

Abstract
Spinal muscular atrophy (SMA) is a common autosomal recessive neurodegenerative disorder in humans. Amongst the earliest signs of neurodegeneration are severe and progressive defects of the neuromuscular synapse. These defects, characterized by poor terminal arborization and immature motor endplates, presumably result in a loss of functional synapses. The slow Wallerian degeneration (Wlds) mutation in rodents has been shown to have a protective effect on mouse models of motor neuron disease by retarding axonal die-back and preventing neuromuscular synapse loss. In this study we tested the effects of the Wlds mutation on the disease phenotype of SMA model mice. Consistent with previous reports, the mutation slows axon and neuromuscular synapse loss following nerve injury in wild-type as well as in SMA mice. However, the synaptic defects found in severely affected SMA patients and model mice persist in the double (Wlds;SMA) mutants. No delay in disease onset was observed and survival was not significantly altered. Finally, Wlds had no effect on the striking phrenic nerve projection defects that we discovered in SMA model mice. Our results indicate that the reported protective effects of Wlds are insufficient to mitigate the neuromuscular phenotype due to reduced SMN protein, and that the mechanisms responsible for distal defects of the motor unit in SMA are unlikely to be similar to those causing neurodegeneration in genetic mutants such as the pmn mouse which is partially rescued by the Wlds protein.
Keywords: Spinal muscular atrophy, Neuromuscular junctions, Wallerian degeneration
There is a growing recognition that defects in the distal segment of a neuron are often a first step in the degenerative process that characterizes neuronal loss due to injury, toxins or as a result of genetic perturbations [3]. There is also an increasing awareness that the mechanisms underlying axonal and synaptic loss may be distinct and separate from those involved in the degeneration of the cyton [7]. This compartmentalization of the neurodegenerative process explains, in part, emerging efforts to stem neuronal degeneration by preventing synapse and axon loss [5,6,8,16,22,23]. Amongst the many neurodegenerative diseases wherein such a strategy may be tested and prove to be effective, spinal muscular atrophy (SMA) stands out for a number of reasons. First, it is a disorder that affects the spinal motor neurons, cells with long axons terminating at neuromuscular synapses. Second, the distal ends of the affected cells lie in the periphery making them relatively accessible to potential therapies compared to cells affected in diseases of the CNS. Third, there is strong evidence that denervation, a process involving a loss of functional synapses, is a key aspect of disease pathology in SMA. Finally, the genetics of the disease have been clearly defined allowing the generation of SMA model mice in which to determine whether protecting axons and synapses mitigates the disease phenotype.
SMA is a pediatric neuromuscular disorder caused by mutations in the SMN1 gene [12]. An almost identical copy gene, SMN2, fails to compensate due to a single exonic nucleotide change that disrupts splicing, resulting in reduced levels of the full-length (FL) SMN transcript [17]. Consequently, patients express reduced levels of the multifunctional SMN protein (reviewed in [20]). It is unclear why this ubiquitously expressed protein selectively affects the neuromuscular system. Introducing the human SMN2 into mice lacking murine Smn (SMN2;Smn−/−) results in animals with a severe form of the human disease [18]. Additional mutant SMN transgenes are capable of modulating the disease phenotype in SMN2;Smn−/− mice [11,19]. We previously demonstrated that neuromuscular weakness in SMA mice appears well before the loss of motor neurons in the spinal cord [9]. We further showed that there is a direct correlation between the onset of weakness and the appearance of defects at the distal end of the motor unit. These defects which are most obvious at the neuromuscular junction (NMJ) involve the pre-synaptic as well as post-synaptic specializations and became apparent as early as post-natal day 2 (P2). Pre-synaptic abnormalities include swellings of neurofilament (NF) protein in pre-terminal axons and nerve terminals, poor terminal arborization and numerous retraction bulb-like structures suggesting denervation. Post-synaptic abnormalities are defined by acetylcholine receptor (AChR) clusters that express significantly higher levels of the fetal (γ) subunit of the receptor than age-matched Controls and fail to mature into the characteristic “pretzels” found in wild-type animals. The severity of these defects presumably results in a loss of functional neuromuscular synapses. Evidence from a more severe animal model of SMA suggests that denervation and synapse loss become apparent as early as embryonic day 18.5 (E18.5) [14]. If these defects contribute to the SMA phenotype and eventually lead to spinal motor neuron loss, protecting the distal end of the motor unit may be beneficial.
