

Abstract
Astrocytes respond to neuronal activity with [Ca2+]i increases after activation of specific receptors. Bergmann glial cells (BGs), astrocytes of the cerebellar molecular layer (ML), express various receptors that can mobilize internal Ca2+. BGs also express Ca2+ permeable AMPA receptors that may be important for maintaining the extensive coverage of Purkinje cell (PC) excitatory synapses by BG processes. Here, we examined Ca2+ signals in single BGs evoked by synaptic activity in cerebellar slices. Short bursts of high-frequency stimulation of the ML elicited Ca2+ transients composed of a small-amplitude fast rising phase, followed by a larger and slower rising phase. The first phase resulted from Ca2+ influx through AMPA receptors, whereas the second phase required release of Ca2+ from internal stores initiated by P2 purinergic receptor activation. We found that such Ca2+ responses could be evoked by direct activation of neurons releasing ATP onto BGs or after activation of metabotropic glutamate receptor 1 on these neurons. Moreover, examination of BG and PC responses to various synaptic stimulation protocols suggested that ML interneurons are likely the cellular source of ATP.
Keywords: cerebellum, neuron–glia interaction, ATP, AMPA receptors, mGluR1, synaptic transmission, interneuron
Introduction
Astrocytes can detect, respond to, and even influence neuronal activity. In particular, synaptic activity induces increases of [Ca2+]i in glial cells in vitro as well as in vivo (Porter and McCarthy, 1996; Araque et al., 2002; Newman, 2005; Perea and Araque, 2005; Serrano et al., 2006; Wang et al., 2006). This is exemplified by Bergmann glial cells (BGs) of the cerebellar cortex. Exogenous application of agonists such as histamine, noradrenaline, glutamate, or ATP (Kirischuk et al., 1995, 1996, 1999; Beierlein and Regehr, 2006) induces intracellular Ca2+ mobilization in BGs. [Ca2+]i elevations in BGs have also been observed during synaptic activity. Such Ca2+ transients are reported to result from activation of α1-adrenergic receptors, nitric oxide production, or activation of P2 purinergic and metabotropic glutamate receptor 1 (mGluR1) receptors (Grosche et al., 1999; Kulik et al., 1999; Matyash et al., 2001; Beierlein and Regehr, 2006).
BGs also express high densities of Ca2+-permeable AMPA receptors (Burnashev et al., 1992). BG AMPA receptors are activated by glutamate released by parallel fiber (PF) and climbing fiber (CF) synapses (Bergles et al., 1997; Clark and Barbour, 1997; Dzubay and Jahr, 1999). Furthermore, such synaptic activation mainly results from release of glutamate from ectopic release sites directly onto BG membranes rather than from glutamate spillover after release at conventional active zones (Matsui and Jahr, 2003, 2004), thereby exposing BG AMPA receptors to fast, high-concentration glutamate transients (Matsui et al., 2005). The function of BG AMPA receptors is not fully understood. However, disruption of the Ca2+ permeability of these receptors leads to significant changes in BG morphology and a decreased coverage of excitatory synapses (Iino et al., 2001). Ca2+ influx through BG AMPA receptors may thus have an important role in the maintenance of synaptic encasement by BG processes that inhibit glutamate diffusion by increasing the local density of glutamate transporters (Chaudhry et al., 1995).
We examined [Ca2+]i transients and electrophysiological currents in BGs after stimulation of the molecular layer (ML). We used a photodiode to detect [Ca2+]i-associated fluorescence changes in single BGs with a fast time resolution. This allowed observation of rapid Ca2+ transients mediated by activation of AMPA receptors as well as slower Ca2+ transients resulting from activation of P2 purinergic receptors. Examination of the potential source of ATP release led us to conclude that BG Ca2+ transients can be evoked after activation of mGluR1 on ML interneurons.
Materials and Methods
Slice preparation.
Postnatal day 14–19 rats were anesthetized with halothane and decapitated, as approved by the Oregon Health and Science University Institutional Animal Care and Use Committee. Parasagittal (250 μm) or coronal/transverse (300–350 μm) cerebellar slices were cut with a vibroslicer (Leica, Nussloch, Germany) in ice-cold solution containing the following (in mm): 119.0 NaCl, 2.5 KCl, 2.0 CaCl2, 1.0 MgCl2, 1.0 NaH2PO4, 26.2 NaHCO3, and 11.0 glucose (saturated with 95% O2/5% CO2). The slices were incubated at ∼34°C for 30 min and were then stored at room temperature. During recordings, the slices were superfused with the above solution with the addition of 100 μm picrotoxin to block GABAA receptors. An in-line heating device (Warner Instruments, Hamden, CT) was used to maintain recording temperature at 32–34°C.
Patch-clamp recordings and Ca2+ measurements.
Slices were visualized with a 60× water immersion objective on an Axioskop FS upright microscope (Zeiss, Thornwood, NY) with infrared Dodt contrast optics (Dodt et al., 2002). BGs were identified by the location of their cell bodies in the Purkinje cell (PC) layer, their low input resistance (10–40 MΩ), and visual inspection of their morphology after filling with the fluorescent dye (see below). Whole-cell recordings were made with an Axopatch 200A patch-clamp amplifier (Molecular Devices, Palo Alto, CA). BGs were recorded using pipettes with resistances of 3–3.5 MΩ filled with an internal solution containing the following (in mm): 100 CsCH3SO3, 30 CsCl, 20 HEPES, 3.1 Mg-ATP, 0.3 Na2-GTP, 4 Na2-phosphocreatine, and 0.1 EGTA. BGs were voltage clamped at −70 to −80 mV. PC recordings were performed using pipettes with resistances of 1.5–2.5 MΩ, filled with an internal solution containing the following (in mm): 140 CsCH3SO3, 10 HEPES, and 1 EGTA. PCs were voltage clamped at 0 mV (not corrected for junction potential).
PFs and CFs were stimulated with a glass microelectrode filled with bath solution using a constant-voltage isolated stimulator (20–200 μs pulse of 50–90 V; Digitimer, Hertfordshire, UK). Stimulations were delivered at a frequency of 0.01–0.02 Hz. Experiments in Figures 1–3, 4C, and 5A were performed on both parasagittal and coronal/transverse slices, and the stimulation electrode was placed in the inner third of the ML, ∼20–30 μm from the recorded cell (referred to in the text and figures as “local ML stimulation”). Experiments in Figures 4 and 6 were performed on coronal/transverse slices exclusively, and the stimulation electrode was placed in the outer half of the ML, 100–250 μm from the recorded cell (referred to in the text and figures as “distal PF stimulation”). Experiments involving CF stimulation (see Fig. 5B) were done on parasagittal slices, and the stimulation electrode was placed in the granule cell (GC) layer close to the PC layer. For pressure applications of ATP, a glass microelectrode (4–5 MΩ) was filled with a solution containing (in mm) 140 NaCl, 1.8 CaCl2, 1.3 MgCl2, 5.0 HEPES, and 1.0 Na2-ATP, and placed in the ML, close to the recorded BG processes. Solution was applied by pressure (∼10 psi, 10 ms pulse duration) with a Picospritzer II unit (General Valve Corporation, Fairfield, NJ) at a frequency of 0.02 Hz.
