

Abstract
Human epidermis elaborates two small cationic, highly hydrophobic antimicrobial peptides (AMP), β-defensin 2 (hBD2), and the carboxypeptide cleavage product of human cathelicidin (hCAP18), LL-37, which are co-packaged along with lipids within epidermal lamellar bodies (LBs) before their secretion. Because of their colocalization, we hypothesized that AMP and barrier lipid production could be coregulated by altered permeability barrier requirements. mRNA and immunostainable protein levels for mBD3 and cathelin-related antimicrobial peptide (CRAMP) (murine homologues of hBD2 and LL-37, respectively) increase 1–8 hours after acute permeability barrier disruption and normalize by 24 hours, kinetics that mirror the lipid metabolic response to permeability barrier disruption. Artificial permeability barrier restoration, which inhibits the lipid-synthetic response leading to barrier recovery, blocks the increase in AMP mRNA/protein expression, further evidence that AMP expression is linked to permeability barrier function. Conversely, LB-derived AMPs are also important for permeability barrier homeostasis. Despite an apparent increase in mBD3 protein, CRAMP−/− mice delayed permeability barrier recovery, attributable to defective LB contents and abnormalities in the structure of the lamellar membranes that regulate permeability barrier function. These studies demonstrate that (1) the permeability and antimicrobial barriers are coordinately regulated by permeability barrier requirements and (2) CRAMP is required for permeability barrier homeostasis.
INTRODUCTION
The cutaneous permeability barrier, which allows survival in the potentially desiccating terrestrial environment, and the epidermal antimicrobial barrier, which prevents the vast majority of exogenous pathogenic microorganisms from colonizing and invading the skin, are considered discrete, protective functions of mammalian skin (Elias et al., 2003). Although permeability barrier homeostasis and the outer antimicrobial shield both localize to the stratum corneum (SC), a unique two-compartment tissue of anucleate, proteinaceous corneocytes embedded in a lipid-enriched extra-cellular matrix (reviewed in Elias and Menon, 1991), the structural and biochemical basis for each differs. While the hydrophobic, extracellular lipids impede transcutaneous water loss (TEWL) (Grubauer et al., 1989a), it is the low water content, acidic surface pH (Fluhr and Elias, 2002), resident microflora, and certain surface-deposited proteins of eccrine and sebaceous gland origin (Schittek et al., 2001; Murakami et al., 2002), that comprise the outermost antimicrobial shield (Schroder and Harder, 2006). Yet, also present within human SC are at least four antimicrobial peptides (AMP), that is the S100 protein, S100A7 (psoriasin), RNase7, the cathelicidin (hCAP18) carboxy-terminal fragment, LL-37, and human β-defensin2 (hBD2) (Elias, 2007) (reviewed in Schröder, 2006). hBD2 and LL-37 serve not only as AMP, but also as distal sensors of the innate immune system (Fulton et al., 1997; Gallo and Huttner, 1998; Gallo et al., 2002; Harder and Schroder, 2005; Lehrer, 2005; Niyonsaba and Ogawa, 2005). Although expressed at low levels under basal conditions, hBD2 and LL-37 are inducible by UV-B, chronic inflammation, pathogen challenge, and/or during wound healing (Frohm et al., 1997; Liu et al., 2002; Heilborn et al., 2003; de Jongh et al., 2005; Mallbris et al., 2005; Sorensen et al., 2005; Weber et al., 2005).
Both hBD2 and LL-37 are packaged within a novel secretory organelle, the epidermal lamellar body (LB) (Oren et al., 2003; Braff et al., 2005), which delivers barrier lipids to the SC interstices (Elias and Menon, 1991). Hence, we hypothesized that these two functions could be linked (Elias, 2005; Elias and Choi, 2005), and explored the inter-relationship between the cutaneous permeability and antimicrobial barriers. We report here that these two functions are coregulated specifically in response to permeability barrier insults; and conversely, that at least one AMP, LL-37, is required for normal permeability barrier homeostasis. Together, these studies demonstrate that these two critical defensive functions of SC are not disparate, but rather interdependent processes.
RESULTS
Constitutive expression of both mBD3 and CRAMP in normal epidermis
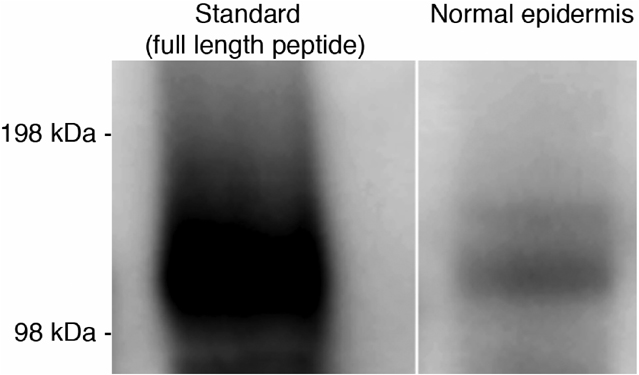
mRNA and protein levels of key synthetic enzymes of cholesterol and fatty acid synthesis increase rapidly in the underlying epidermis after acute permeability barrier disruption (Feingold, 1991). Hence, we first assessed the kinetics of changes in epidermal cathelin-related antimicrobial peptide (CRAMP)/mBD3 mRNA and protein levels after acute permeability barrier disruption by repeated tape stripping. Prior reports claim that both hBD2 and LL-37 are not expressed in normal human epidermis (eg, de Jongh et al., 2005). Yet, murine epidermis clearly demonstrates readily detectible levels of both mouse β-defensin and CRAMP protein in the outer epidermis by immunofluorescence (Figure 1a and e). To insure that our antibodies were detecting these peptides, we assessed CRAMP expression in CRAMP−/− epidermis, and found no detectible signal, while the mBD3 antibody labels a single band in Western blots of extracts of normal mouse epidermis (Figures S2 and S3).
Figure 1. Acute permeability barrier disruption by tape stripping (TS) stimulates CRAMP and mBD3 expression; upregulation is blocked by occlusion.

Flanks of normal hairless mice (n=3 in all groups) were sequentially tape stripped until TEWL≥10-fold higher than in untreated controls. Tape-stripped (TS) sites on some animals were immediately occluded with a vapor-impermeable (Latex) membrane. (a–h) Frozen sections (8 μm) from biopsies of CRAMP and mBD3 were immunostained as described in Materials and Methods. (i and j) mRNA was isolated from epidermis and quantitated by reverse transcription–PCR, using 18S RNA as standard (see Materials and Methods). (b and f) The levels of CRAMP and mBD3 are at baseline, likely replenished via rapid secretion of preformed lamellar body contents. (i and j) As expected, there is no significant increase in mRNA levels at this time point. At 4 hours after TS, expression of CRAMP and mBD3 increases (c and g), which is blocked by occlusion (d and h). This correlates with increased mRNA levels for both CRAMP and mBD3 at the same time point (i and j). Bars=50 μm.
