14, suggesting an important role of TNF-α signaling in hepatocarcinogenesis. Binding of TNF-α and TNFR in hepatocytes can mediate various biological effects including cell proliferation, apoptosis, or even cell death. In addition, nuclear factor-kappaB (NF-κB), one important transcriptional factor regulated by TNF-α in hepatocytes, has been shown to be closely associated with liver neoplastic progression, and mediates transcriptional regulation of multiple genes involved in cellular transformation, proliferation, and survival 15. TNF-α mediated NF-κB activation could be one of the essential components of the TNF proliferative pathway with fundamental importance in carcinogenesis 16, 17. So far, there is no information regarding whether the occurrence of chronic inflammation in NASH pathogenesis and its related pathophysiological changes (e.g., oxidative stress and NF-κB activation) can promote chemical carcinogen-initiated hepatocarcinogenesis.
Diethylnitrosamine (DEN), a specific genotoxic hepatocarcinogen, is well-known for its initiating effects to induce hepatocyte DNA damage and mutation 18. In the present study, we investigated the potential promoting effects of NASH induced by a high fat diet 19, 20 on DEN-initiated hepatic carcinogenesis in a rat model. Specifically, we determined the incidence of precancerous lesions such as preneoplastic foci in the liver and explored the potential cellular mechanisms involved.
Materials and methods
Animals and diets
Eight-week old male Sprague-Dawley rats (Charles River Co., Wilmington, MA) were given a single i.p. injection of 30 mg DEN/kg body weight after one week of chow diet adaptation. The dosage of DEN (30 mg/kg body weight) is not considered to be necrogenic or carcinogenic in the absence of other promoting agents 21. The rats were then randomly divided into two groups (n=6) and fed ad libitum with either Lieber-DeCarli liquid control diet (CD, 35% energy from fat) or high-fat diet (HFD, 71% energy from fat) (Dyets Inc., Bethlehem, PA) for six weeks. The diet compositions have been described previously 20. As the liquid diet provides an adequate amount of fluid, extra water was not given. Rats were housed individually in temperature and humidity controlled rooms and kept on a 12-hour light: dark cycle. The Institutional Animal Care and Use Committee at the USDA Human Nutrition Research Center on Aging approved the animal protocol. Body weight of each rat was measured weekly. After killing, the liver was promptly excised and removed. Two small portions of liver at the right lobe were fixed in 10% neutral-buffered formalin for histological examination, and the remaining liver was snap frozen in liquid nitrogen and stored at −80°C for subsequent analysis.
Histological examination
Formalin-fixed and paraffin-embedded liver tissues were processed routinely for hematoxylin and eosin (H&E) staining. Liver histology was examined and graded according to the magnitude of steatosis, inflammation and hepatocyte ballooning degeneration, as described before 22, 23. Briefly, the degree of steatosis was graded 0–4 based on the average percent of fat-accumulated hepatocytes per field at ×200 magnification under H&E staining (Grading 0 = <5%, 1 = 5~25%, 2 = 26~50%, 3 = 51~75%, 4 = >75%). Inflammation was evaluated by counting the number of a mixed population of inflammatory cells, which mainly constitutes mononuclear inflammatory cells, in 10 random fields at ×200 magnification. The mean of these numbers was calculated and reported as inflammatory cells per mm2. Hepatocellular ballooning degeneration was evaluated as either negative (absent) or positive (present).
Immunohistochemical staining
Precancerous lesions such as preneoplastic foci from fixed liver tissue were visualized by immunostaining of placental form glutathione-S-transferase (P-GST). Hepatocyte proliferation was semi-quantified by immunohistochemical analysis of proliferating cell nuclear antigen (PCNA). Briefly, sections (5 μm thick) were cut from formalin-fixed and paraffin embedded liver samples. After a standard dehydration-rehydration procedure, liver sections were incubated with 3% H2O2 for 15 min to quench endogenous peroxidase activity. The sections were then heated using a steamer for 20 min in 10 mM sodium citrate (pH 6.0) buffer to retrieve antigen. The routine biotin-streptavidin immunohistochemical method consisted of sequential incubations in goat or horse serum blocking solution, polyclonal anti-P-GST (Novocastra Laboratory, UK) or monoclonal anti-PCNA (clone PC10, Dako Co-operation, Carpinteria, CA, USA), biotinylated goat anti-rabbit or horse anti-mouse IgG, and streptavidin conjugated to a horseradish peroxidase label. The liver specimens were finally treated with diaminobenzidine substrate and then counterstained with hematoxylin. All liver sections were viewed under light microscopy by two independent investigators. The number of either P-GST positive preneoplastic foci or P-GST positive single hepatocyte in a liver specimen from each rat was counted in random 15 fields at ×100 magnification. The incidence refers to the percentage of rats bearing P-GST positive foci or single hepatocyte from each group; while the multiplicity represents the average number of P-GST positive foci or single hepatocyte in rats from each group. For PCNA staining, a total of 10 randomly selected fields were screened, and PCNA positive cells with dark brown nucleus were expressed as the number of PCNA (+) cells per 100 hepatocytes.