Figure 1.
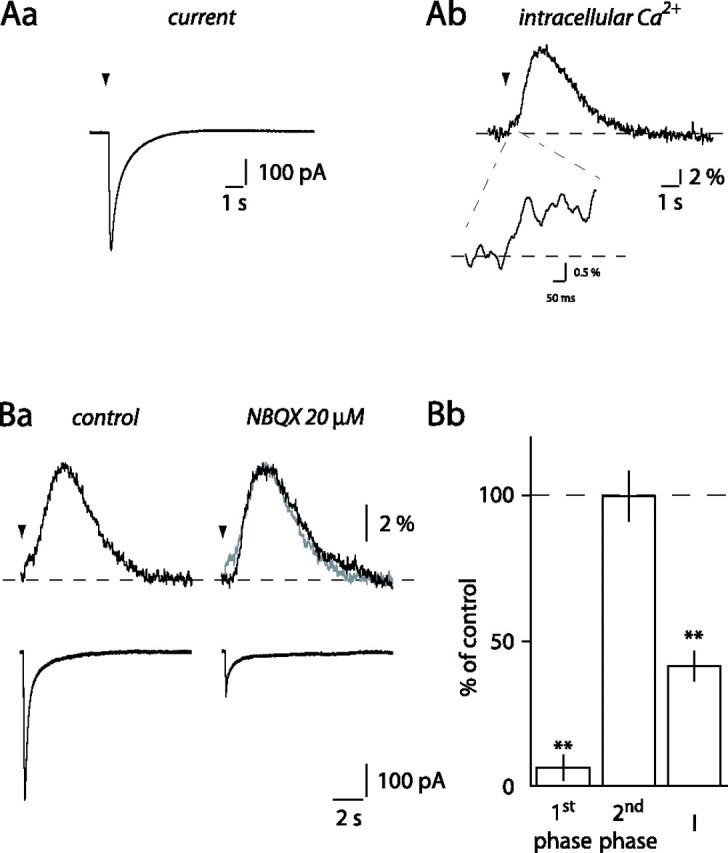
ML stimulation evokes [Ca2+]i elevations in BG. A, Whole-cell current (Aa) and simultaneous Ca2+ transients (Ab) evoked in a BG by local ML stimulation (arrowhead). Inset, Magnification of the first phase of the Ca2+ transient. Ba, Bath application of 20 μm NBQX inhibited the current and the first phase of the Ca2+ transient evoked in a BG by local ML stimulation. Control (gray) and NBQX traces were superimposed on the right to highlight the block of the first phase by NBQX. Bb, Histogram of the effect of NBQX on currents (I) and Ca2+ transients in 10 different BGs. **p < 0.01.
Figure 2.
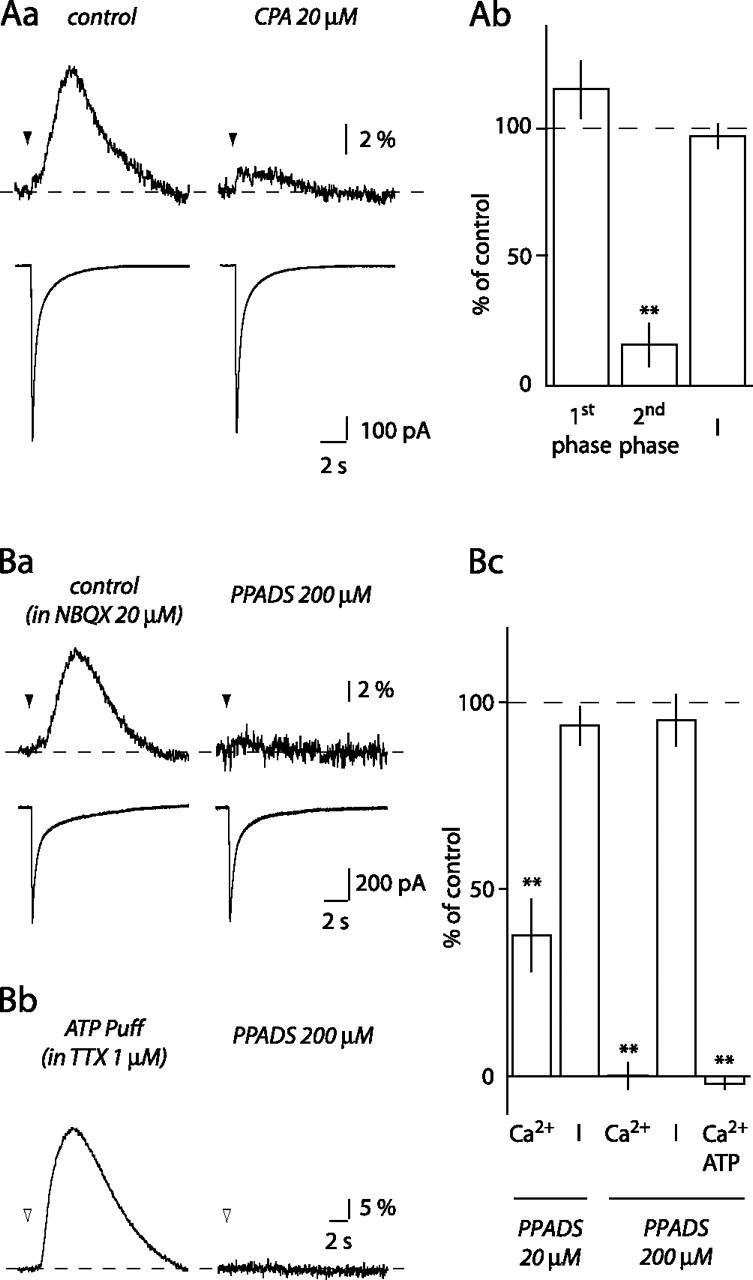
Ca2+ transient second phase involves Ca2+ release from internal stores and P2 receptor activation. Aa, Depleting internal Ca2+ stores with 20 μm CPA inhibited the second phase of the BG Ca2+ transients but did not affect the first phase or the whole-cell current. Ab, Histogram of the effect of CPA (n = 6). Ba, PPADS (200 μm) blocked the Ca2+ transient recorded in the presence of NBQX. Bb, Ca2+ transients evoked by pressure application of 1 mm ATP (open arrowhead) and their blockade by 200 μm PPADS. Bc, Histogram of the effect of 20 μm (n = 9) and 200 μm PPADS (n = 4) on Ca2+ transients (Ca2+) and currents (I) evoked in BGs in the presence of NBQX and of 200 μm PPADS on Ca2+ transients elicited by pressure application of ATP (Ca2+ ATP; n = 4). **p < 0.01.
Figure 3.

Role of mGluR1 in the generation of BG Ca2+ transients. A, CPCCOet did not affect BG currents or Ca2+ transients evoked by local ML stimulation. B, TBOA enhanced the BG Ca2+ transient. The enhancement was blocked by CPCCOet. C, Histogram summarizing the effect of CPCCOet alone (n = 5), TBOA alone, and CPCCOet in the presence of TBOA (n = 6) on BG currents (I) and Ca2+ transients. *p < 0.05.