Permeability barrier disruption upregulates mBD3 and CRAMP expression
Prior studies have shown that acute barrier disruption (TEWL rates >10 × normal) removes not only extracellular lipids (Grubauer et al., 1989a), but it also co-extracts both preformed mBD3 and CRAMP from SC (Elias and Choi, 2005). By 1–2 hours after acute disruption, substantial repletion of both CRAMP and mBD3 can be detected (Figure 1b and f), a finding that correlates with the known rapid secretion of the pre-formed pool of preformed LB contents from the stratum granulosum (SG) after acute disruption (Menon et al., 1992; Elias et al., 1998). By 4 hours after tape stripping, CRAMP and mBD3 protein levels surpass pretreatment levels, increasing still further at 8 hours, with immunodetectable protein declining toward pretreatment levels by 24 hours (8 and 24 hours data not shown). As seen in Figure 1i and j, CRAMP and mBD3 mRNA levels also increase between 1 and 4 hours after tape stripping, but mBD3 mRNA levels increase more rapidly than do mRNA levels for CRAMP (Figure 1i and j). Thus, permeability barrier disruption by tape stripping is followed by rapid reappearance of both AMP protein and mRNA in the epidermis, which precisely parallels the kinetics of the lipid metabolic response to permeability barrier disruption (Feingold, 1991).
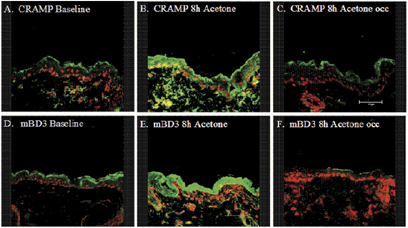
To assess whether increased AMP expression can be attributed to permeability barrier perturbation, independent of method, we next assessed changes in AMP expression after equivalent degrees of barrier disruption, induced by repeated acetone wipes. Solvent-induced barrier disruption increases levels of both mBD3 and CRAMP immunostainable protein, again between baseline and 8 hours (Figure S1B and E vs A and D), as following tape stripping (c.f., Figure 1).
Artificial permeability barrier restoration blocks AMP upregulation
To assess directly the link between AMP expression and permeability barrier function, we next restored permeability barrier homeostasis artificially by application of a vapor-impermeable Latex® membrane, immediately after either tape stripping or acetone treatment. As with lipid metabolic responses, artificial barrier restoration blocks the expected, tape stripping- and acetone-induced increases in both mBD3 and CRAMP protein immunostaining and mRNA expression (Figure 1d and h, i and j; Figure S1C and F). Thus, acute alterations in permeability barrier status, regardless of type, specifically regulate expression of these AMP.
Permeability barrier homeostasis is abnormal in CRAMP knockout epidermis
Whereas the above studies demonstrate that permeability barrier requirements regulate both mBD3 and CRAMP expression, the converse possibility, that is, AMP are required for permeability barrier homeostasis was next addressed, assessing permeability barrier function in CRAMP knockout (k.o.) mice, which display defective resistance to group A Streptococcus pyogenes (Nizet et al., 2001). Although CRAMP is not detectable in CRAMP k.o. mice (Figure S3E), immunostaining of mBD3 in CRAMP−/− epidermis greatly exceeds immunolabelling of this AMP in wild-type (wt) mice (Figure S3B vs A). Under basal conditions, SC hydration, surface pH, and basal permeability barrier function, assessed as TEWL, are all comparable in CRAMP−/− and wt mice (Table S1). To assess whether the permeability barrier is impaired in CRAMP−/− mice, even under basal conditions, we utilized an alternate, ultrastructural method to assess permeability barrier function, that is, visualization of the outward movement of the low-molecular-weight, watersoluble, electron-dense tracer, colloidal lanthanum nitrate (Figure 2, curved arrows). While perfused tracer fails to reach the SG–SC interface in wt mice (Figure 2a and b), lanthanum moves above the SG–SC interface, breaching the SC in CRAMP−/− mice (Figure 2c). Yet, tracer does not penetrate across the full thickness of the SC of CRAMP−/− mice, consistent with normal TEWL levels in CRAMP−/− epidermis. These results demonstrate that CRAMP−/− mice display subtle barrier abnormality, even under basal conditions.
Figure 2. Soluble tracer moves up to and through SC interstices in CRAMP −/− epidermis.
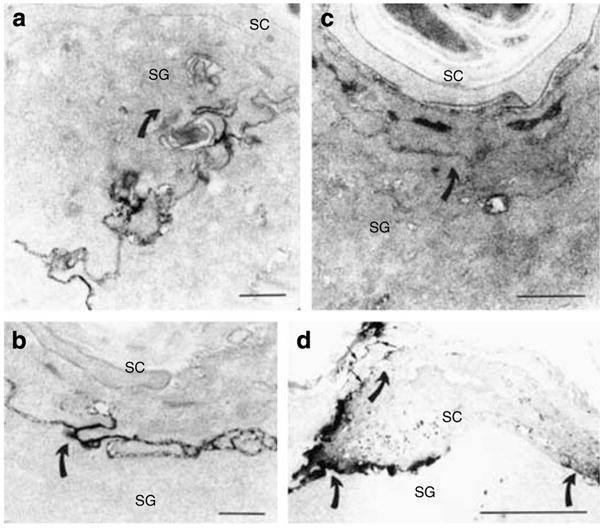
In wt epidermis, lanthanum tracer outward egress (indicated by direction of curved arrows) is blocked at level of SG (a), while tracer breaches the SG–SC interstices, and focally even above that level, in CRAMP−/− epidermis (c). Six hours after acute barrier disruption by tape stripping, tracer egress is again impeded at level of outer SG in wt epidermis (b, curved arrows), indicating restoration of normal barrier function. Yet, abundant tracer still traverses the entire SC, primarily via the interstices, in CRAMP−/− epidermis (d, arrows). (a–d) Osmium tetroxide post-fixation. Bars=1 μm (a and c); 0.5 μm (b); and 5 μm (c).
Defective permeability barrier function typically is accentuated when the kinetics of permeability barrier recovery is assessed after acute disruption (“cutaneous stress test”) (Feingold et al., 1991). The kinetics of barrier recovery are delayed significantly in k.o. versus wt mice at 3 hours (P<0.001), and apparently also at 6 hours (P<0.07) (Figure 3). Thus, while the SC of CRAMP−/− mice provides a permeability barrier sufficient for survival under basal conditions, it demonstrates subtle abnormalities even under these non-stressed conditions; and absence of CRAMP provokes a marked abnormality in permeability barrier homeostasis.
Figure 3. CRAMP k.o. mice display a significant delay in permeability barrier recovery after tape stripping.