Quantification of cell apoptosis
Apoptotic hepatocytes were identified in rat liver by the Terminal dUTP Nick End Labeling (TUNEL) assay. An In Situ Cell Death Detection Kit, TMR red (Roche Diagnostics, Indianapolis, IN) was used for detection of DNA strand breaks in apoptotic cells using a fluorescence microscope. Briefly, deparaffinized liver sections were boiled in 100mM citrate buffer (pH=6.0) for 20 min and subsequently in 3% H2O2 for 15 min at room temperature to quench endogenous peroxidase activity. After these pretreatments, TMR-dUTP and TdT were incorporated into the slides and incubated for 1 hour at 37°C in the dark. Tissue sections were viewed under a fluorescence microscope. The number of cells with TUNEL-positive nuclei was determined by manual counting from 20 randomly selected fields at ×400 magnification per liver sample. Results were expressed as the average number of TUNEL positive cells/per microscopic field.
Hepatic lipid peroxidation
Lipid peroxides are unstable and decompose to form a complex series of compounds. Malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) are major aldehydic metabolites of lipid peroxidation and measurement of them has been used as a reliable indicator of lipid peroxidation. A lipid peroxidation colorimetric microplate assay (Oxford Biochemical Research Inc, Oxford MI) was used to assay MDA plus 4-HNE in the liver.
Gene expression by Real-time PCR
Total RNA was isolated from the liver by TriPure Isolation Reagent (Roche Diagnostics, Indianapolis, IN) according to the instructions. cDNA was then prepared from the RNA samples using M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA) and an automated thermal cycler (Bio-Rad Laboratories, Hercules, CA). After quantification and qualification, the PCR reaction for mRNA detection was carried out in each well using 20 μL reaction mixture containing 10 μL 2X SYBR Green Supermix, 0.4 μL of 10 μmol/L primer mix (including forward and reverse primers) and 2.5 μL cDNA diluted in Rnase-free water. Cycling conditions were 50°C for 2 min and 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min. Gene-specific primer sequences were designed using the Primer Express version 2.0 software (Applied Biosystems, Foster City, CA). PCR results were then normalized to the levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and calculated by reference to the average value for the control group using the comparative Ct method. For each sample and each gene, PCR reactions were carried out in duplicate and repeated twice. Gene expression was analyzed using the following pairs of primers: TNF-α (forward, CCAGACCCTCACACTCAGATCA; reverse, 5’-TCCGCTTGGTGGTTTGCTA-3’), cyclinD1 (forward, TTCGTGGCCTCTAAGATGAAGG; reverse, TGAGCTTGTTCACCAGAAGCAG) and GAPDH (forward, AGTGCCAGCCTCGTCTCATAG; reverse, CCTTGACTGTGCCGTTGAACT).
Protein concentrations by Western Blotting
Whole liver homogenate and nuclear proteins were prepared from liver samples as previously described 24. Liver protein extracts (40 μg each) were resolved on sodium dodecyl sulfate--polyacrylamide (SDS-PAGE) gel electrophoresis. After blocking the membrane, immunoblotting was performed based on the manufacturer’s instruction for each primary antibody against total and phosphorylated ERK, total and phosphorylated JNK, total and phosphorylated p38, cleaved caspase-3 (Cell Signaling Technology, Inc), cyclinD1 and nuclear NF-κB p65 subunit (Santa Cruz Biotechnology, Inc). Membranes were then incubated with the secondary antibodies against rabbit or mouse (Bio-Rad Laboratory, Hercules, CA). Anti-GAPDH antibody (Chemicon International, Inc, CA) was used as internal control for equal loading of proteins.