Figure 4.
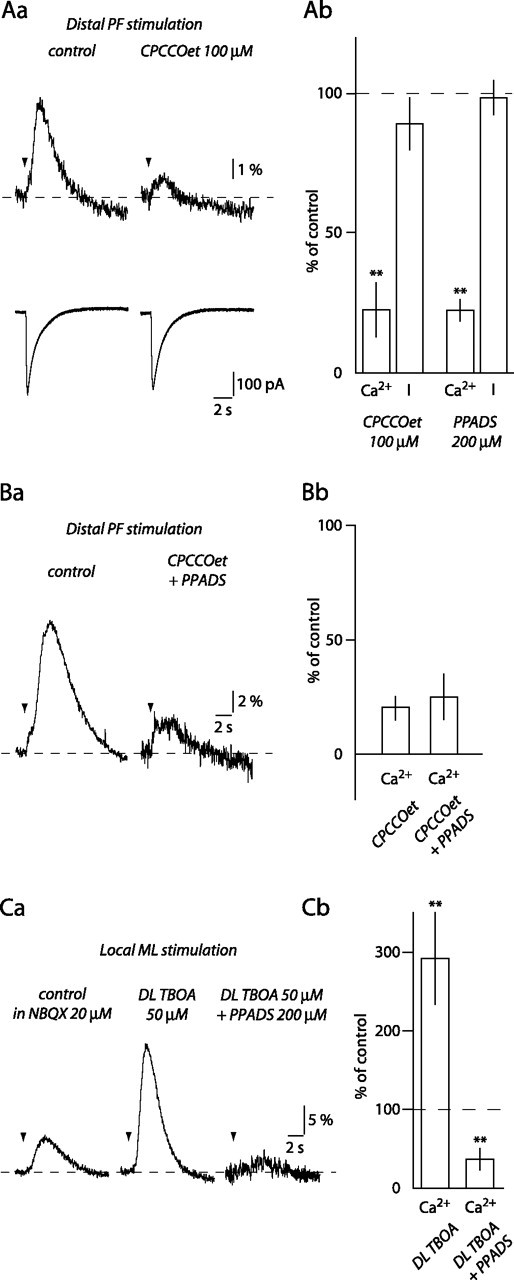
Distal PF stimulation evokes mGluR1-mediated Ca2+ transients in BGs. Aa, CPCCOet inhibits the second phase of BG Ca2+ transient evoked by distal stimulation of PFs without affecting the current (I). Ab, Histogram of the effects of CPCCOet (n = 5) and 200 μm PPADS (n = 5) on BG currents and Ca2+ transients evoked in the same conditions. Ba, Coapplication of CPCCOet and PPADS did not inhibit BG Ca2+ transients evoked by distal PF stimulation more than CPCCOet alone. Bb, Histogram of the effect of CPCCOet and of CPCCOet plus PPADS in six cells. Ca, The effect of TBOA and TBOA plus PPADS on BG Ca2+ transients evoked by local ML stimulation. Cb, Histogram summarizing results of the experiment shown in Ca. **p < 0.01.
Figure 5.

Neither BGs nor PCs are likely sources for ATP release. A, TTX blocked BG Ca2+ transients and whole-cell currents evoked by local ML stimulation. Histogram of the effect of TTX on the Ca2+ transient first and second phases and on whole-cell currents (I; n = 5). B, Neither single CF stimulations (left) nor short CF trains (right) evoked Ca2+ signals in BGs (top traces), but both elicited whole-cell currents (bottom traces). Insets show Ca2+ transients evoked in the corresponding cells when the stimulating electrode was moved to the ML. **p < 0.01.
Figure 6.
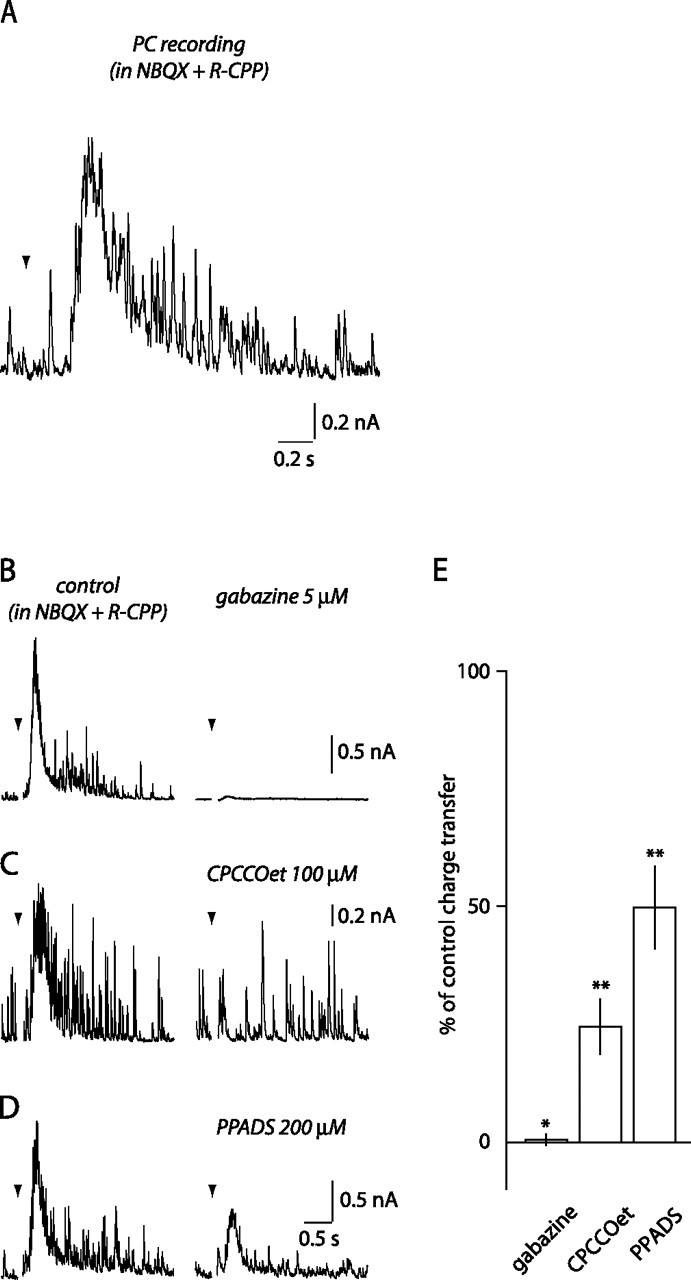
Disynaptic GABA release via activation of mGluR1 on ML interneurons. A, Disynaptic IPSC evoked in a PC by distal PF stimulation (10 at 100 Hz; arrowhead) in NBQX and R-CPP. B–D, The effects of gabazine (B; n = 5), CPCCOet (C; n = 6), and PPADS (D; n = 5) on the disynaptic IPSC. E, Histogram summary. *p < 0.05, **p < 0.01.