Wlds (Slow Wallerian degeneration) is a chimeric mutant gene in mice that encodes nicotinamide mononucleotide adenylyltransferase (Nmnat1; a NAD+-synthesizing enzyme) fused in-frame via a novel 18-amino sequence to the N-terminus of the ubiquitination enzyme Ube4b [2]. Wlds mice are indistinguishable from their wild-type littermates but exhibit delayed Wallerian degeneration [13]. In particular, distal axons and neuromuscular synapses of these mice remain morphologically and functionally intact for as long as 2 weeks following axotomy, a finding consistent with delayed atrophy of denervated Wlds muscle [1]. The Wlds gene has also been shown to retard axon and NMJ loss in dying-back axonopathies [5,6,23]. Although molecular mechanisms to explain these effects are unclear, the observations suggest a clear rationale for testing the effect of Wlds on other neuropathies. In this study, we assessed the phenotypic and cellular consequences of expressing the Wlds gene in mouse models of SMA. In contrast to other models of neurodegeneration in which Wlds provided a significant neuroprotective effect, we found that the chimeric gene did not alter the SMA phenotype. This was reflected in a persistence of distal axonal and NMJ defects in the double (Wlds;SMA) mutants. Our analysis also revealed a novel and striking axonal phenotype in the diaphragms of severely affected SMA mice which is not rescued by Wlds. The new phenotype suggests that insufficient SMN protein may impair appropriate signaling between the nerve and its target muscle and could explain early mortality in the most severe instances of the disease.
Wlds mice were obtained from Harlan, Bicester, UK and back-crossed to FVB/N mice (Jackson Labs, Bar Harbor, ME, USA) over 10 successive generations. The Wlds gene was identified by PCR using the following primers: 5′-CGTTGGCTCTAAGGACAGCAC-3′; 5′-CTGCAGCCCCCACCCCTT-3′ while SMA carriers were genotyped as previously described [11,18]. RNA expression of the Wlds gene was assessed by RT-PCR using a 5′-TGTAGGTCAACACCACCAAC-3′ forward primer and a 5′-TTCCCACGTATCCACTT CCA-3′ reverse primer.
To determine whether the Wlds phenotype was maintained on the FVB/N strain background, 2-month-old animals with or without the gene were anesthetized with 2.5% avertin (8 μl/g), the right sciatic nerve exposed and transected before suturing the wound. Five or ten days later, the animals were euthanized and the distal stump of the transected nerve dissected out. The contralateral nerve served as a Control. Nerve segments were processed for Toluidene blue staining as previously described [9] to examine individual axons. Transections in neonates were carried out following anesthesia on wet ice. Two days later, the mice were euthanized and relevant muscles isolated to analyze NMJs and intramuscular nerves by immunohistochemistry as described elsewhere [9]. To examine the effect of Wlds on E18.5 SMA embryos, timed pregnancies were set up and fetal gastrocnemius, intercostal and diaphragm muscles harvested for NMJ analysis. Analysis of the phrenic nerve was carried out on the distal 600 μm portion the sternal branch. AChR clusters and nerves were visualized using labeled α-bungarotoxin (Molecular Probes, Eugene, OR, USA) and anti-NF-160 kDa antibody (Chemicon, Temecula, CA, USA) respectively. Terminal axon lengths and endplate band width were measured using SPOT advanced image analysis software (Diagnostic Instruments, Starling Heights, MI, USA). Differences in means between samples were calculated using the two-tailed Student's t-test and all data presented as mean ± S.E.M. Kaplan–Meier curves were used to determine survival. All animal work was carried out according to institutional guidelines.