Statistical Analysis
The incidence of preneoplastic foci was evaluated using the Fisher Exact test. All group values are presented as Mean ± Standard error of the mean (SEM) and compared using unpaired student t-test between DEN+CD and DEN+HFD groups at a significance level of P < 0.05.
Results
General appearance
There was no mortality of rats from both DEN+HFD and DEN+CD groups in response to a low dose of DEN administration, regardless of the amount of dietary fat. The body weight did not show any significant difference between the two groups at either the initial or final study period. There were also no significant differences between the two groups in the liver weight and liver index at the end of this experiment.
Histopathological lesions
Both a greater accumulation of fat droplets and infiltration of mainly mononuclear inflammatory cells were readily observed in the liver of rats fed the HFD as compared with mild steatosis and inflammation from the CD group (Figure 1). The ballooning degeneration of hepatocytes, a characteristic pathological feature of NASH, was only detected in response to HFD treatment but not to the CD group. These typical histological features were further confirmed by using a magnitude grading of each hepatic lesion (Table 1). The average percent of fat-accumulated hepatocytes was significantly higher in DEN+HFD group than that in DEN+CD group, which was accompanied by more than 3-fold greater amount of inflammatory cells.
Figure 1.
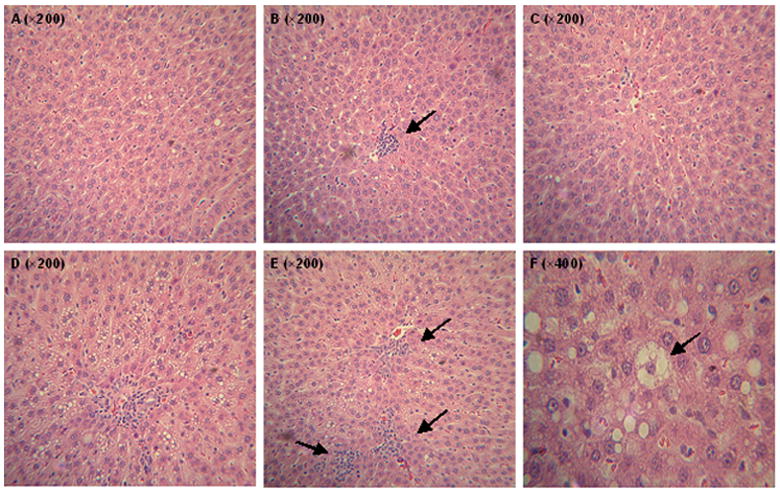
Representative histological features (H&E staining by light microscopy) of NASH induced by a high-fat diet. Feeding of Control Diet caused mild steatosis (A), scattered focal collection of mononuclear inflammatory cells in the liver (B), and no occurrence of cell necrosis (C); in contrast, severe steatosis with infiltration of mononuclear inflammatory cells (D), abundant infiltration of mononuclear cells in addition to small portion of polymorphonuclear cell infiltrates (E), and ballooning degeneration of hepatocytes (F) were found in the livers of rats fed HFD after six weeks.
Table 1.
Summary of histopathological lesions in the liver1
| Group | Steatosis2 | Inflammatory cells (per mm2) | Ballooning degeneration of hepatocytes |
|---|---|---|---|
| DEN+CD | 0.4 ± 0.1 | 4.5 ± 1.7 | − |
| DEN+HFD | 1.6 ± 0.3* | 16.9 ± 4.3* | + |
Means±SD are shown, n=6 animals per group (* P < 0.05)
These values are averaged from the grading score of steatosis, which was graded 0–4 based on the average percent of fat-accumulated hepatocytes per field at ×200 magnification under H&E staining (Grading 0 = <5%, 1 = 5~25%, 2 = 26~50%, 3 = 51~75%, 4 = >75%)
Precancerous lesions in the liver
P-GST has been widely used as an early marker for hepatic carcinogenesis in rodent models 25. Single hepatocytes that express P-GST may belong to a population of initiated cells which can develop into hepatic foci 26. After administration of an initiating dose of DEN, single P-GST positive hepatocytes were found in both groups (Figure 2A) with same incidence but showed a higher multiplicity (number of lesions per animal) in DEN+HFD group (Table 2). More importantly, P-GST positive foci (Figure 2B, C and D) were detected in almost 70% of rats from HFD-fed group, with a multiplicity of 3.6 ± 0.6, while none were found in the CD-fed group (Table 2).