To measure changes in [Ca2+]i, 0.2 mm Fluo-5F, a Ca2+-sensitive dye (Kd of 2.3 μm; Invitrogen, Carlsbad, CA), was added to the internal solution. The dye was excited using a 100 W halogen lamp driven by an ATE 15-15 M power supply (Kepco, Flushing, NY). Excitation and emission light was filtered using an XF-100-2 filter set (excitation 480/40 nm, 505 nm dichroic long pass, emission 535/50 nm; Omega, Brattleboro, VT). [Ca2+]i-dependent variations in single-cell fluorescence were detected using a laboratory-built photodiode system mounted on the direct port of the microscope. The cathode of the photodiode was connected to a custom-built amplifier circuit. Photodiode currents were further amplified using a model 440 Instrumentation Amplifier (Brownlee Precision, San Jose, CA). Changes in [Ca2+]i were expressed as a percentage change in fluorescence relative to background fluorescence (ΔF/F).
The peak amplitude of the first phase of the Ca2+ transients was measured between 150 and 200 ms after the beginning of the stimu-lation. After blockade by 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) or by TTX, the first phase was measured by averaging amplitude within 100–200 ms after stimulation, to reduce baseline noise. After drug application, the second phase amplitude was measured at the peak or averaged 600 ms around the control response peak time when a peak could not be distinguished.
PC responses to PF stimulation were analyzed by measuring the current integral (charge transfer) 1.5 s after the end of the stimulation. This time window included both the outward envelope current and the increased IPSC frequency (see Fig. 6). The baseline charge transfer measured 1.5 s before stimulation was then subtracted from this measurement to estimate the effect of 7-(hydroxy-imino) cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOet) and pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS) more accurately.
Electrophysiological and Ca2+ signals were filtered at 2 and 1 kHz, respectively, and digitized at 2 kHz with an ITC-18 interface (InstruTech, Port Washington, NY), using acquisition software written by J. S. Diamond in Igor Pro software (WaveMetrics, Lake Oswego, OR). Ca2+ signals were subsequently digitally filtered. Data analysis was performed with Axograph 4.6 and Axograph X (Molecular Devices), and statistical analysis was performed with Kaleidagraph (Synergy Software, Reading, PA) using Student's t test. Values are given as the mean ± SEM. Traces shown in Figures 1–5 are averages of three to four sweeps. PC recording traces shown in Figure 6 are single sweeps.
Results
Local ML stimulation (10 at 100 Hz) elicited large and slowly decaying currents in BGs (Fig. 1Aa). These currents were associated with transient increases in [Ca2+]i that were composed of a small-amplitude, relatively fast rising increase (Fig. 1Ab, inset), followed by a slower rising, larger-amplitude phase (Fig. 1Ab). The fast and slow rising [Ca2+]i increases will be referred to as first and second phases, respectively. A first phase could be measured in 46 of 57 cells after local ML stimulation. The second phase peaked 2.9 ± 0.1 s after the beginning of the stimulation (n = 57) (Fig. 1Ab). The following experiments address the pharmacology and the origin of these Ca2+ transients.
BG AMPA receptors mediate the first phase of the Ca2+ transients
Because BGs express high densities of Ca2+-permeable AMPA receptors (Burnashev et al., 1992), we first tested whether these receptors mediated the BG Ca2+ transients. NBQX (20 μm), an AMPA/kainate receptor antagonist, inhibited the first phase of the Ca2+ transient to 6.3 ± 4.2% of control (p < 0.0001; n = 10) (Fig. 1B), indicating that the first phase was mediated by activation of Ca2+-permeable AMPA receptors in BGs. Currents recorded in BGs were also inhibited by NBQX to 41.3 ± 5.0% of control (p = 0.0001). The NBQX-resistant current is attributable to synaptic activation of glutamate transporters and to potassium released as a result of PF activation (Clark and Barbour, 1997). The second phase of the Ca2+ transients, however, remained unaffected by the application of NBQX (99.8 ± 8.5% of control; p = 0.38) (Fig. 1B). This result indicates that the second phase is not attributable to Ca2+-induced Ca2+ release after Ca2+ entry through BG AMPA receptors. It also implies that another receptor/factor is involved in the generation of the second phase of the BG Ca2+ transients.
The second phase Ca2+ signal involves Ca2+ release from internal stores
The slow rise time of the second phase of the Ca2+ transients is consistent with the release of Ca2+ from internal stores. Indeed, cyclopiazonic acid (CPA) (20 μm), a blocker of Ca2+ uptake into the endoplasmic reticulum by Ca2+-ATPase pumps (Seidler et al., 1989), inhibited the second phase to 15.9 ± 8.4% of control (p = 0.009; n = 6) but did not significantly change the amplitude of the first phase (115.9 ± 11.2%; p = 0.25) or that of the current (97.2 ± 4.7%; p = 0.69) (Fig. 2A). These data suggest that the slow [Ca2+]i increase seen in BGs after local ML stimulation results from Ca2+ release from internal stores.
The second phase Ca2+ signal is mediated by P2 receptor activation
We then investigated the identity of the receptor involved in the generation of the second phase of BG Ca2+ transients. Neither an α1 adrenoceptor antagonist nor a nitric oxide synthase inhibitor affected the second phase of the Ca2+ transients (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) in contrast to previously described synaptically induced Ca2+ signals in BGs (Kulik et al., 1999; Matyash et al., 2001).
It has been shown previously that ATP can mobilize Ca2+ from internal stores in BGs (Kirischuk et al., 1995; Beierlein and Regehr, 2006). To determine whether ATP was involved in generating the second phase of the Ca2+ signal, we tested the effect of PPADS, a P2 receptor antagonist. In the presence of NBQX, 200 μm PPADS blocked the Ca2+ transients to 0.1 ± 3.5% of their control amplitude (p = 0.001), leaving the current amplitude unchanged (95.3 ± 6.8%; p = 0.47; n = 4) (Fig. 2Ba,Bc). A lower concentration of PPADS (20 μm) also significantly decreased the BG Ca2+ transient amplitude, albeit to a lesser extent (37.7 ± 9.7%; p = 0.003; n = 9) (Fig. 2Bc). In the absence of NBQX, 200 μm PPADS inhibited the second phase to the same extent as in the presence of NBQX (5.0 ± 4.0% of control; p = 0.008; n = 4). These data suggest that the second phase of BG Ca2+ transients requires P2 receptor activation.
Application of exogenous ATP mimicked the effect of local ML stimulation. In the presence of 1 μm TTX, 10 ms pressure applications of 1 mm ATP induced large, slow rising [Ca2+]i increases (Fig. 2Bb). These ATP-induced Ca2+ transients had large amplitudes (31.1 ± 3.6%, ΔF/F), rose slowly (peak at 4.0 ± 0.3 s; n = 11), and were blocked by 200 μm PPADS (−2.0 ± 1.4% of control; p = 0.009; n = 4) (Fig. 2Bb,Bc).