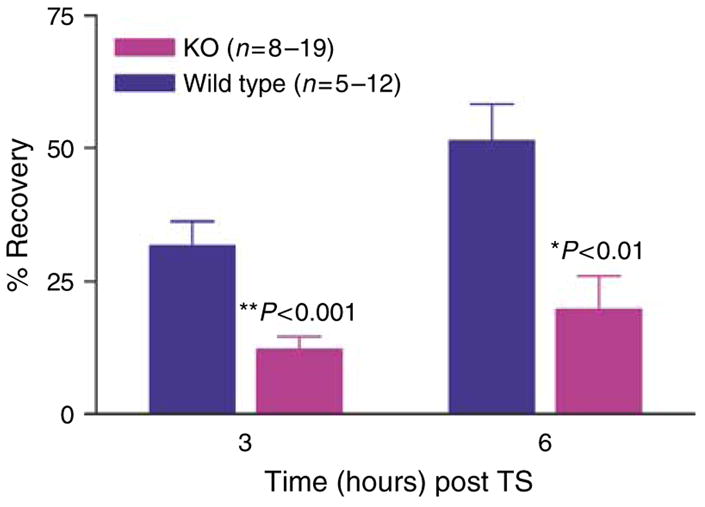
Barrier recovery after tape stripping was compared in 6-week old CRAMP (Cnl-1-) k.o. versus wt mice (29). Mice were shaved 24 hours before tape stripping, which was repeated until TEWL rates ≥10-fold normal levels. The extent of permeability barrier recovery, as percent of the original abnormality, was assessed 3 and 6 hours after tape stripping.
Abnormalities in the lamellar body secretory system in CRAMP−/− epidermis
The structural basis for the permeability barrier abnormality in CRAMP−/− mice can be attributed to distinctive abnormalities in LB contents and post-secretory membrane structure under basal conditions, and after acute barrier disruption, in parallel with delayed barrier recovery kinetics (c.f., Figure 3). Although the quantities (density) of LB appear comparable in k.o. versus wt epidermis (Figure 4c, wt not shown), the internal lamellar cargo of LB is often sparse in k.o. epidermis (Figure 4c, arrows vs inset (=wt)). Although these vacuolated LB appear to undergo exocytosis normally, they form highly abnormal, electron-lucent vacuoles/clefts at the SG–SC interface (Figure 4a, asterisks) and c, double arrows vs 4b, wt; Figure S4C and D). Despite the paucity of lamellar contents in some LB in CRAMP epidermis, other proteins (eg, the hydrolytic enzyme, acid lipase) are packaged and secreted from LB normally (Figure S4A and B). These results demonstrate structural abnormalities in LB contents in CRAMP−/− epidermis.
Figure 4. Abnormalities LB secretory system in CRAMP −/− epidermis.

(a, c, and e) CRAMP−/− epidermis. (b and d, inset) wt epidermis. Under basal conditions, CRAMP−/− epidermis reveals abnormalities in contents of individual LB (c, single arrows). In contrast, wt epidermis has LB with replete contents (d, inset, arrowheads). In the CRAMP−/− mouse epidermis, there are non-lamellar clefts within secreted LB contents at the SG–SC interface (a, asterisks; c and e, double arrows). In wt epidermis, transformation of secreted LB contents into lamellar bilayers already occurs at SG–SC interface, as demonstrated in (b) (arrows). (a and b) Ruthenium textroxide post-fixation; (c and d) osmium tetroxide post-fixation. Bars=0.2 μm (a and b); 1 μm (c and e); and 0.1 μm (d, inset).
Using ruthenium tetroxide post-fixation, it is possible to visualize the post-secretory, structural transformation of secreted LB contents into the “mature” lamellar membrane bilayers that regulate the permeability barrier (Elias and Menon, 1991). Whereas formation of mature lamellar membranes begins at the SG–SC interface (Figures 4b and 5b), and transformation is completed by the next (=second) interspace in wt epidermis, the formation of “mature” lamellae is both delayed (Figure 5c, open arrows) and incomplete, that is, non-transformed LB contents persist high into the SC interstices in k.o. mice (Figure 5a, open arrows). Even when mature lamellar structures eventually form (Figure 5a, solid arrows), they appear foreshortened, and fail to fully engorge the SC interstices (Figure 5a vs b, solid arrows in wt). Thus, absence of CRAMP leads to the formation of defective lamellar bilayers.
Figure 5. Delayed and incomplete formation of lamellar bilayers in CRAMP −/− epidermis.

In wt epidermis under basal conditions, secreted LB contents transform into lamellar bilayers within the SG–SC interface (b, solid arrows). In contrast, secreted lamellar contents remain partially untransformed several layers above SG-SC interface (a and c, open arrows), within SC interstices of CRAMP−/− mice. Moreover, lamellar bilayers, when present in SC of k.o. mice, fail to fill interstices (a, solid arrows). (a–c) Ruthenium textroxide post-fixation. Bars=0.2 μm.
To assess whether these abnormal-appearing lamellar membranes account for defective permeability barrier homeostasis, we next assessed the pathway of lanthanum tracer egress through the SC interstices of CRAMP−/− versus wt mice. By 6 hours after acute barrier disruption, wt SC completely excludes lanthanum (Figure 2b), as under basal conditions (c.f.; Figure 2a), and consistent with the >50% barrier recovery in wt (and normal) murine epidermis at 6 hours (c.f.; Figure 3). In contrast, tracer still fully permeates the SC interstices in CRAMP−/− epidermis at 6 hours (Figure 2d), consistent with the delay in permeability barrier recovery at this time point (c.f., Figure 3). Since the tracer moves across the SC of k.o. mice solely through extracellular domains (c.f.; Figure 2d), abnormal lamellar membrane architecture accounts for the permeability barrier abnormality (c.f.; Figure 5). Together, these studies provide a structural basis for the permeability barrier abnormality in CRAMP−/− mice, demonstrating that CRAMP is critical for the formation of functionally competent, extracellular lamellar bilayers.
DISCUSSION
We assessed the relationship between permeability barrier function and antimicrobial defense in mammalian epidermis, because barrier function can be perturbed incrementally by well-defined methods, such as solvent/detergent treatment or tape stripping (Feingold, 1991). The SC of mammalian epidermis, which subserves several protective functions (Elias et al., 2003; Elias, 2005), comprises a heterogeneous structure of anucleate corneocytes embedded in a lipid-enriched, extracellular matrix (Elias and Friend, 1975; Elias, 1983; Elias and Menon, 1991), important for the permeability barrier. The extracellular matrix also contains two AMP, which contribute to antimicrobial defense. Both hBD2 and LL-37, the human homologues of mBD3 and CRAMP, are sequestered in LB (Oren et al., 2003; Braff et al., 2005), and their secretion is supported by a membrane pattern of immunostaining for hBD2 in human SC (Huh et al., 2002).
Although the constitutive expression of both of these peptides is low (Frohm et al., 1997; Liu et al., 2002; Heilborn et al., 2003; de Jongh et al., 2005; Mallbris et al., 2005; Sorensen et al., 2005; Weber et al., 2005), substantial protein is present in the outer layers of unperturbed murine epidermis, consistent with their dual roles in antimicrobial defense (Elias, 2007), and the innate immune system (Schroder, 1999; Gallo et al., 2002; Ganz, 2002; Izadpanah and Gallo, 2005; Lehrer, 2005). Finally, the increased severity of cutaneous streptococcal infections in CRAMP−/− mice clearly demonstrates that constitutive levels of CRAMP are critical for antimicrobial defense (Nizet et al., 2001).