To determine the effect of the Wlds gene on the SMA phenotype, Wlds;SMA double mutant mice (Wlds+/−;SMN2+/+;SMNΔ7+/−;Smn−/−) were generated. Since Wlds(C57BL/Ola) and SMA carriers (FVB/N) are on different genetic backgrounds which can mask genuine Wlds effects in their mixed background progeny, we first derived congenic FVB/N Wlds mice. Next, we undertook to ensure that the originally reported phenotype was preserved on the new genetic background. Two-month-old mice with or without the Wlds gene were subjected to sciatic nerve transection and the number of intact axons distal to the lesion were quantified 5 and 10 days later. Wallerian degeneration was significantly delayed in Wlds mice but not wild-type littermates (Fig. S1A and B) indicating that strain differences do not abolish the Wlds phenotype. Furthermore, as expected and in keeping with previous reports, the Wlds gene is ubiquitously expressed and at relatively high levels in neural tissue (Fig. S1C). Next, we introduced that Wlds gene into SMA carrier mice and then crossed these animals (Wlds+/−;SMN2+/+;Smn+/−) to SMN2+/+;SMNΔ7+/+;Smn+/− mice to generate SMA mutants with or without the Wlds gene. At birth, SMA mice were significantly smaller than Control littermates irrespective of whether they carried the Wlds gene or not (Fig. 1A). Additionally, there was no difference in weight between SMA and Wlds;SMA pups (SMA, 1.20 ± 0.02 g; Wlds;SMA, 1.23 ± 0.02 g; Control, 1.37 ± 0.01 g; P < 0.05 for Control vs. SMA and Control vs. Wlds;SMA; P > 0.37 for SMA vs. Wlds;SMA, n ≥ 10 for each group). Disease progression in SMA mice with and without the Wlds gene was virtually identical (Fig. 1B) and there was no significant difference in survival (SMA, 6.1 ± 0.6 days; Wlds;SMA, 7.2 ± 0.9 days; P > 0.3; n ≥ 10; also see Fig. 1C). These results indicate that the Wlds gene neither delays disease onset nor enhances survival in SMA model mice.
Fig. 1.
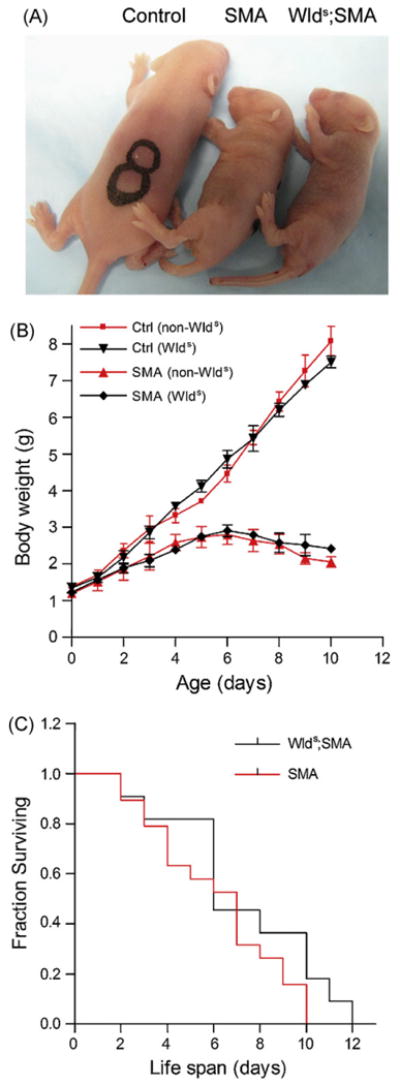
The effect of the Wlds gene on the SMA phenotype. (A) Gross morphology at P5 and (B) body weights of Control (SMN2+/+;SMNΔ7+/−;Smn+/+) and SMA mice with or without Wlds indicating that the chimeric gene has no effect on the overt phenotype or disease progression in mutant animals. (C) Kaplan–Meier survival curve analysis indicates that the presence of the Wlds gene in SMA mice does not significantly alter survival. The log-rank test was used to statistically assess survival in the two groups.