PPADS may have effects on signaling cascades independently of P2 receptor binding (Shehnaz et al., 2000) (see also Singaravelu and Deitmer, 2006). We assessed the validity of the previous results by using a different P2 receptor antagonist. The P2Y receptor antagonist reactive blue-2 (50 μm) decreased the amplitude of BG Ca2+ transients evoked by local ML stimulation as well as the amplitude of BG Ca2+ transients elicited by 10 ms pressure applications of 1 mm ATP onto BG processes (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Together, our results indicate that the second phase of BG Ca2+ transients results from activation of P2Y receptors by ATP or one of its metabolites.
Involvement of mGluR1 in the generation of the second phase of BG Ca2+ signals
High-frequency stimulation of PFs in the cerebellum has been shown to activate mGluR1 in PCs (Batchelor et al., 1994) as well as in interneurons (Karakossian and Otis, 2004). Moreover, activation of mGluR1 leads to Ca2+ release from internal stores in PCs and BGs, via production of IP3 (Finch and Augustine, 1998; Takechi et al., 1998; Beierlein and Regehr, 2006). However, in our hands, application of 100 μm CPCCOet, an mGluR1 antagonist, did not alter the first or second phase of Ca2+ transients evoked by local ML stimulation (first phase, 99.0 ± 9.8%, p = 0.92, n = 4; second phase, 110.6 ± 15.4%, p = 0.82, n = 5) nor BG currents (101.2 ± 4.2%; p = 0.38) (Fig. 3A,C). It may be that glutamate transporters, densely expressed by BGs (Chaudhry et al., 1995), prevented the activation of mGluR1 during local ML stimulation. Indeed, in a background of NBQX, the glutamate transporter blocker dl-threo-β-benzyloxyaspartic acid (TBOA) (50 μm) increased the amplitude of BG Ca2+ transients (313.7 ± 77.7% of control; p = 0.028; n = 6) (Fig. 3B,C). TBOA also decreased the current amplitude (84.4 ± 4.0%; p = 0.011) (Fig. 3B,C), reflecting the inhibition of electrogenic glutamate transporters activated during stimulation (Bergles and Jahr, 1997; Clark and Barbour, 1997; Matsui and Jahr, 2004). The addition of CPCCOet decreased the amplitude of BG Ca2+ transients back to control values (86.0 ± 15.8%; p = 0.53 when compared with control) but had no additional effect on the current amplitude (87.2 ± 4.9% of control) (Fig. 3B,C). This observation indicates that mGluR1 can participate in the generation of BG Ca2+ transients when glutamate transporters are inhibited. However, in control conditions, mGluR1 are not responsible for the second phase of BG Ca2+ transients evoked by local ML stimulation. We also investigated the involvement of mGluR5, which have been reported to mobilize intracellular calcium in astrocytes (Wang et al., 2006). 2-Methyl-6-(phenylethynyl)-pyridine, a selective mGluR5 antagonist, did not significantly alter BG Ca2+ transients, even in the presence of TBOA (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) (Beierlein and Regehr, 2006). mGluR5, therefore, do not play any significant role in BG Ca2+ transients.
Distant stimulation of PFs reveals involvement of mGluR1
Electrical stimulation of the ML can directly evoke action potentials not only in PFs but also in interneurons (Delaney and Jahr, 2002) and possibly in PCs. Thus, the source of ATP could be any of these cells or BGs themselves. In addition, PF stimulation can evoke bursting in ML interneurons and PCs via mGluR1 activation (Batchelor et al., 1994; Karakossian and Otis, 2004) as well as Ca2+ signals in BGs. To address whether mGluR1-mediated activation of interneurons, PCs, or BGs results in release of ATP, we recorded BGs in coronal/transverse slices and stimulated the ML 100–250 μm from the recording site to decrease direct activation of cells nearby the recorded BG (distal PF stimulation). Distal PF stimulation (10 at 100 Hz) evoked currents and biphasic Ca2+ transients in BGs that were similar to those evoked by local ML stimulation. The second phase of the Ca2+ transients peaked at 2.5 ± 0.2 s (n = 34) (Fig. 4Aa), and a first phase could be distinguished in 32 of 34 cells.
In contrast to the results with local ML stimulation, 100 μm CPCCOet inhibited the second phase of BG Ca2+ transients (22.6 ± 9.7% of control; p = 0.002; n = 5), without affecting the BG currents (89.2 ± 9.3%; p = 0.49) or the first phase of the Ca2+ signals (86.7 ± 12.1%; p = 0.44) (Fig. 4A). Thus, the second phase Ca2+ transients evoked by distal PF stimulation require mGluR1 activation. In addition, these Ca2+ transients also require P2 receptor activation. Application of 200 μm PPADS inhibited the second phase of BG Ca2+ transients evoked by distal PF stimulation to 24.2 ± 3.9% of control (p = 0.009) without changing the amplitude of BG currents (98.5 ± 6.1%; p = 0.52; n = 5) (Fig. 4Ab).
Neither CPCCOet nor PPADS entirely blocked the amplitude of the second phase of BG Ca2+ transients, suggesting that P2 receptor activation and mGluR1 activation might represent parallel pathways both leading to [Ca2+]i increases in BGs. However, we found that coapplication of PPADS and CPCCOet did not decrease the second phase of BG Ca2+ transients further than CPCCOet alone (CPCCOet, 20.2 ± 5.2% of control; CPCCOet plus PPADS, 24.9 ± 10.0% of control; p = 0.58; n = 6) (Fig. 4B). Thus, CPCCOet prevented any additional blockade by PPADS. Because BG Ca2+ signals evoked by local ML stimulation were also blocked by PPADS but not by CPCCOet (Figs. 2, 3), activation of mGluR1 after distal PF stimulation is upstream of the activation of P2 receptors on BGs. We hypothesized that distal PF stimulation activates mGluR1 expressed by secondary cells, and this leads to ATP release and activation of P2 receptors on BGs, whereas local ML stimulation evokes direct release of ATP from its source.
We predict, based on this hypothesis, that activation of mGluR1 by local ML stimulation in the presence of TBOA (as shown in Fig. 3) will enhance release of ATP and activation of P2 receptors on BGs. As we described previously, TBOA significantly increased the amplitude of BG Ca2+ transients evoked by local ML stimulation (291.8 ± 58.2% of control; p = 0.007; n = 6) (Fig. 4C). The addition of PPADS not only blocked the TBOA enhancement but inhibited BG Ca2+ transients to 36.4 ± 7.2% of the pre-TBOA control amplitude (n = 6; p = 0.0004 when compared with control). This indicates that activation of mGluR1 can promote ATP release and activation of P2 receptors on BGs after local ML stimulation as well. The Ca2+ transient remaining in the presence of TBOA and PPADS could result from activation of mGluR1 located on BGs, assuming that they express these receptors.
Together, these results indicate that glutamate released from PFs can activate mGluR1 expressed on cells interposed between PFs and BGs. This mGluR1 activation causes ATP release onto P2 receptors expressed by BGs.