Figure 2.
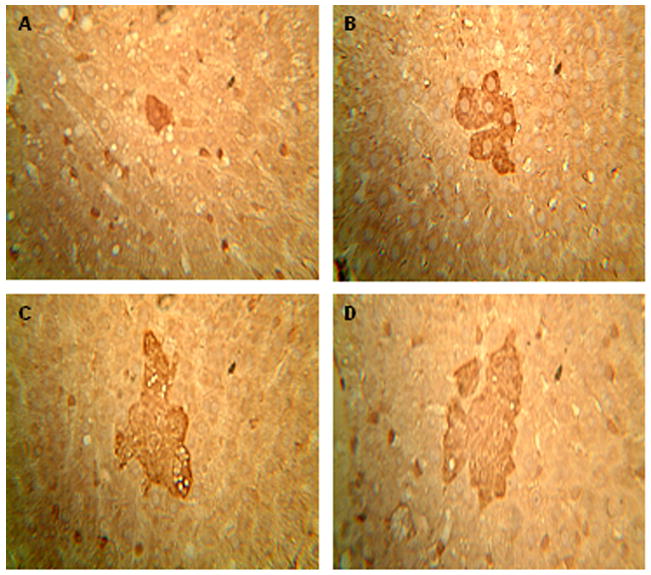
Preneoplastic lesions of P-GST positive foci in the liver. An immunohistochemical assay was applied to each formalin-fixed and paraffin-embedded liver samples from both groups. A cluster of ≥ six single P-GST positive hepatocytes was defined as P-GST positive foci. (A) shows the normal liver after CD feeding while (B), (C) and (D) represent typical foci observed under ×400 magnification from DEN+HFD group. *P < 0.05 between DEN+CD and DEN+HFD groups.
Table 2.
P-GST positive single hepatocyte and foci1
| Incidence |
Multiplicity |
|||
|---|---|---|---|---|
| Single | Foci | Single | Foci | |
| DEN+CD | 4/6 (66.7%) | 0 | 1.5 ± 0.4 | 0 |
| DEN+HFD | 4/6 (66.7%) | 4/6 (66.7%) | 2.5 ± 0.3 | 3.6 ± 0.6* |
Means±SD are shown, n=6 animals per group (* P < 0.05)
Cell proliferation and apoptosis
The average number of PCNA (+) hepatocytes was 60% greater (P < 0.05) in the DEN+HFD group compared to that in DEN+CD group (Figure 3A). The increase of PCNA was associated with a greater (P < 0.05) cellular cyclinD1 protein concentration in DEN+HFD group (Figure 3B). In contrast to cell proliferation, neither apoptotic hepatocytes nor the cleaved caspase-3 protein showed a significant difference between these two groups (data not shown).
Figure 3.
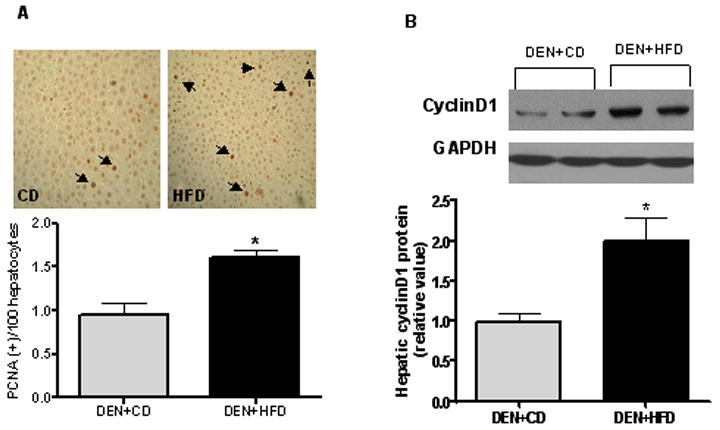
Assessment of cell proliferation. (A) Proliferating cell nuclear antigen (PCNA) expression was examined using immunostaining. PCNA positive hepatocytes were counted from 10 randomly selected fields at ×100 magnification. (B) CyclinD1, an essential regulator for cell cycle at G1-S phase, was measured by western blotting from whole liver homogenate. GAPDH was chosen as internal control for protein equal loading. Values are expressed as means±SEM. *P < 0.05 between DEN+CD and DEN+HFD groups.