Previously we showed that the earliest neuromuscular defects in SMA mice are observed at the NMJ and that the onset of these defects parallels the onset of disease symptoms [9]. To determine if Wlds has any effect on this cellular phenotype, we examined the distal axons and NMJs in the gastrocnemius muscles of SMA, Wlds;SMA and Control littermates. To simultaneously rule out the possibility that reduced SMN abolishes the delayed Wallerian degeneration effect in Wlds transgenic mice, we transected the right sciatic nerves of the animals at P3 while leaving the contralateral side intact. Distal nerves and NMJs were examined 48 h later at P5. Consistent with our previous findings, an examination of the NMJs in the non-axotomized muscle of SMA mice revealed severe synaptic defects characterized by NF aggregates in the nerve terminals and pre-terminal axons, poor terminal arborization and motor end-plates of reduced size (Fig. 2A and B). In comparison, few if any NMJs in Control littermates with or without Wlds exhibited these abnormalities. An analysis of the same (non-axotomized) NMJs in muscle from Wlds;SMA mice revealed defects strikingly similar to those in SMA mutants lacking the Wlds gene (Fig. 2A and B). Together, these results indicate first, that Wlds is unable to attenuate the NMJ defects resulting from reduced SMN protein and second, that the chimeric gene does not on its own cause abnormalities of the neuromuscular synapse.
Fig. 2.
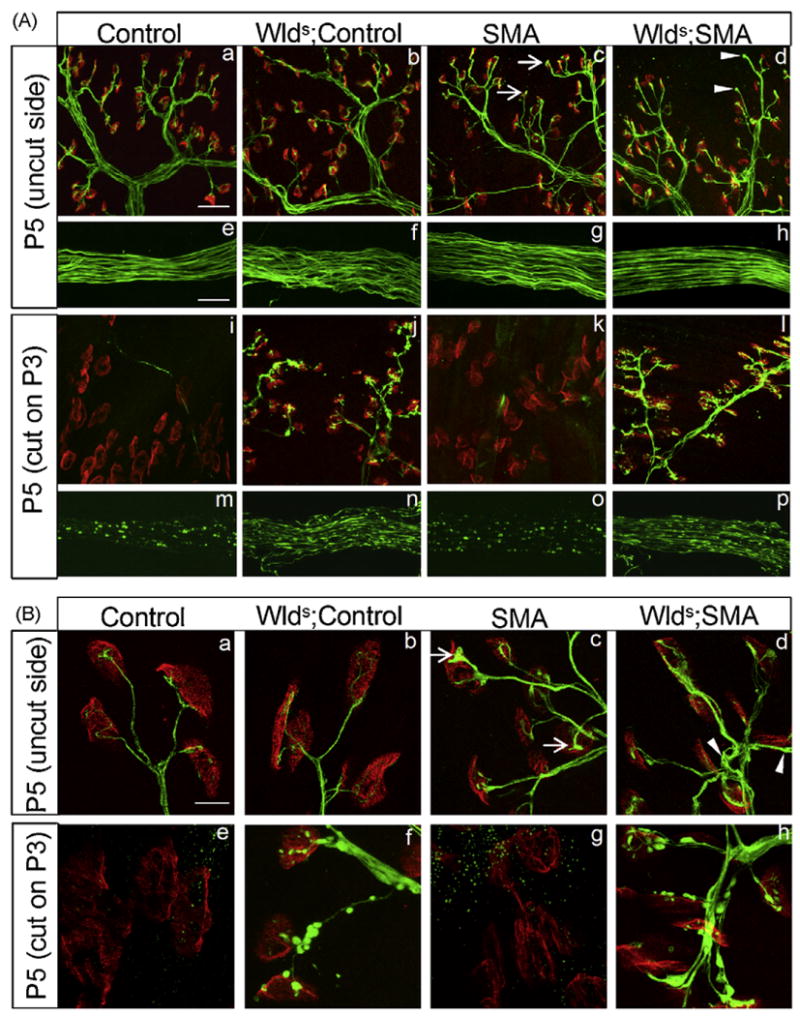
The Wlds gene does not mitigate NMJ defects in SMA mice despite delaying Wallerian degeneration. (A) NMJs in the gastrocnemius muscle of Control (SMN2+/+;SMNΔ7+/−;Smn+/+) and SMA mice with or without Wlds were immunohistologically analyzed following transection of the sciatic nerve. The contralateral nerve was left intact. NMJ defects as assessed by NF aggregates in pre-terminal axons and nerve terminals, poor terminal arborization and AChR clusters of reduced complexity seen in SMA mice (c, arrows) persisted in Wlds;SMA mice (d, arrowheads). Unoccupied AChR clusters were not significantly increased in either group of mutants. Following transection, severe Wallerian degeneration, characterized by fragmented axons (m and o) and denervated endplates (i and k), was seen in mice lacking Wlds while Control (j and n) as well as SMA (l and p) mice carrying the mutation were protected. (B) Higher magnification images indicate NF swellings in SMA (c, arrows) as well as in Wlds;SMA (d, arrowheads) mice. Synpase loss following transection in mice lacking Wlds (e and g) is marked by enlarged AChR clusters. Scale bars: a–d, i–l = 40 μm and e–h, m–p = 25 μm.