Source of ATP
ATP activating BG P2 receptors after local ML stimulation or distal PF stimulation could be released from BG, PF, PC, or ML interneurons. We first examined the involvement of BGs. Direct stimulation of astrocytes has been shown to trigger Ca2+ waves that can propagate from cell to cell (for review, see Haydon, 2001). BG Ca2+ transients could thus result from direct electrical stimulation of BGs. However, the Ca2+ transients as well as the currents recorded in BGs evoked by local ML stimulation (Fig. 5A) were inhibited by TTX (1 μm), indicating that generation of neuronal action potentials preceded BG activation. TTX decreased the first and second phases to −0.1 ± 7.2% (p < 0.0001) and 14.7 ± 9.7% (p = 0.003; n = 5) of control, respectively. Simultaneously recorded currents were diminished to 4.7 ± 0.7% of control (p = 0.002; n = 5) (Fig. 5A). The Ca2+ transient remaining in TTX could be explained by ATP release from neurons independently of sodium channel activation or by release of ATP from non-neuronal cells. Nevertheless, this result indicates that the second phase of the Ca2+ transient requires, for the most part, the generation of action potentials and, therefore, cannot be explained by the propagation of Ca2+ waves after direct stimulation of BGs.
PFs are an unlikely source of ATP because BG Ca2+ transients evoked by distal PF stimulation required activation of mGluR1. This implies that ATP must be released from a cell type expressing mGluR1. It has been reported that ATP can be co-packaged with GABA in synaptic vesicles (Jo and Schlichter, 1999; Jo and Role, 2002), making the GABAergic stellate, basket, and Purkinje cells possible sources for its release. These cells express mGluR1 that can be activated after short high-frequency PF stimulation (Batchelor et al., 1994; Karakossian and Otis, 2004). In addition, mGluR1 on PCs can also be activated after CF stimulation (Dzubay and Otis, 2002). We therefore investigated whether Ca2+ transients could be evoked in BGs after CF stimulation. Figure 5B illustrates that, in the presence of 100 μm TBOA, activation of a CF with a single pulse failed to evoke Ca2+ transients in all but 1 of 11 BGs, whereas it readily evoked currents in these cells. Moreover, in six of six recordings, when the stimulating electrode was subsequently placed in the ML, Ca2+ transients could be readily evoked (Fig. 5B, inset). Even train stimulation (four at 20 Hz) did not evoke Ca2+ signals in BGs (Fig. 5B) (n = 6), whereas local ML stimulation evoked transients in five of five of these cells (Fig. 5B, inset). Such stimulation protocols efficiently activate PC mGluR1 (Dzubay and Otis, 2002), yet, in our recording conditions, they did not evoke Ca2+ transients in BGs. PCs are, therefore, unlikely the source of ATP release. Instead, this suggests that activation of ML interneurons may lead to ATP release onto BGs.
mGluR1-mediated activation of ML interneurons
In cerebellar slices, GABA release from ML interneurons can be induced by direct electrical stimulation of these neurons or synaptically, by glutamate released from PFs (Delaney and Jahr, 2002). If ATP and GABA are coreleased, both should be released by either direct stimulation of ML interneurons or synaptic activation of mGluR1 expressed by ML interneurons. To test this, we monitored release from ML interneurons by recording IPSCs in PCs. Brief trains of PF stimulations evoke AMPA and NMDA receptor-mediated EPSCs in ML interneurons (Carter and Regehr, 2000). We therefore used 20 μm NBQX and 10 μm 3-(R-2-carboxypiperazin-4-yl)-propyl-1-phosphoninc acid (R-CPP) to block AMPA and NMDA receptors, respectively, to isolate mGluR1-mediated excitation of ML interneurons. These conditions have been used previously to record mGluR1-mediated bursting in ML interneurons (Karakossian and Otis, 2004) and did not affect the second phase of the BG [Ca2+]i increase (supplemental Fig. S4, available at www.jneurosci.org as supplemental material) (Beierlein and Regehr, 2006). Because brief high-frequency stimulation of PFs also evokes mGluR1-mediated currents in PCs (Kim et al., 2003), PCs were recorded at 0 mV to diminish the contribution of these synaptic events in the recorded cells. As illustrated in Figure 6A, distal PF stimulation (10 at 100 Hz) in these conditions evoked late onset (100–200 ms after the train) slow outward currents in PCs. This current was blocked by the GABAA receptor antagonist gabazine (5 μm; 0.5 ± 1.2% of control; p = 0.014; n = 5) (Fig. 6B,E). Thus, even in the presence of ionotropic glutamate receptor antagonists, trains of PF stimulation can evoke interneuron activation and release of GABA onto PCs. In the following experiments, we examined whether the slow GABAA receptor-mediated current in PCs and the BG Ca2+ transients had similar pharmacology.
As with the BG Ca2+ transient, CPCCOet (100 μm) inhibited the charge transfer induced in PCs by distal PF stimulation (24.5 ± 5.7% of control; p = 0.0001; n = 6) (Fig. 6C,E). In five of six recordings, CPCCOet abolished the slow GABAergic current, leaving individual IPSCs on a flat baseline (Fig. 6C). This result indicates that activation of mGluR1 is responsible for GABA release onto PCs. We also tested the effect of PPADS on the slow IPSC recorded in PCs. PPADS at 200 μm decreased, but did not block, the slow IPSC evoked by distal PF stimulation (49.8 ± 8.7% of control charge transfer; p = 0.005; n = 5) (Fig. 6D,E), suggesting an additional site of action than CPCCOet. Indeed, activation of P2 receptors has been shown previously to increase IPSC amplitude and frequency in PCs via presynaptic and postsynaptic mechanisms (Brockhaus et al., 2004; Casel et al., 2005; Saitow et al., 2005). Blockade of this P2 receptor-mediated potentiation of GABAA currents could explain the effect of PPADS. Thus, ATP release from ML interneurons after mGluR1 activation may have two effects: potentiation of GABAergic inhibition of PCs and Ca2+ elevation in BGs. Because of the kinetic and pharmacological similarity of the disynaptic responses in PCs and the BG Ca2+ transients, we conclude that ML interneurons represent a likely candidate as a source for ATP release.
Discussion
We find that ML stimulation evokes Ca2+ transients in BGs. These responses consist of two phases displaying distinct kinetics and amplitudes. Both phases require generation of action potentials, revealing neuronal involvement. The fast rising, small-amplitude first phase results from activation of BG AMPA receptors. The second phase involves Ca2+ release from internal stores and is likely mediated by activation of P2Y receptors. The involvement of mGluR1 receptors in the second phase of the Ca2+ transients depends on the stimulation protocol: both mGluR1 and P2 receptors play a role with distal PF stimulation, although only P2 receptors are required with local ML stimulation. Beierlein and Regehr (2006) recently reported Ca2+ transients in BGs displaying similar pharmacology. Using a CCD camera, they observed [Ca2+]i increases slowly spreading along BG processes after ML stimulation. In contrast to our results, however, they did not detect Ca2+ transients mediated by AMPA receptors. This discrepancy may result from technical differences. We measure Ca2+ transients with a photodiode, which allows faster sampling (2 kHz vs 5–20 Hz in the study of Beierlein and Regehr), making detection of the relatively short AMPA receptor-mediated transients possible. The use of a photodiode in our study, however, does not provide any spatial information about the two phases of the [Ca2+]i increases in BG processes. AMPA receptor-mediated Ca2+ transients presumably occur in BG processes surrounding active excitatory synapses (Xu-Friedman et al., 2001; Matsui et al., 2005). The slow phase of the signal, however, propagates along BG processes (Beierlein and Regehr, 2006). The slow phase of the Ca2+ transients in our recordings represents the spatial summation of the Ca2+ waves spreading along BG processes.