MAPK signaling and oxidative stress
To further explore the potential mechanisms responsible for above changes in cell growth, we examined the response of both oxidative stress and MAPK signaling pathway in this model. The protein concentrations of phosphorylated ERK1/2 were significantly increased (more than doubled), while phos-p38 was significantly decreased in the HFD group after DEN treatment as compared to those in CD group despite similar concentrations found for their total protein levels (Figure 4A and B). Total and phosphorylated JNK were not significantly different between the two groups (data not shown). There were significantly increased amounts of lipid peroxidation end products (MDA plus 4-HNE) in the DEN+HFD group as compared to those in DEN+CD group (Figure 5).
Figure 4.
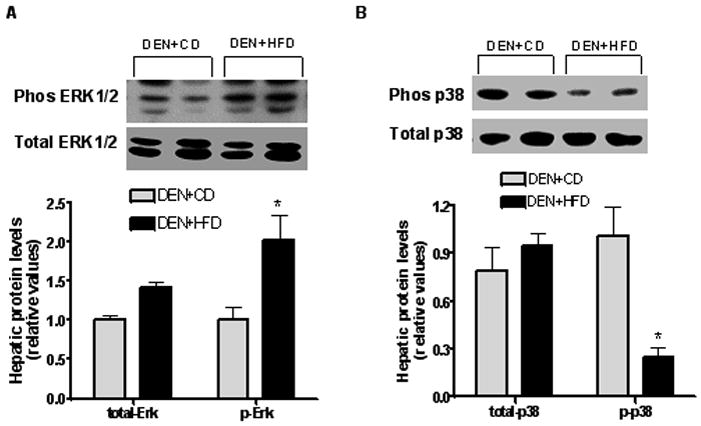
MAPK activation to CD and HFD feeding. Protein concentrations of both total and phosphorylated ERK (A) and p38 (B) in the liver were examined by western blotting from both groups. GAPDH was chosen as internal control for protein equal loading. Values are expressed as means±SEM. *P < 0.05 between DEN+CD and DEN+HFD groups.
Figure 5.
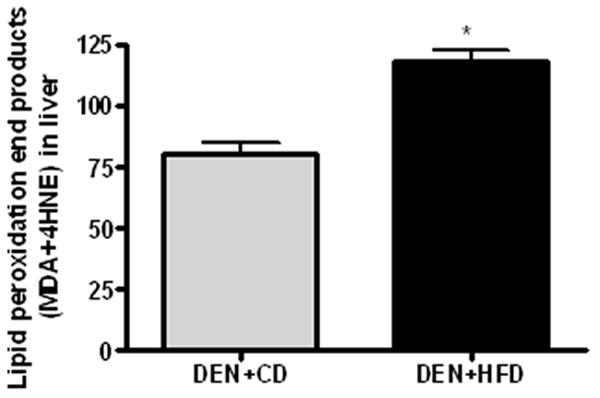
Lipid peroxidation in the liver. Lipid peroxidation end products MDA and 4-HNE was measured by using LPO 586 assay in the liver of rats from both groups. Values are expressed as means±SEM. *P < 0.05 between DEN+CD and DEN+HFD groups.
TNF-α and NF-κB
There were significantly greater TNF-α mRNA level (Figure 6A) and nuclear concentration of NF-κB p65 subunits (3.5-fold of control) in the livers of rats from the DEN+HFD group as compared to those from the DEN+CD group (Figure 6B).
Figure 6.
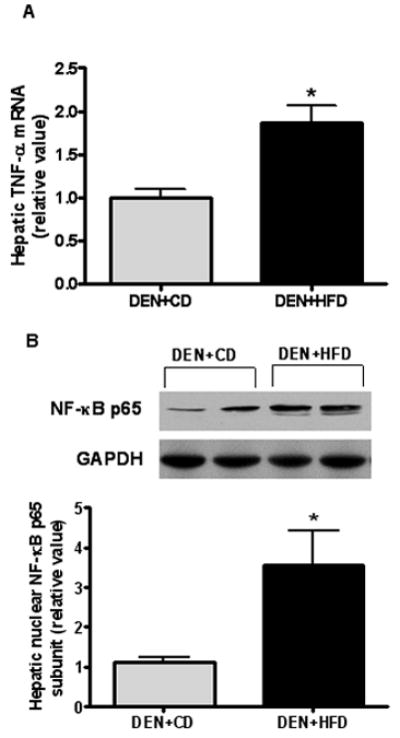
TNF-α and NF-κB activation in the liver. (A) Proinflammatory cytokine TNF-α mRNA expression in the liver was determined by real-time PCR. GAPDH was used as the internal control for adjustification. (B) Nuclear protein was extracted from the liver and specific antisense for NF-κB p65 functional subunit was used to detect its protein level in the nucleus. GAPDH was used for equal protein loading. Values are expressed as means±SEM. *P < 0.05 between DEN+CD and DEN+HFD groups.