We next examined NMJs of the gastrocnemius innervated by the injured nerve in the various groups of mice. As expected, distal axons were relatively well preserved and AChR clusters remained innervated in Control mice carrying the Wlds gene. In contrast, the vast majority of endplates in non-Wlds Control littermates were devoid of nerve endings and there was clear evidence of extensive Wallerian degeneration of the distal axons (Fig. 2A and B). Interestingly, our findings in the mutant animals mirrored those in the Controls with widespread denervation of NMJs and degeneration of axons in SMA but not Wlds;SMA mice. Furthermore, the characteristic dispersal of AChRs following neonatal transection [25] was suppressed in Controls as well as in SMA mice carrying the Wlds gene (Fig. S2). Collectively, these results indicate that the Wlds phenotype is maintained in FVB/N neonates including animals expressing greatly reduced levels of the SMN protein.
To validate our findings described above, we tested the effect of Wlds on a different but related mouse model of SMA [18]. These mice carry only the human SMN2 transgene on a murine Smn null background (SMN2;Smn−/−). On a pure FVB/N background they are more severely affected than previously reported [18]. Indeed, death frequently occurs at P0 (S. Kariya and U. Monani, unpublished observation). Given the severity of this phenotype, we examined the effect of Wlds in E18.5 mutant embryos. Timed pregnancies were set up to generate Wlds;SMA progeny (Wlds+/+;SMN2+/+;Smn−/−) on the one hand, and SMA (SMN2+/+;Smn−/−) mutants on the other. An examination of the embryos revealed no appreciable difference in gross morphology or body weight between the various genotypes (Table S1). We next asked if SMA mutant embryos displayed NMJ defects similar to those described above and whether Wlds had a mitigating effect on the abnormalities. An analysis of the distal gastrocnemius muscle indicated no abnormalities in the SMA mutants. We therefore examined the more proximally located diaphragm which was previously shown to exhibit NMJ defects early in the disease [9]. Interestingly, we found that NF varicosities at the nerve terminals of the phrenic nerve axons are indeed present in mutant embryos at E18.5 (Fig. S3). To further characterize nerve defect in this muscle, we quantified the number of motor end-plates in a defined area of the sternal branch of the phrenic nerve, the mean distance of these endplates from the nerve trunk, the average number of terminal axons on either side of the nerve trunk and mean length of these axons from the nerve trunk. The parameters were first compared between SMA mutants and Controls (SMN2+/+;Smn+/+). No differences were found between the numbers of axon terminals or endplates and the distances of the endplates from the nerve trunk (data not shown). However, we found that the average length of the axon terminals from the nerve trunk was ∼50% longer in the mutants than in Control littermates (Fig. 3A and B). Wlds had no effect on this novel phenotype which was absent in the two other muscles (gastrocnemius and intercostals) that we examined (data not shown). Nor were the NF varicosities reduced in the double mutants, a result consistent with our findings in the Wlds;SMN2;SMNΔ7;Smn−/− mice. Taken together, this data confirms our conclusions that Wlds fails to attenuate the distal axonal and NMJ defects seen in SMA mice.