Several results here support the hypothesis that the permeability and antimicrobial barriers of the SC may not be discrete (Elias, 2005; Elias and Choi, 2005), but rather coregulated and even interdependent functions; (1) the AMP metabolic response parallels the lipid metabolic response to acute permeability barrier disruption. When the permeability barrier is perturbed by solvent treatment or tape stripping, a sequential, lipid-synthetic response ensues in the underlying epidermis that rapidly restores normal permeability barrier homeostasis (Feingold, 1991). Acute barrier perturbation is followed sequentially by rapid secretion (within 30 minutes) of much of the preformed pool of LB (Menon et al., 1992; Elias et al., 1998); transcriptional upregulation of epidermal cholesterol, fatty acid, and ceramide synthesis (1–9 hours) (Holleran et al., 1991; Proksch et al., 1993; Ottey et al., 1995); further LB production (2–6 hours) (Menon et al., 1992); increased production of lipid hydrolases, necessary to generate lamellar membranes (9–12 hours) (Holleran et al., 1994); and increased DNA synthesis (16–24 hours) (Proksch et al., 1991). Yet, permeability barrier disruption not only removes lipids (Grubauer et al., 1989b), but it also initiates a parallel, rapid loss of AMP from SC (Elias and Choi, 2005). We show here that subsequent changes in AMP protein and mRNA levels upregulate in parallel with the lipid metabolic response. While some AMP protein is restored rapidly to the SC, presumably by secretion of the preformed pool of LB (≤60 minutes) (Menon et al., 1992; Elias et al., 1998). mRNA and protein for both AMPs begin to increase 1–4 hours after acute disruption, peaking by 4–8 hours, and returning to basal levels by 24 hours in parallel with permeability barrier normalization after acute perturbations (Menon et al., 1985; Grubauer et al., 1989b). In addition, similar kinetics of AMP production occur, independent of the method of barrier disruption, that is, tape stripping or organic solvent treatment. The most specific evidence that AMP expression is linked inextricably to the permeability barrier is shown by the blockade of the expected upregulation of AMP expression following artificial restoration of the permeability barrier with a vapor-impermeable membrane. Blockage of the increase in epidermal lipid synthesis and secretion by occlusion is the “gold standard” that links metabolic responses in the underlying epidermis specifically to permeability barrier function (Feingold, 1991). Thus, the parallel AMP metabolic and lipid metabolic responses comprise initial evidence that AMP expression is regulated by changes in permeability barrier status.
Further evidence for a link between permeability barrier function and AMP expression is supported by reports that the human homologues of these AMP, which are cargo within LB, are upregulated in psoriasis (Frohm et al., 1997; Ong et al., 2002b; de Jongh et al., 2005; Harder and Schroder, 2005), a disorder characterized by permeability barrier abnormalities that parallel disease severity (Ghadially et al., 1996).
The intimate link between these two functions is likely due to the coassembly of AMP (Oren et al., 2003; Braff et al., 2005), with barrier lipid precursors (Grayson et al., 1983, 1985), within nascent LB. Coassembly of lipids and AMP could be interdependent, because protein delivery to epidermal LB requires prior lipid deposition (Rassner et al., 1999). Finally, the interrelationship of these two functions is shown further by the fact that certain SC lipids, such as free fatty acids and ceramide metabolite (sphingosine), display potent antimicrobial activity (Miller et al., 1988; Bibel et al., 1992). Finally, secreted LB contents provide a formidable physical barrier (Elias, 2007) that both excludes exogenous pathogens, and conversely offers the path of least resistance for penetration of most microbes through compromised SC (Miller et al., 1988). Thus, when the lamellar matrix is abnormal (as in atopic dermatitis) (Fartasch et al., 1993; Chamlin et al., 2002), pathogens can penetrate due to the more “porous” nature of the SC interstices.
Utilizing CRAMP−/− mice, which display defective, cutaneous antimicrobial defense, particularly against S. pyogenes (Nizet et al., 2001), we also showed the converse, that is, at least one AMP, CRAMP, is required for normal permeability barrier function. Thus, in addition to its dual role as an antimicrobial agent and signal of the innate immune system, CRAMP displays an additional, critical structural role. Although CRAMP−/− mice display normal levels of TEWL under basal conditions, tracer perfusion studies demonstrate subtle functional abnormalities, even under these conditions. More importantly, after the stress of acute barrier disruption, CRAMP−/− mice demonstrate a highly significant delay in barrier recovery kinetics, consistent with a role for this AMP in the maintenance of permeability barrier homeostasis. This so-called “cutaneous stress test” reveals pathology in several clinical settings, where basal function is (misleadingly) normal (Ghadially et al., 1995; Altemus et al., 2001; Garg et al., 2001; Fluhr et al., 2004). Importantly, where the “cutaneous stress test” reveals abnormalities, it is usually possible to delineate the metabolic and/or structural basis for defective function, because the “stress test” amplifies the functional abnormalities (Elias, 2005). Accordingly, abnormal permeability barrier function correlates with abnormalities in both LB contents and in the extracellular lamellar membrane structure in CRAMP−/− epidermis. It is possible that CRAMP interacts with more hydrophobic, LB-derived, pro-barrier lipids in a manner that facilitates the dispersion or organization of secreted lipids into the lamellar bilayers that subserve the permeability barrier (Elias and Menon, 1991). Since CRAMP, like mBD3 and certain other AMP, is a low-molecular- weight, highly hydrophobic peptide (Schroder, 1999; Ganz, 2002; Izadpanah and Gallo, 2005), it could provide critical, amphiphilic constituents that maintain the extracellular, lamellar membrane system, while still remaining bioavailable, because it does not completely integrate into the hydrophobic core of lipid bilayers (Henzler Wildman et al., 2003). Pertinently, the apparent, compensatory upregulation of mBD3 protein in CRAMP−/− mice does not suffice to correct the barrier abnormality, suggesting a specific requirement for CRAMP for permeability barrier homeostasis. Finally, two class II nuclear hormone receptor ligands, retinoids and 1,25(OH)2 vitamin D3, display divergent effects on epidermal differentiation (Elias et al., 1981; Pillai and Bikle, 1991), with parallel regulation of hCAP and hBD2 expression, consistent with the link between epithelial integrity and AMP expression in extracutaneous tissues (Weber et al., 2005; Zasloff, 2005; Nenci et al., 2007). For example, abnormal mucosal permeability leads to inflammatory bowel disease in transgenic mice with deletion of the antimicrobial cytokine, resistin-like molecule-β (Hogan et al., 2006), and a similar relationship may pertain to patients deficient in CARD15 (Buhner et al., 2006), α-defensin, or REGIIIγ production (Strober, 2006). Since LL-37 and hBD2 are also produced by airway epithelial cells (Bals et al., 1998a, b), and since these peptides fail to upregulate in atopic dermatitis (Ong et al., 2002a), a deficiency in LL-37/hBD2 could weaken respiratory epithelial integrity, sufficient to allow infectious triggers of asthma.