Discussion
Six-week feeding of a HFD induced typical histological features of NASH and a significantly higher incidence and multiplicity of precancerous markers of preneoplastic P-GST positive foci in rat liver. These lesions were associated with greater lipid peroxidation, activation of TNF-α/NF-κB signaling and MAPK related hepatocyte proliferation. These data, for the first time, demonstrate that a high-fat diet induced NASH promotes the chemical initiated early hepatocarcinogenesis in an animal model. Given the essential role of oxidative stress in NASH pathogenesis and its close association with hepatic carcinogenesis, the high oxidative stress in HFD induced NASH, reflected by increased lipid peroxidation in the liver, may contribute to the neoplastic process. The increased oxidative stress observed in the DEN+HFD group may result in the activation of ERK by hydrogen peroxides as previously reported 27. ERK activation, in turn, acts as a survival factor to promote PCNA and cyclinD1 expression. Interestingly, some evidence also suggests that ERK activation can mediate some protection against free radical induced cell apoptosis in primary hepatocytes 28, which might partly explain the lower but non-significant cellular apoptosis in the DEN+HFD group. In contrast to previous findings showing that p38 MAPK could be activated by environmental stress 29, 30, HFD feeding in our study increased cellular oxidative stress but remarkably decreased activated p38 in the liver. Our data are in agreement with results from a recent study reporting that a decreased expression of p38 MAPK was found in both P-GST positive foci and foci-bearing liver tissue extract, as compared with the control group 31. Similarly, decreased p38 activity also led to high tumorigenesis by enhancing proliferation of fibroblasts 32. On the other hand, in mice expressing the oncogenes ErbB2 or Ha-Ras, activation of p38 was found to be able to inhibit mammary carcinogenesis 33, 34. One of the mechanisms proposed for the potential tumor suppressing effect of p38 activation was its inhibition on ERK activation 35. The opposite expression pattern in our study seems to support a conflicting effect between activation of ERK and p38 in terms of the induction of preneoplastic foci in this model. Interestingly, besides regulation of cell proliferation, activated p38 cascade was also reported to account for the resistance of cell to apoptosis, leading to unrestricted cell growth 36.
The higher inflammatory response in this model, as identified by abundant infiltration of inflammatory cells and higher gene expression of TNF-α in the DEN+HFD group, may provide another mechanistic link between NASH and pre-malignancy in the liver. NF-κB was more highly induced by the HFD than the CD in rat liver along with the presence of preneoplastic foci, which is in agreement with previous results supporting a causal link between inappropriate or persistent activation of NF-κB and liver neoplastic progression 37. The mechanisms by which NF-κB activation can promote neoplasm are due to its causing cell proliferation through the direct regulation of a downstream molecule like cyclin D1 38 that is significantly greater in the HFD-fed rat liver than that in the CD group. In addition, since NF-κB activation may provide a protective role against apoptosis 39, greater induction of NF-κB in the DEN+HFD group in this model could also antagonize the pro-apoptotic response elicited by oxidative stress or its mediated DNA damage, thereby favoring survival of hepatocytes harboring genetic mutations. However, inconsistent with the observations from our model, Karin and co-workers found that mice lacking IKKβ only in hepatocytes, leading to inactivation of NF-κB, exhibited a marked increase in hepatocarcinogenesis initiated by DEN 40. Although it is difficult to make a direct comparison of data from a genetically modified animal model with that induced by diet in our model, these results do support the concept that NF-κB is essentially involved in hepatocellular carcinogenesis.