AMPA receptor-mediated Ca2+ transients
The role of Ca2+ signals in astrocytes is not fully understood. As shown here, synaptic activation results in Ca2+ transients in BGs composed of two distinct phases involving at least three different receptors. The two phases may serve different purposes. Ca2+-permeable AMPA receptors are expressed all along BG processes, including the small processes that engulf excitatory synapses (Matsui et al., 2005). They can be activated by glutamate released from CFs and PFs (Bergles et al., 1997; Clark and Barbour, 1997; Dzubay and Jahr, 1999; Matsui and Jahr, 2003, 2004), and we show here that they can mediate detectable Ca2+ transients in BGs. It was reported previously that disruption of the Ca2+ permeability of these receptors causes a decrease in the encasement of excitatory synapses by BG processes (Iino et al., 2001). Because they express high densities of glutamate transporters (Chaudhry et al., 1995), retraction of BG processes results in a slowing of glutamate clearance. Ca2+ entry through BG AMPA receptors may thus play a role in the maintenance of glial coverage of synapses and, consequently, in the efficiency of glutamate clearance.
In addition to a potential role in BG morphology, a recent study suggests that BG AMPA receptors may play a role in GC migration during postnatal development (Kim et al., 2005). The authors propose a model in which BG AMPA receptor activation promotes production of d-serine by activating glial serine racemase that in turn coactivates GC NMDA receptors and facilitates GC migration along BG processes. Whether Ca2+ entry through BG AMPA receptors is involved in this process remains to be seen.
Origin of the second phase of BG Ca2+ transients
The two stimulation protocols used in this study result in slow BG Ca2+ transients with different pharmacology. Whereas PPADS inhibits the second phase of all BG Ca2+ transients, CPCCOet only inhibits those evoked by distal PF stimulation, leaving locally stimulated events unaltered. In addition, PPADS and CPCCOet do not have additive effects on Ca2+ transients evoked by distal PF stimulation. Finally, mGluR1-mediated Ca2+ transients recorded in the presence of TBOA are sensitive to PPADS. These findings suggest a sequential role of mGluR1 and P2 receptors in the generation of the second phase of BG Ca2+ transients.
Local ML stimulation likely results in direct electrical stimulation of the cells that eventually release ATP. Because Ca2+ transients evoked by local ML stimulation are sensitive to TTX, direct stimulation of BGs is unlikely to participate in the generation of these events. We cannot fully rule out BGs as an ultimate source of ATP. If this were the case, however, release of a factor other than glutamate would be needed upstream of ATP release because neither NBQX nor CPCCOet blocks the slow Ca2+ transients evoked by local ML stimulation.
Distal PF stimulation, conversely, cannot activate the cells releasing ATP directly but rather only by release of glutamate from PFs. Moreover, CPCCOet inhibits Ca2+ transients evoked by distal PF stimulation, implicating an mGluR1-expressing cell type and ruling out ATP release from PFs. We also find that CF stimulation, although it is sufficient to evoke mGluR1-mediated currents in PCs (Dzubay and Otis, 2002), fails to induce Ca2+ transients in BGs, even in the presence of TBOA, making PCs an unlikely contributor to BG Ca2+ signals. This finding led us to hypothesize that ML interneurons are the source of ATP release. In favor of this hypothesis, GABAergic neurons such as ML interneurons have been shown to corelease GABA and ATP (Jo and Schlichter, 1999; Jo and Role, 2002). Moreover, we also find that synaptic activation of mGluR1 on ML interneurons (Karakossian and Otis, 2004) is sufficient to cause GABA release onto PCs. For all of these reasons, ML interneurons appear a likely candidate as a source for ATP release.
Physiological implications of the second phase of BG Ca2+ transients
Slow Ca2+ transients in astrocytes, such as those we and others have described in BGs (Grosche et al., 1999; Kulik et al., 1999; Matyash et al., 2001; Beierlein and Regehr, 2006), are thought to promote release of active substances. For example, ATP after its degradation into adenosine can control synaptic release of glutamate as well as synaptic plasticity (Pascual et al., 2005; Serrano et al., 2006). In addition, it has been reported that release of glutamate from astrocytes can activate NMDA receptors on pyramidal cells (Fellin et al., 2004; Perea and Araque, 2005) and modulates excitatory transmission (Fiacco and McCarthy, 2004) in the hippocampus. However, Beierlein and Regehr (2006) found no evidence that BG Ca2+ transients play any role in short-term modulation of transmission at PF synapses or promote release of glutamate on adjacent neurons in the cerebellum.
Astrocytic Ca2+ transients are also thought to participate in the regulation of cerebral blood flow during brain activity. Both in vitro and in vivo data suggest that [Ca2+]i increases in astrocytic end feet induce constriction (Mulligan and MacVicar, 2004) or dilation (Zonta et al., 2003; Takano et al., 2006) of arterioles. Because Ca2+ transients propagate along BG processes (Beierlein and Regehr, 2006), it is possible that they can affect the diameter of cerebellar arterioles. BG Ca2+ transients could thus be part of the signaling pathways that modulate cerebellar blood flow. Given our findings, it is tempting to speculate that activation of mGluR1 on ML interneurons signals network activity to BGs to modulate blood flow accordingly. Interestingly, a recent study reported that, in the cerebellum, application of exogenous glutamate on slices modifies arteriole diameter (Rancillac et al., 2006). Whether BG Ca2+ transients and P2 receptors are involved in this phenomenon remains to be investigated.
Footnotes
This work was supported by National Institutes of Health Grant NS40056 (C.E.J.). R.P. was supported by Fondation Recherche Médicale and Bourses Lavoisier du Ministère des Affaires Étrangères. We thank members of the Jahr laboratory for valuable comments and suggestions.