Fig. 3.
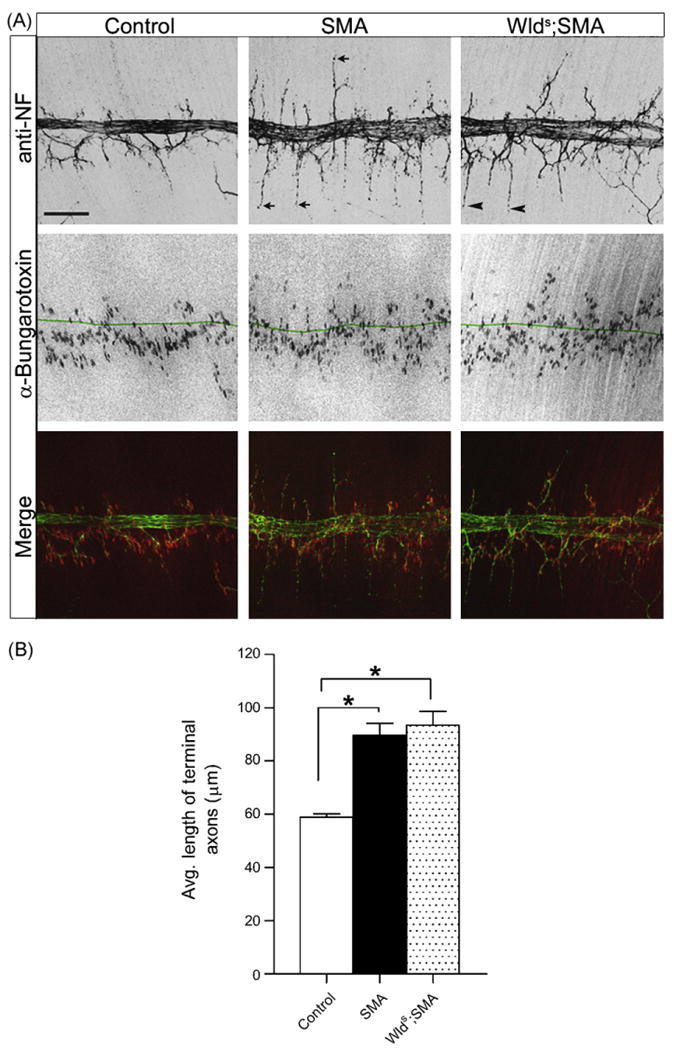
Axon projection defects in severe SMA. (A) Phrenic nerve axons in SMN2+/+;Smn−/− E18.5 embryos with or without Wlds project past AChR clusters (arrows in SMA; arrowheads in Wlds;SMA) and are significantly longer than those in Control (SMN2+/+;Smn+/+) littermates. Note: green line in middle row panels represents the midline of the nerve trunk. Note: green line in middle panel represents nerve trunk midline. (B) Quantification of increased length of mutant axon terminals in SMA and Wlds;SMA mice. *Significant difference with P < 0.001, Student's t-test, n ≥ 3. Scale bar= 120 μm.