Taken together, in the present study, chemically initiated hepatic carcinogenesis has been demonstrated to be promoted by high-fat diet induced NASH in a rat model. The increased cell growth mainly resulted from upregulated cell proliferation rather than impaired cell apoptosis. The MAPK cascade in response to high oxidative stress and elevated TNF-α/NF-κB signaling could contribute to this neoplastic process.
Acknowledgments
The authors thank Dr. Donald E. Smith for his assistance on animal care and handling.
The work was supported by NIH grant R01CA104932, Tufts Cancer Center pilot project award 004-08PP and US Department of Agriculture, under agreement of NO 1950-51000-064S. Any opinions, findings, conclusion, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of National Institute of Health and the U.S. Department of Agriculture.
Abbreviations used are
- CD
control diet
- DEN
diethylnitrosamine
- ERK
extracellular signal-regulated kinase
- 4-HNE
4-hydroxynonenal
- GAPDH
glyceraldehydes-3-phosphate dehydrogenase
- HFD
high-fat diet
- JNK
c-Jun NH2-terminal kinase
- LPO
lipid peroxidation
- MAPK
mitogen-activated protein kinase
- MDA
malondialdehyde
- NASH
nonalcoholic steatohepatitis
- PCNA
proliferating cell nuclear antigen
- P-GST
placental form of glutathione S-transferase
- TNF-α
tumor necrosis factor-α
Footnotes
Author Disclosures: Yan Wang, Lynne M. Ausman, Robert M. Russell, Andrew S. Greenberg and Xiang-Dong Wang have no conflicts of interest.
References
- 1.Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. 1990;11:74–80. doi: 10.1002/hep.1840110114. [DOI] [PubMed] [Google Scholar]
- 2.Schoen RE, Tangen CM, Kuller LH, Burke GL, Cushman M, Tracy RP, Dobs A, Savage PJ. Increased blood glucose and insulin, body size, and incident colorectal cancer. J Natl Cancer Inst. 1999;91:1147–54. doi: 10.1093/jnci/91.13.1147. [DOI] [PubMed] [Google Scholar]
- 3.Bugianesi E. Non-alcoholic steatohepatitis and cancer. Clin Liver Dis. 2007;11:191–207. doi: 10.1016/j.cld.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 4.McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:521–33. doi: 10.1016/j.cld.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Gupte P, Amarapurkar D, Agal S, Baijal R, Kulshrestha P, Pramanik S, Patel N, Madan A, Amarapurkar A, Hafeezunnisa Non-alcoholic steatohepatitis in type 2 diabetes mellitus. J Gastroenterol Hepatol. 2004;19:854–8. doi: 10.1111/j.1440-1746.2004.03312.x. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Monzon C, Martin-Perez E, Iacono OL, Fernandez-Bermejo M, Majano PL, Apolinario A, Larranaga E, Moreno-Otero R. Characterization of pathogenic and prognostic factors of nonalcoholic steatohepatitis associated with obesity. J Hepatol. 2000;33:716–24. doi: 10.1016/s0168-8278(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 7.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 8.Talarmin H, Rescan C, Cariou S, Glaise D, Zanninelli G, Bilodeau M, Loyer P, Guguen-Guillouzo C, Baffet G. The mitogen-activated protein kinase kinase/extracellular signal-regulated kinase cascade activation is a key signalling pathway involved in the regulation of G(1) phase progression in proliferating hepatocytes. Mol Cell Biol. 1999;19:6003–11. doi: 10.1128/mcb.19.9.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 10.Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007;1773:1358–75. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Sawa T, Ohshima H. Nitrative DNA damage in inflammation and its possible role in carcinogenesis. Nitric Oxide. 2006;14:91–100. doi: 10.1016/j.niox.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pessayre D, Mansouri A, Fromenty B. Nonalcoholic steatosis and steatohepatitis. V. Mitochondrial dysfunction in steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2002;282:G193–9. doi: 10.1152/ajpgi.00426.2001. [DOI] [PubMed] [Google Scholar]
- 14.Knight B, Yeoh GC, Husk KL, Ly T, Abraham LJ, Yu C, Rhim JA, Fausto N. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J Exp Med. 2000;192:1809–18. doi: 10.1084/jem.192.12.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arsura M, Cavin LG. Nuclear factor-kappaB and liver carcinogenesis. Cancer Lett. 2005;229:157–69. doi: 10.1016/j.canlet.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest. 2001;107:241–6. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirillova I, Chaisson M, Fausto N. Tumor necrosis factor induces DNA replication in hepatic cells through nuclear factor kappaB activation. Cell Growth Differ. 1999;10:819–28. [PubMed] [Google Scholar]