MATERIALS AND METHODS
Murine models
Female hairless mice (Skh1/Hr), 8 weeks of age, were purchased from Charles River Laboratories (Wilmington, MA). CRAMP k.o. mice (Cnl-1-) and wt mice were prepared on a 127/SvJ background (Nizet et al., 2001). All animal protocols were approved by the Animal Research Committee, SFVAMC. Mice were maintained in a temperature- and humidity-controlled room, and given standard laboratory food and tap water ad libitum. Mice were subjected to acute barrier disruption by either tape stripping or acetone wipes, and left either air-exposed or immediately covered with a vapor-impermeable (Latex) membrane (Grubauer et al., 1989a).
Functional studies
Surface pH was measured under basal conditions with a flat, glass surface electrode from Mettler-Toledo (Columbus, OH), attached to a pH meter (PH 900; Courage and Khazaka). SC hydration was quantitated as changes in electrical capacitance in arbitrary units (Corneometer CM 820; Courage and Khazaka, Köln, Germany) in the basal state of both wt and CRAMP−/− mice. The mean of three separate measurements on each animal was utilized for statistical comparisons. TEWL in the basal state was measured as p.p.m./cm2/hour with an electrolytic water analyzer (Meeco, Warrington, PA) (Grubauer et al., 1989a). Barrier recovery kinetics were compared by measuring TEWL immediately after 3, 6, and 24 hours following acute barrier disruption by either tape stripping or acetone treatment (Grubauer et al., 1989a).
Tissue preparation, protein, and RNA isolation
Skin samples were obtained immediately after, 4, 8, or 24 hours following tape stripping or acetone treatment of normal hairless mice, with or without prior occlusion, as above, and from CRAMP−/− versus wt littermates. Epidermis was obtained by EDTA separation, and total RNA was extracted with a commercially available kit, RNeasy Mini RNA isolation kit from Qiagen (Valencia, CA). RNA solution (50 μl) was reverse-transcribed to cDNA (Horikoshi et al., 1992), and real-time quantitative PCR was performed for mBD3, CRAMP, and 18S (internal control) in triplicate on an ABI 7900 machine, using the SYBR green detection kit from Applied Biosystems (Foster City, CA), (Tirmenstein et al., 2000). Primer sequences were as follows: mBD3 fw: ATTTCTCCTGGTGCTCGTGT 3, rev: GGAACTCCACAACTGCCAAT; and 18S fw: GTAACCCGTTGAACCCCATT, rev: CCATCCAATCGGTAGTAGCG. The mBD3 and 18S PCR primers were synthesized by Biosearch Technologies (Novato, CA), while murine cathelicidin primers were from Richard Gallo. Standard reaction volumes were 20 μl with 250 ng cDNA and 125 nM of mBD3, 500 nM of 18S, or 1 μM final concentration of cathelicidin primers. Initial steps of reverse transcription–PCR (RT–PCR) were 2 minutes at 50°C, followed by a 10 minutes at 95°C. Cycles (n=40) consisted of a 15-second melt at 95°C, followed by a 300-second annealing/extension at 60°C. Threshold (Ct) analysis for all samples was set at 0.50 relative fluorescence units. Quantitative polymerase chain reaction (QPCR) data were analyzed using the method (Winer et al., 1999; Schmittgen et al., 2000).
Western immunoblotting
For Western blotting, low-molecular-weight, hydrophobic, cationic peptides were extracted, and separated from full-thickness mouse skin in 10mM EDTA (Schroder, 2001). Extractions were performed in an acidic buffer (30% acetonitrile, 0.1% formic acid; pH<3); extracts were homogenized on ice, centrifuged at 14,000 r.p.m. for 30 minutes at 4 °C, and supernatants were re-centrifuged for 15 minutes before protein fractionation. An equal amount of extracted protein in NuPAGE sample buffer in water was heated at 85 °C, without reducing agents, followed by loading of equal amounts of samples from each experimental group onto 12% tricine gels (Invitrogen, Carlsbad, CA), using β-actin as an internal standard. After electrophoresis, proteins were transferred from gels onto polyvinylidene difluoride membranes and electrophoresed for 1 hour in tricine/glycine transfer buffer, followed by immunoblotting with the rabbit anti-mouse mBD3 antibody (Alpha Diagnostics, San Antonio, TX). Antibody binding to mBD3 was detected with the Western-Breeze chemiluminescence kit, following the manufacturer’s protocol (Invitrogen).
Electron microscopy
Skin biopsies were taken from CRAMP−/− and wt mice in the basal state, and processed as described for 0.5% ruthenium tetroxide (RuO4) and 2% aqueous osmium tetroxide (OsO4), post-fixation (Hou et al., 1991). Samples then were dehydrated in graded ethanol solutions and embedded in an Epon-epoxy mixture. Prior to post-fixation, some samples were processed for ultrastructural cytochemistry, utilizing triacylglycerol lipase as an alternate marker of protein assembly and secretion from LB (Rassner et al., 1999). Some fresh biopsies were submerged en bloc in 4% colloidal lanthanum nitrate, which serves as an electron-dense tracer of water movement through the epidermis (Hanley et al., 1997; Elias et al., 2002), and post-fixed in osmium tetroxide. Ultrathin sections were examined, with or without further lead citrate contrasting, in Zeiss 10A electron microscope, operated at 60 kV.
Immunofluorescence and immunohistochemistry
Freshly obtained skin biopsies were snap frozen in liquid nitrogen, and stored in a tissue-embedding medium. Frozen sections (8 μm) were soaked in acetone for 10 minutes, washed in phosphate-buffered saline, and blocked with 4% BSA and 0.05% cold fish gelatin in phosphate-buffered saline for 30 minutes. Slides were incubated overnight at 4°C with CRAMP (from Richard Gallo) or mBD3 primary antibodies, followed by incubation with an FITC-conjugated goat anti-rabbit, secondary antibody (Alpha Diagnostic, no. 5) for 40 minutes at room temperature. Slides were counter-stained with propidium iodide for nuclear visualization and examined on a Leica TCS-SP confocal microscope.
Data were expressed as means ±SE. Statistical analyses were performed using paired and unpaired Student’s t-tests.
Supplementary Material
Figure S1. Acute permeability barrier disruption by an alternative method (acetone treatment) stimulates CRAMP/mBD3 expression; upregulation again is blocked by occlusion.
{kind=link}
Figure S2. Western immunoblotting reveals a single band corresponding to mBD3 in normal mouse epidermis.
{kind=link}
Figure S3. Compensatory increase in mBD3 protein in CRAMP−/− epidermis.