Defects in the distal axons and at the NMJs are amongst the first signs of motor neuron pathology in SMA model mice [9]. These defects likely result in a loss of functional neuromuscular synapses which in turn lead to motor neuron cell death. The Wlds mutation in mice has been shown not only to delay Wallerian degeneration following axotomy but also to protect axons and synapses from degenerating due to toxins or genetic mutations. In this study, we assessed whether the protective effects of Wlds benefit mice with SMA. We conclude that although Wallerian degeneration is delayed in Wlds;SMA mice following nerve injury, the Wlds mutation does not attenuate the SMA phenotype or the NMJ defects characteristic of this motor neuron disease. Similar findings were reported [21] while this manuscript was being prepared although the authors used a single mouse model—the originally reported mixed background SMN2+/+;SMNΔ7+/−;Smn−/− animals [11] and examined the proximal rather than distal compartments of the motor neuron. The lack of any protective effect ought not to be completely unexpected. While Wlds has been beneficial in a number of mouse models of neurodegeneration, there are prominent examples where the effect has either been modest [6,16] or completely lacking [26]. Even in instances where it was demonstrated to be of benefit, the underlying mechanisms are not clear. Attempts to understand the mechanisms underlying Wlds-mediated protection have implicated numerous proteins involved in regulating mitochondrial stability and degeneration [27]. Additionally, RNA microarray analyses have revealed changes in axonal transport genes in Wlds mice [24]. Non-somatic mitochondrial defects [10] and axonal transport perturbations [4] lie at the basis of many neurodegenerative diseases. If Wlds does indeed alter synaptic mitochondrial responses and/or axonal transport to confer its neuroprotective effects, it is possible that neither process underlies motor neuron disease in SMA. However, concrete evidence of this will require further investigation.
Perhaps the more interesting outcome of this study was the striking phrenic nerve projection defects we discovered in the most severely affected SMA mice. Axon pathfinding defects have been previously described in a fish model of SMA [15] but were not found in SMA mice [14]. Our findings are in contrast to those recently reported [14]. One explanation for the different results is the severity of the phenotypes of the mice used in the two studies, the fully congenic mutants being considerably more severe [9]. Indeed, we have previously shown that in E18.5 SMA mutants carrying the SMNΔ7 transgene which reduces SMA severity, there were no signs of phrenic nerve projection defects [9]. This is consistent with the hypothesis that SMN has numerous functions that are affected in a hierarchical fashion depending on SMN levels. Our data showing an increase in mean length of the phrenic nerve axons but no significant increase in the distance of the motor endplates from the nerve trunk (endplate band width) suggests that the axons overshoot the AChR clusters, implying either an inability of the muscle to signal appropriately and/or a failure of the nerve to respond to cues secreted by the muscle. Although it is unclear which, if either, mechanism serves to explain our findings, the phenotype may be indicative of a novel function of the SMN protein in muscle and/or nerve. Furthermore, it could account for pre-natal or early neonatal death in the most severely affected SMA model mice and human patients.
Supplementary Material
Acknowledgments
We thank O. Leykekhman, C. Neeb and C. Caine for technical help and G.-H. Park for expert assistance with imaging software. This work was funded by grants from the Families of SMA, SMA Foundation, Muscular Dystrophy Association of America and NINDS (R01 NS057482) to U.M.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.neulet.2008.10.107.