- 18.Sarma DS, Rao PM, Rajalakshmi S. Liver tumour promotion by chemicals: models and mechanisms. Cancer Surv. 1986;5:781–98. [PubMed] [Google Scholar]
- 19.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, DeCarli LM. Acarbose attenuates experimental non-alcoholic steatohepatitis. Biochem Biophys Res Commun. 2004;315:699–703. doi: 10.1016/j.bbrc.2004.01.116. [DOI] [PubMed] [Google Scholar]
- 20.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, DeCarli LM. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502–9. doi: 10.1093/ajcn/79.3.502. [DOI] [PubMed] [Google Scholar]
- 21.Dragan YP, Hully JR, Nakamura J, Mass MJ, Swenberg JA, Pitot HC. Biochemical events during initiation of rat hepatocarcinogenesis. Carcinogenesis. 1994;15:1451–8. doi: 10.1093/carcin/15.7.1451. [DOI] [PubMed] [Google Scholar]
- 22.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–9. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 23.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–74. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 24.Chung J, Chavez PR, Russell RM, Wang XD. Retinoic acid inhibits hepatic Jun N-terminal kinase-dependent signaling pathway in ethanol-fed rats. Oncogene. 2002;21:1539–47. doi: 10.1038/sj.onc.1205023. [DOI] [PubMed] [Google Scholar]
- 25.Sato K, Kitahara A, Satoh K, Ishikawa T, Tatematsu M, Ito N. The placental form of glutathione S-transferase as a new marker protein for preneoplasia in rat chemical hepatocarcinogenesis. Gann. 1984;75:199–202. [PubMed] [Google Scholar]
- 26.Cameron RG. Identification of the putative first cellular step of chemical hepatocarcinogenesis. Cancer Lett. 1989;47:163–7. doi: 10.1016/0304-3835(89)90086-4. [DOI] [PubMed] [Google Scholar]
- 27.Guyton KZ, Gorospe M, Wang X, Mock YD, Kokkonen GC, Liu Y, Roth GS, Holbrook NJ. Age-related changes in activation of mitogen-activated protein kinase cascades by oxidative stress. J Investig Dermatol Symp Proc. 1998;3:23–7. [PubMed] [Google Scholar]
- 28.Conde de la Rosa L, Schoemaker MH, Vrenken TE, Buist-Homan M, Havinga R, Jansen PL, Moshage H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44:918–29. doi: 10.1016/j.jhep.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 29.Raingeaud J, Gupta S, Dickens M, Han J. Pro-inflammatory Cytokines and Environmental Stress Cause p38 Mitogen-activated Protein Kinase Activation by Dual Phosphorylation on Tyrosine and Threonine. J Biol Chem. 1995;270:7420–6. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 30.Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–7. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 31.Kucharczak J, Simmons MJ, Fan Y, Gelinas C. To be, or not to be: NF-kappaB is the answer--role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;22:8961–82. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 32.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–78. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G, Nebreda AR, Anderson CW, Kallioniemi A, Fornace AJ, Jr, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–5. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 34.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, Appella E, Fornace AJ., Jr Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 35.Li SP, Junttila MR, Han J, Kahari VM, Westermarck J. p38 Mitogen-activated protein kinase pathway suppresses cell survival by inducing dephosphorylation of mitogen-activated protein/extracellular signal-regulated kinase kinase1,2. Cancer Res. 2003;63:3473–7. [PubMed] [Google Scholar]
- 36.Iyoda K, Sasaki Y, Horimoto M, Toyama T, Yakushijin T, Sakakibara M, Takehara T, Fujimoto J, Hori M, Wands JR, Hayashi N. Involvement of the p38 mitogen-activated protein kinase cascade in hepatocellular carcinoma. Cancer. 2003;97:3017–26. doi: 10.1002/cncr.11425. [DOI] [PubMed] [Google Scholar]
- 37.Szlosarek PW, Balkwill FR. Tumour necrosis factor alpha: a potential target for the therapy of solid tumours. Lancet Oncol. 2003;4:565–73. doi: 10.1016/s1470-2045(03)01196-3. [DOI] [PubMed] [Google Scholar]
- 38.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 39.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 40.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]