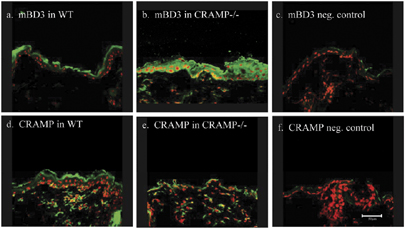{kind=link}
Figure S4. Normal content and secretion of another lamellar body protein in CRAMP−/− epidermis.
{kind=link}
Table S1. CRAMP−/− mice display no significant differences in either hydration, surface pH, or basal TEWL.
Acknowledgments
These studies were supported by NIH Grants AR19098, AR39448(PP), AI059311, and the Medical Research Service, Department of Veterans Affairs, and a NIH post-doctoral fellowship (AR07175-28) to Dr Aberg. We gratefully acknowledge the excellent editorial assistance of Ms Joan Wakefield and Jerelyn Magnusson.
Abbreviations
- AMP
antimicrobial peptides
- BD
β-defensin
- CRAMP
cathelin-related antimicrobial peptide
- hBD2
human β-defensin2
- hCAP
human cathelicidin
- k.o
knockout
- LB
lamellar body
- SC
stratum corneum
- SG
stratum granulosum
- TEWL
transcutaneous water loss
- wt
wild type
Footnotes
A preliminary report of these studies was presented at the Society for Investigative Dermatology annual meeting, May, 2006 (J Invest Dermatol 126:60, 2006a)
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Altemus M, Rao B, Dhabhar FS, Ding W, Granstein RD. Stress-induced changes in skin barrier function in healthy women. J Invest Dermatol. 2001;117:309–17. doi: 10.1046/j.1523-1747.2001.01373.x. [DOI] [PubMed] [Google Scholar]
- Bals R, Wang X, Wu Z, Freeman T, Bafna V, Zasloff M, et al. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest. 1998a;102:874–80. doi: 10.1172/JCI2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bals R, Wang X, Zasloff M, Wilson JM. The peptide antibiotic LL-37/hCAP- 18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci USA. 1998b;95:9541–6. doi: 10.1073/pnas.95.16.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel DJ, Aly R, Shinefield HR. Antimicrobial activity of sphingosines. J Invest Dermatol. 1992;98:269–73. doi: 10.1111/1523-1747.ep12497842. [DOI] [PubMed] [Google Scholar]
- Braff MH, Di Nardo A, Gallo RL. Keratinocytes store the antimicrobial peptide cathelicidin in lamellar bodies. J Invest Dermatol. 2005;124:394–400. doi: 10.1111/j.0022-202X.2004.23443.x. [DOI] [PubMed] [Google Scholar]
- Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, et al. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut. 2006;55:342–7. doi: 10.1136/gut.2005.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamlin SL, Kao J, Frieden IJ, Sheu MY, Fowler AJ, Fluhr JW, et al. Ceramide-dominant barrier repair lipids alleviate childhood atopic dermatitis: changes in barrier function provide a sensitive indicator of disease activity. J Am Acad Dermatol. 2002;47:198–208. doi: 10.1067/mjd.2002.124617. [DOI] [PubMed] [Google Scholar]
- de Jongh GJ, Zeeuwen PL, Kucharekova M, Pfundt R, van der Valk PG, Blokx W, et al. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J Invest Dermatol. 2005;125:1163–73. doi: 10.1111/j.0022-202X.2005.23935.x. [DOI] [PubMed] [Google Scholar]
- Elias PM. Epidermal lipids, barrier function, and desquamation. J Invest Dermatol. 1983;80(Suppl):44s–9s. doi: 10.1038/jid.1983.12. [DOI] [PubMed] [Google Scholar]
- Elias PM. Stratum corneum defensive functions: an integrated view. J Invest Dermatol. 2005;125:183–200. doi: 10.1111/j.0022-202X.2005.23668.x. [DOI] [PubMed] [Google Scholar]
- Elias PM. The skin barrier as an innate immune element. Semin Immunopathol. 2007;29:3–14. doi: 10.1007/s00281-007-0060-9. [DOI] [PubMed] [Google Scholar]
- Elias PM, Choi EH. Interactions among stratum corneum defensive functions. Exp Dermatol. 2005;14:719–26. doi: 10.1111/j.1600-0625.2005.00363.x. [DOI] [PubMed] [Google Scholar]
- Elias PM, Cullander C, Mauro T, Rassner U, Komuves L, Brown BE, et al. The secretory granular cell: the outermost granular cell as a specialized secretory cell. J Investig Dermatol Symp Proc. 1998;3:87–100. doi: 10.1038/jidsymp.1998.20. [DOI] [PubMed] [Google Scholar]
- Elias PM, Feingold KR, Fluhr JW. Skin as an organ of protection, Fitzpatrick’s Dermatology in General Medicine. 6. New York, NY: 2003. pp. 107–18. [Google Scholar]
- Elias PM, Friend DS. The permeability barrier in mammalian epidermis. J Cell Biol. 1975;65:180–91. doi: 10.1083/jcb.65.1.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias PM, Fritsch PO, Lampe M, Williams ML, Brown BE, Nemanic M, et al. Retinoid effects on epidermal structure, differentiation, and permeability. Lab Invest. 1981;44:531–40. [PubMed] [Google Scholar]
- Elias PM, Menon GK. Structural and lipid biochemical correlates of the epidermal permeability barrier. Adv Lipid Res. 1991;24:1–26. doi: 10.1016/b978-0-12-024924-4.50005-5. [DOI] [PubMed] [Google Scholar]
- Elias PM, Schmuth M, Uchida Y, Rice RH, Behne M, Crumrine D, et al. Basis for the permeability barrier abnormality in lamellar ichthyosis. Exp Dermatol. 2002;11:248–56. doi: 10.1034/j.1600-0625.2001.110308.x. [DOI] [PubMed] [Google Scholar]
- Fartasch M, Bassukas ID, Diepgen TL. Structural relationship between epidermal lipid lamellae, lamellar bodies and desmosomes in human epidermis: an ultrastructural study. Br J Dermatol. 1993;128:1–9. doi: 10.1111/j.1365-2133.1993.tb00138.x. [DOI] [PubMed] [Google Scholar]
- Feingold KR. The regulation and role of epidermal lipid synthesis. Adv Lipid Res. 1991;24:57–82. doi: 10.1016/b978-0-12-024924-4.50007-9. [DOI] [PubMed] [Google Scholar]
- Feingold KR, Man MQ, Proksch E, Menon GK, Brown BE, Elias PM. The lovastatin-treated rodent: a new model of barrier disruption and epidermal hyperplasia. J Invest Dermatol. 1991;96:201–9. doi: 10.1111/1523-1747.ep12461153. [DOI] [PubMed] [Google Scholar]
- Fluhr JW, Elias PM. Stratum corneum pH: formation and function of the “acid mantle”. Exog Dermatol. 2002;1:163–75. [Google Scholar]
- Fluhr JW, Mao-Qiang M, Brown BE, Hachem JP, Moskowitz DG, Demerjian M, et al. Functional consequences of a neutral pH in neonatal rat stratum corneum. J Invest Dermatol. 2004;123:140–51. doi: 10.1111/j.0022-202X.2004.22726.x. [DOI] [PubMed] [Google Scholar]