References
- Araque A, Martin ED, Perea G, Arellano JI, Buno W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J Neurosci. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor AM, Madge DJ, Garthwaite J. Synaptic activation of metabotropic glutamate receptors in the parallel fibre-Purkinje cell pathway in rat cerebellar slices. Neuroscience. 1994;63:911–915. doi: 10.1016/0306-4522(94)90558-4. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Regehr WG. Brief bursts of parallel fiber activity trigger calcium signals in Bergmann glia. J Neurosci. 2006;26:6958–6967. doi: 10.1523/JNEUROSCI.0613-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Dzubay JA, Jahr CE. Glutamate transporter currents in Bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci USA. 1997;94:14821–14825. doi: 10.1073/pnas.94.26.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockaus J, Dressel D, Herold S, Deitmer JW. Purinergic modulation of synaptic input to Purkinje neurons in rat cerebellar brain slices. Eur J Neurosci. 2004;19:2221–2230. doi: 10.1111/j.0953-816X.2004.03325.x. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Khodorova A, Jonas P, Helm PJ, Wisden W, Monyer H, Seeburg PH, Sakmann B. Calcium-permeable AMPA-Kainate receptors in fusiform cerebellar glial cells. Science. 1992;256:1566–1570. doi: 10.1126/science.1317970. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casel D, Brockhaus J, Deitmer JW. Enhancement of spontaneous synaptic activity in rat Purkinje neurons by ATP during development. J Physiol (Lond) 2005;568:111–122. doi: 10.1113/jphysiol.2005.091371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Clark BA, Barbour B. Currents evoked in Bergmann glial cells by parallel fibre stimulation in rat cerebellar slices. J Physiol (Lond) 1997;502:335–350. doi: 10.1111/j.1469-7793.1997.335bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney AJ, Jahr CE. Kainate receptors differentially regulate release at two parallel fiber synapses. Neuron. 2002;36:475–482. doi: 10.1016/s0896-6273(02)01008-5. [DOI] [PubMed] [Google Scholar]
- Dodt HU, Eder M, Schierloh A, Zieglgansberger W. Infrared-guided laser stimulation of neurons in brain slices. Sci STKE. 2002;2002:PL2. doi: 10.1126/stke.2002.120.pl2. [DOI] [PubMed] [Google Scholar]
- Dzubay JA, Jahr CE. The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J Neurosci. 1999;19:5265–5274. doi: 10.1523/JNEUROSCI.19-13-05265.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzubay JA, Otis TS. Climbing fiber activation of metabotropic glutamate receptors on cerebellar purkinje neurons. Neuron. 2002;36:1159–1167. doi: 10.1016/s0896-6273(02)01052-8. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signaling by inositol-1,4,5-triphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci. 1999;2:139–143. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- Haydon PG. Glia: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K, Ozawa S. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–929. doi: 10.1126/science.1058827. [DOI] [PubMed] [Google Scholar]
- Jo YH, Role LW. Coordinate release of ATP and GABA at in vitro synapses of lateral hypothalamic neurons. J Neurosci. 2002;22:4794–4804. doi: 10.1523/JNEUROSCI.22-12-04794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YH, Schlichter R. Synaptic corelease of ATP and GABA in cultured spinal neurons. Nat Neurosci. 1999;2:241–245. doi: 10.1038/6344. [DOI] [PubMed] [Google Scholar]
- Karakossian MH, Otis TS. Excitation of cerebellar interneurons by group I metabotropic glutamate receptors. J Neurophysiol. 2004;92:1558–1565. doi: 10.1152/jn.00300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PM, Aizawa H, Kim PS, Huang AS, Wickramasinghe SR, Kashani AH, Barrow RK, Huganir RL, Ghosh A, Snyder SH. Serine racemase: activation by glutamate neurotransmission via glutamate receptor interacting protein and mediation of neuronal migration. Proc Natl Acad Sci USA. 2005;102:2105–2110. doi: 10.1073/pnas.0409723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature. 2003;426:285–291. doi: 10.1038/nature02162. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Moller T, Voitenko N, Kettenmann H, Verkhratsky A. ATP-induced cytoplasmic calcium mobilization in Bergmann glial cells. J Neurosci. 1995;15:7861–7871. doi: 10.1523/JNEUROSCI.15-12-07861.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Tuschick S, Verkhratsky A, Kettenmann H. Calcium signaling in mouse Bergmann glial cells mediated by α1-adrenoreceptors and H1 histamine receptors. Eur J Neurosci. 1996;8:1198–1208. doi: 10.1111/j.1460-9568.1996.tb01288.x. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Kirchhoff F, Matyash V, Kettenmann H, Verkhratsky A. Glutamate-triggered calcium signaling in mouse Bergmann glial cells in situ: role of inositol-1,4,5-triphosphate-mediated intracellular calcium release. Neuroscience. 1999;92:1051–1059. doi: 10.1016/s0306-4522(99)00067-6. [DOI] [PubMed] [Google Scholar]
- Kulik A, Haentzsch A, Luckermann M, Reichelt W, Ballanyi K. Neuron–glia signaling via α1-adrenoceptor-mediated Ca2+ release in Bergmann glial cells in situ. J Neurosci. 1999;19:8401–8408. doi: 10.1523/JNEUROSCI.19-19-08401.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Jahr CE. Ectopic release of synaptic vesicles. Neuron. 2003;40:1173–1183. doi: 10.1016/s0896-6273(03)00788-8. [DOI] [PubMed] [Google Scholar]
- Matsui K, Jahr CE. Differential control of synaptic and ectopic vesicular release of glutamate. J Neurosci. 2004;24:8932–8939. doi: 10.1523/JNEUROSCI.2650-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Jahr CE, Rubio ME. High-concentration rapid transients of glutamate mediate neural-glial communication via ectopic release. J Neurosci. 2005;25:7538–7547. doi: 10.1523/JNEUROSCI.1927-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyash V, Filippov V, Mohrhagen K, Kettenmann H. Nitric oxide signals parallel fiber activity to Bergmann glial cells in the mouse cerebellar slice. Mol Cell Neurosci. 2001;18:664–670. doi: 10.1006/mcne.2001.1047. [DOI] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Newman EA. Calcium increase in retinal glial cells evoked by light-induced neuronal activity. J Neurosci. 2005;25:5502–5510. doi: 10.1523/JNEUROSCI.1354-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci. 2005;25:2192–2203. doi: 10.1523/JNEUROSCI.3965-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancillac A, Rossier J, Guille M, Tong XK, Geoffroy H, Amatore C, Arbault S, Hamel E, Cauli B. Glutamatergic control of microvascular tone by distinct GABA neurons in the cerebellum. J Neurosci. 2006;26:6997–7006. doi: 10.1523/JNEUROSCI.5515-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitow F, Murakoshi T, Suzuki H, Konishi S. Metabotropic P2Y purinoceptor-mediated presynaptic and postsynaptic enhancement of cerebellar GABAergic transmission. J Neurosci. 2005;25:2108–2116. doi: 10.1523/JNEUROSCI.4254-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehnaz D, Torres B, Balboa MA, Insel PA. Pyridoxal-phosphate-6-azophenyl-2′,4′-disulfonate (PPADS), a putative P2Y(1) receptor antagonist, blocks signaling at a site distal to the receptor in Madin-Darby canine kidney-D(1) cells. J Pharmacol Exp Ther. 2000;292:346–350. [PubMed] [Google Scholar]
- Singaravelu K, Deitmer JW. Calcium mobilization by nicotinic acid adenine dinucleotide phosphate (NAADP) in rat astrocytes. Cell Calcium. 2006;39:143–153. doi: 10.1016/j.ceca.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Takechi H, Eilers J, Konnerth A. A new class of synaptic response involving calcium release in dendritic spines. Nature. 1998;396:757–760. doi: 10.1038/25547. [DOI] [PubMed] [Google Scholar]
- Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, Kang J, Takano T, Nedergaard M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci. 2006;9:816–823. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Harris KM, Regehr WG. Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J Neurosci. 2001;21:6666–6672. doi: 10.1523/JNEUROSCI.21-17-06666.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]