References
- 1.Brown MC, Booth CM, Lunn ER, Perry VH. Delayed response to denervation in C57Bl/Ola mice. Neuroscience. 1991;43:279–283. doi: 10.1016/0306-4522(91)90435-q. [DOI] [PubMed] [Google Scholar]
- 2.Conforti L, Tarlton A, Mack TGA, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conforti L, Adalbert R, Coleman MP. Neuronal death: where does the end begin? Trends Neurosci. 2007;30:159–166. doi: 10.1016/j.tins.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 4.De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- 5.Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- 6.Fischer LR, Culver DG, Davis AA, Tennant P, Wang M, Coleman M, Asress S, Adalbert R, Alexander GM, Glass JD. The Wlds gene modestly prolongs survival in the SOD1G93A fALS mouse. Neurobiol Dis. 2005;19:293–300. doi: 10.1016/j.nbd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Gillingwater TH, Ribchester RR. Compartmental neurodegeneration and synaptic plasticity in the Wlds mutant mouse. J Physiol. 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillingwater TH, Haley JE, Ribchester RR, Horsburgh K. Neuroprotection after transient global cerebral ischaemia in Wlds mutant mice. J Cereb Blood Flow Metab. 2004;24:62–66. doi: 10.1097/01.WCB.0000095798.98378.34. [DOI] [PubMed] [Google Scholar]
- 9.Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwong JQ, Beal MF, Manfredi G. The role of mitochondria in inherited mitochondrial neurodegenerative disease. J Neurochem. 2006;6:1659–1675. doi: 10.1111/j.1471-4159.2006.03990.x. [DOI] [PubMed] [Google Scholar]
- 11.Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 12.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 13.Mack TGA, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 14.McGovern VL, Gavrilina TO, Beattie CE, Burghes AH. Embryonic motor axon development in the severe SMA mouse. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn189. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McWhorter ML, Monani UR, Burghes AH, Beattie CE. Knockdown of the survival motor neuron (smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;162:919–931. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mi W, Beirowski B, Gillingwater TH, Adalbert R, Wagner D, Grumme D, Osaka H, Conforti L, Arnhold S, Addicks K, Wada K, Ribchester RR, Coleman MP. The slow Wallerian degeneration gene, Wlds, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain. 2005;128:405–416. doi: 10.1093/brain/awh368. [DOI] [PubMed] [Google Scholar]
- 17.Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 18.Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 19.Monani UR, Pastore MT, Gavrilina TO, Jablonka S, Le TT, Andreassi C, DiCocco JM, Lorson C, Androphy EJ, Sendtner M, Morris GE, Burghes AH. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Rose FF, Meehan W, Coady TH, Garcia VB, Garcia ML, Lorson CL. The Wallerian degeneration slow (Wlds) gene does not attenuate disease in a mouse model of spinal muscular atrophy. Biochem Biophys Res Commun. 2008 doi: 10.1016/j.bbrc.2008.07.130. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sajadi A, Schneider BL, Aebischer P. Wlds mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol. 2004;14:326–330. doi: 10.1016/j.cub.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 23.Samsam M, Mi W, Wessig C, Zielasek J, Toyka KV, Coleman MP, Martini R. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model of myelin-related axonopathy. J Neurosci. 2003;23:2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simonin Y, Perrin FE, Kato AC. Axonal involvement in the Wlds neuroprotective effect: analysis of pure motoneurons in a mouse model protected from motor neuron disease at a pre-symptomatic age. Eur J Neurosci. 2006;8:2269–2274. doi: 10.1111/j.1471-4159.2006.04366.x. [DOI] [PubMed] [Google Scholar]
- 25.Slater CR. Neural influence on the postnatal changes in acetylcholine receptor distribution at nerve-muscle junctions in the mouse. Dev Biol. 1982;94:23–30. doi: 10.1016/0012-1606(82)90064-1. [DOI] [PubMed] [Google Scholar]
- 26.Vande Velde C, Garcia ML, Yin X, Trapp BD, Cleveland DW. The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. Neuromol Med. 2004;5:193–204. doi: 10.1385/NMM:5:3:193. [DOI] [PubMed] [Google Scholar]
- 27.Wishart TM, Paterson JM, Short DM, Meredith S, Robertson KA, Sutherland C, Cousin MA, Dutia MB, Gillingwater TH. Differential proteomics analysis of synaptic proteins identifies potential cellular targets and protein mediators of synaptic neuroprotection conferred by the slow Wallerian degeneration (Wlds) gene. Mol Cell Proteomics. 2007;8:1318–1330. doi: 10.1074/mcp.M600457-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.