- Frohm M, Agerberth B, Ahangari G, Stahle-Backdahl M, Liden S, Wigzell H, et al. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J Biol Chem. 1997;272:15258–63. doi: 10.1074/jbc.272.24.15258. [DOI] [PubMed] [Google Scholar]
- Fulton C, Anderson GM, Zasloff M, Bull R, Quinn AG. Expression of natural peptide antibiotics in human skin. Lancet. 1997;350:1750–1. doi: 10.1016/S0140-6736(05)63574-X. [DOI] [PubMed] [Google Scholar]
- Gallo RL, Huttner KM. Antimicrobial peptides: an emerging concept in cutaneous biology. J Invest Dermatol. 1998;111:739–43. doi: 10.1046/j.1523-1747.1998.00361.x. [DOI] [PubMed] [Google Scholar]
- Gallo RL, Murakami M, Ohtake T, Zaiou M. Biology and clinical relevance of naturally occurring antimicrobial peptides. J Allergy Clin Immunol. 2002;110:823–31. doi: 10.1067/mai.2002.129801. [DOI] [PubMed] [Google Scholar]
- Ganz T. Immunology. Versatile defensins. Science. 2002;298:977–9. doi: 10.1126/science.1078708. [DOI] [PubMed] [Google Scholar]
- Garg A, Chren MM, Sands LP, Matsui MS, Marenus KD, Feingold KR, et al. Psychological stress perturbs epidermal permeability barrier homeostasis: implications for the pathogenesis of stress-associated skin disorders. Arch Dermatol. 2001;137:53–9. doi: 10.1001/archderm.137.1.53. [DOI] [PubMed] [Google Scholar]
- Ghadially R, Brown BE, Sequeira-Martin SM, Feingold KR, Elias PM. The aged epidermal permeability barrier. Structural, functional, and lipid biochemical abnormalities in humans and a senescent murine model. J Clin Invest. 1995;95:2281–90. doi: 10.1172/JCI117919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadially R, Reed JT, Elias PM. Stratum corneum structure and function correlates with phenotype in psoriasis. J Invest Dermatol. 1996;107:558–64. doi: 10.1111/1523-1747.ep12582813. [DOI] [PubMed] [Google Scholar]
- Grayson S, Johnson-Winegar AD, Elias PM. Isolation of lamellar bodies from neonatal mouse epidermis by selective sequential filtration. Science. 1983;221:962–4. doi: 10.1126/science.6879194. [DOI] [PubMed] [Google Scholar]
- Grayson S, Johnson-Winegar AG, Wintroub BU, Isseroff RR, Epstein EH, Jr, Elias PM. Lamellar body-enriched fractions from neonatal mice: preparative techniques and partial characterization. J Invest Dermatol. 1985;85:289–94. doi: 10.1111/1523-1747.ep12276826. [DOI] [PubMed] [Google Scholar]
- Grubauer G, Elias PM, Feingold KR. Transepidermal water loss: the signal for recovery of barrier structure and function. J Lipid Res. 1989a;30:323–33. [PubMed] [Google Scholar]
- Grubauer G, Feingold KR, Harris RM, Elias PM. Lipid content and lipid type as determinants of the epidermal permeability barrier. J Lipid Res. 1989b;30:89–96. [PubMed] [Google Scholar]
- Hanley K, Jiang Y, Crumrine D, Bass NM, Appel R, Elias PM, et al. Activators of the nuclear hormone receptors PPARalpha and FXR accelerate the development of the fetal epidermal permeability barrier. J Clin Invest. 1997;100:705–12. doi: 10.1172/JCI119583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder J, Schroder JM. Psoriatic scales: a promising source for the isolation of human skin-derived antimicrobial proteins. J Leukoc Biol. 2005;77:476–86. doi: 10.1189/jlb.0704409. [DOI] [PubMed] [Google Scholar]
- Heilborn JD, Nilsson MF, Kratz G, Weber G, Sorensen O, Borregaard N, et al. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J Invest Dermatol. 2003;120:379–89. doi: 10.1046/j.1523-1747.2003.12069.x. [DOI] [PubMed] [Google Scholar]
- Henzler Wildman KA, Lee DK, Ramamoorthy A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry. 2003;42:6545–58. doi: 10.1021/bi0273563. [DOI] [PubMed] [Google Scholar]
- Hogan SP, Seidu L, Blanchard C, Groschwitz K, Mishra A, Karow ML, et al. Resistin-like molecule beta regulates innate colonic function: barrier integrity and inflammation susceptibility. J Allergy Clin Immunol. 2006;118:257–68. doi: 10.1016/j.jaci.2006.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleran WM, Feingold KR, Man MQ, Gao WN, Lee JM, Elias PM. Regulation of epidermal sphingolipid synthesis by permeability barrier function. J Lipid Res. 1991;32:1151–8. [PubMed] [Google Scholar]
- Holleran WM, Takagi Y, Menon GK, Jackson SM, Lee JM, Feingold KR, et al. Permeability barrier requirements regulate epidermal beta-glucocerebrosidase. J Lipid Res. 1994;35:905–12. [PubMed] [Google Scholar]
- Horikoshi S, Fukuda K, Ray PE, Sawada M, Bruggeman LA, Klotman PE. A PCR method for the quantitative assessment of mRNA for laminin A, B1, and B2 chains. Kidney Int. 1992;42:764–9. doi: 10.1038/ki.1992.345. [DOI] [PubMed] [Google Scholar]
- Hou SY, Mitra AK, White SH, Menon GK, Ghadially R, Elias PM. Membrane structures in normal and essential fatty acid-deficient stratum corneum: characterization by ruthenium tetroxide staining and X-ray diffraction. J Invest Dermatol. 1991;96:215–23. doi: 10.1111/1523-1747.ep12461361. [DOI] [PubMed] [Google Scholar]
- Huh WK, Oono T, Shirafuji Y, Akiyama H, Arata J, Sakaguchi M, et al. Dynamic alteration of human beta-defensin 2 localization from cytoplasm to intercellular space in psoriatic skin. J Mol Med. 2002;80:678–84. doi: 10.1007/s00109-002-0373-z. [DOI] [PubMed] [Google Scholar]
- Izadpanah A, Gallo RL. Antimicrobial peptides. J Am Acad Dermatol. 2005;52:381–90. doi: 10.1016/j.jaad.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Lehrer RI. In defense of skin. J Invest Dermatol. 2005;125:viii–x. doi: 10.1111/j.0022-202X.2005.23769.x. discussion x–xi. [DOI] [PubMed] [Google Scholar]
- Liu AY, Destoumieux D, Wong AV, Park CH, Valore EV, Liu L, et al. Human beta-defensin-2 production in keratinocytes is regulated by interleukin-1, bacteria, and the state of differentiation. J Invest Dermatol. 2002;118:275–81. doi: 10.1046/j.0022-202x.2001.01651.x. [DOI] [PubMed] [Google Scholar]
- Mallbris L, Edstrom DW, Sundblad L, Granath F, Stahle M. UVB upregulates the antimicrobial protein hCAP18 mRNA in human skin. J Invest Dermatol. 2005;125:1072–4. doi: 10.1111/j.0022-202X.2005.23872.x. [DOI] [PubMed] [Google Scholar]
- Menon GK, Feingold KR, Elias PM. Lamellar body secretory response to barrier disruption. J Invest Dermatol. 1992;98:279–89. doi: 10.1111/1523-1747.ep12497866. [DOI] [PubMed] [Google Scholar]
- Menon GK, Grayson S, Elias PM. Ionic calcium reservoirs in mammalian epidermis: ultrastructural localization by ion-capture cytochemistry. J Invest Dermatol. 1985;84:508–12. doi: 10.1111/1523-1747.ep12273485. [DOI] [PubMed] [Google Scholar]
- Miller SJ, Aly R, Shinefeld HR, Elias PM. In vitro and in vivo antistaphylococcal activity of human stratum corneum lipids. Arch Dermatol. 1988;124:209–15. [PubMed] [Google Scholar]
- Murakami M, Ohtake T, Dorschner RA, Schittek B, Garbe C, Gallo RL. Cathelicidin antimicrobial peptide expression in sweat, an innate defense system for the skin. J Invest Dermatol. 2002;119:1090–5. doi: 10.1046/j.1523-1747.2002.19507.x. [DOI] [PubMed] [Google Scholar]
- Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–61. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- Niyonsaba F, Ogawa H. Protective roles of the skin against infection: implication of naturally occurring human antimicrobial agents beta-defensins, cathelicidin LL-37 and lysozyme. J Dermatol Sci. 2005;40:157–68. doi: 10.1016/j.jdermsci.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, et al. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414:454–7. doi: 10.1038/35106587. [DOI] [PubMed] [Google Scholar]
- Ong PY, Hamid QA, Travers JB, Strickland I, Al Kerithy M, Boguniewicz M, et al. Decreased IL-15 may contribute to elevated IgE and acute inflammation in atopic dermatitis. J Immunol. 2002a;168:505–10. doi: 10.4049/jimmunol.168.1.505. [DOI] [PubMed] [Google Scholar]
- Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002b;347:1151–60. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- Oren A, Ganz T, Liu L, Meerloo T. In human epidermis, beta-defensin 2 is packaged in lamellar bodies. Exp Mol Pathol. 2003;74:180–2. doi: 10.1016/s0014-4800(02)00023-0. [DOI] [PubMed] [Google Scholar]
- Ottey KA, Wood LC, Grunfeld C, Elias PM, Feingold KR. Cutaneous permeability barrier disruption increases fatty acid synthetic enzyme activity in the epidermis of hairless mice. J Invest Dermatol. 1995;104:401–4. doi: 10.1111/1523-1747.ep12665893. [DOI] [PubMed] [Google Scholar]
- Pillai S, Bikle DD. Epidermal vitamin D metabolism, function, and regulation. Adv Lipid Res. 1991;24:321–41. doi: 10.1016/b978-0-12-024924-4.50015-8. [DOI] [PubMed] [Google Scholar]
- Proksch E, Feingold KR, Man MQ, Elias PM. Barrier function regulates epidermal DNA synthesis. J Clin Invest. 1991;87:1668–73. doi: 10.1172/JCI115183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proksch E, Holleran WM, Menon GK, Elias PM, Feingold KR. Barrier function regulates epidermal lipid and DNA synthesis. Br J Dermatol. 1993;128:473–82. doi: 10.1111/j.1365-2133.1993.tb00222.x. [DOI] [PubMed] [Google Scholar]
- Rassner U, Feingold KR, Crumrine DA, Elias PM. Coordinate assembly of lipids and enzyme proteins into epidermal lamellar bodies. Tissue Cell. 1999;31:489–98. doi: 10.1054/tice.1999.0050. [DOI] [PubMed] [Google Scholar]
- Schittek B, Hipfel R, Sauer B, Bauer J, Kalbacher H, Stevanovic S, et al. Dermcidin: a novel human antibiotic peptide secreted by sweat glands. Nat Immunol. 2001;2:1133–7. doi: 10.1038/ni732. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription–polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- Schroder JM. Epithelial antimicrobial peptides: innate local host response elements. Cell Mol Life Sci. 1999;56:32–46. doi: 10.1007/s000180050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder JM. Isolation and purification of chemokines from natural sources. Mol Biotechnol. 2001;18:71–7. doi: 10.1385/MB:18:1:71. [DOI] [PubMed] [Google Scholar]
- Schroder JM, Harder J. Antimicrobial skin peptides and proteins. Cell Mol Life Sci. 2006;63:469–86. doi: 10.1007/s00018-005-5364-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen OE, Thapa DR, Rosenthal A, Liu L, Roberts AA, Ganz T. Differential regulation of beta-defensin expression in human skin by microbial stimuli. J Immunol. 2005;174:4870–9. doi: 10.4049/jimmunol.174.8.4870. [DOI] [PubMed] [Google Scholar]
- Strober W. Immunology. Unraveling gut inflammation. Science. 2006;313:1052–4. doi: 10.1126/science.1131997. [DOI] [PubMed] [Google Scholar]
- Tirmenstein MA, Nicholls-Grzemski FA, Schmittgen TD, Zakrajsek BA, Fariss MW. Characterization of nitric oxide production following isolation of rat hepatocytes. Toxicol Sci. 2000;53:56–62. doi: 10.1093/toxsci/53.1.56. [DOI] [PubMed] [Google Scholar]
- Weber G, Heilborn JD, Chamorro Jimenez CI, Hammarsjo A, Torma H, Stahle M. Vitamin D induces the antimicrobial protein hCAP18 in human skin. J Invest Dermatol. 2005;124:1080–2. doi: 10.1111/j.0022-202X.2005.23687.x. [DOI] [PubMed] [Google Scholar]
- Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270:41–9. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
- Zasloff M. Sunlight, vitamin D, and the innate immune defenses of the human skin. J Invest Dermatol. 2005;125:xvi–I. doi: 10.1111/j.0022-202X.2005.23924.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Acute permeability barrier disruption by an alternative method (acetone treatment) stimulates CRAMP/mBD3 expression; upregulation again is blocked by occlusion.
Figure S2. Western immunoblotting reveals a single band corresponding to mBD3 in normal mouse epidermis.
Figure S3. Compensatory increase in mBD3 protein in CRAMP−/− epidermis.
Figure S4. Normal content and secretion of another lamellar body protein in CRAMP−/− epidermis.
Table S1. CRAMP−/− mice display no significant differences in either hydration, surface pH, or basal TEWL.