

Abstract
Determining protein – small molecule binding affinity is a key component of present-day rational drug discovery. To circumvent the time, labor, and materials costs associated with experimental protein – small molecule binding assays, a variety of structure-based computational methods have been developed for determining protein – small molecule binding affinities. These methods can be placed in one of two classes: accurate but slow (Class 1), and fast but approximate (Class 2). Class 1 methods, which explicitly take into account protein flexibility and include an atomic-level description of solvation, are capable of quantitatively reproducing experimental protein – small molecule absolute binding free energies. However, Class 1 computational requirements make screening thousands to millions of small molecules against a protein, as required for rational drug design, infeasible for the foreseeable future. Class 2 methods, on the other hand, are sufficiently fast to perform such inhibitor screening, yet they suffer from limited descriptions of protein flexibility and solvation, which in turn limit their ability to select and rank-order small molecules by computed binding affinities. This review presents an overview of Class 1 and Class 2 methods, avenues of research in Class 2 methods aimed at bringing them closer to Class 1 accuracy, and intermediate approaches that incorporate features of both Class 1 and Class 2 methods.
Keywords: protein ligand binding, virtual screening, docking, free energy perturbation, thermodynamic integration, implicit solvent, Poisson-Boltzmann, generalized Born
Introduction
Atomic resolution structures of proteins and protein – small molecule complexes have enabled structure-based approaches to drug design. Instead of blindly assaying the binding of small molecule libraries to a protein in the search for a “hit,” protein structural information is now commonly used to “rationally” guide the discovery and optimization of ligands. A major contributor to the rational discovery process are computational approaches to protein – small molecule binding. Using structural information, they can be applied to determine protein – small molecule binding affinities and thereby reduce time, labor, and materials associated with experiments. On one end of the spectrum are computationally expensive (Class 1) methods based on atomically-detailed representations of the protein, small molecule, and solvent. Class 1 methods are capable of reproducing experimental absolute binding free energies, but are too slow to be used for computational high-throughput screening. On the other end of the spectrum are methods fast enough to screen thousands to millions of compounds in a matter of days to weeks (Class 2). However, such speed requires simplifying assumptions in the underlying models representing the protein and the solvent, which compromise the accuracy of the results. This review presents an overview of recent developments in Class 1 and Class 2 methods for computational protein – small molecule binding, including recent examples of their application. Also discussed is research in Class 2 methods to include protein flexibility and solvation effects. This research is aimed at addressing the two major approximations in Class 2 methods, namely the treatment of the protein as a rigid body and the treatment of the solvent as a continuum or mean-field.
Class 1 methods: Accurate but slow
Class 1 methods are based on an accurate structure of the protein – small molecule complex, such as that determined from X-ray crystallography or NMR spectroscopy, in combination with molecular dynamics (MD) simulations [1,2]. In MD simulations all atoms of the protein, the small molecule, and both structural and bulk water molecules, are explicitly represented using a “force field” [3,4]. Atomic forces are calculated from the force field and the atomic positions, and these forces are numerically integrated to create a trajectory of the motion of the system in time, which constitutes an MD simulation. Importantly, MD simulations can be done for a system under conditions analogous to laboratory or physiological temperature and pressure [5,6], allowing for direct comparison of computed and experimentally determined binding affinities. The resulting binding energy from a Class 1 study represents a true free energy, one that includes both energetic and entropic contributions from all components in the system.
The first Class 1 method is termed “annihilation.” Two systems are simultaneously simulated using MD. System 1 initially contains the protein – small complex in water and System 2 initially only water molecules. During MD on System 1, the small molecule interactions are uncoupled in stages from the rest of the system, until the small molecule becomes invisible to the rest of the system, i.e. is “annihilated.” During MD on System 2, the small molecule is gradually coupled with the system until it fully interacts with the water molecules (Figure 1a). In simple terms, the small molecule is transferred from System 1 to System 2 by deleting it from the protein – small molecule complex and putting it in the all-water system. The uncoupling of small molecule interactions with System 1 and the coupling with System 2 are done in discrete stages, and a free energy change ΔGstage is computed for each stage. Formalisms for doing this are Free Energy Perturbation and Thermodynamic Integration [7]. Summing the ΔGstage values gives the total free-energy change ΔGtotal for transferring the small molecule from System 1 to System 2, with the caveat that standard states need to be properly defined and that correction terms for translation and rotation of the uncoupled small molecule in System 1 are included in ΔGtotal [8].
Figure 1.
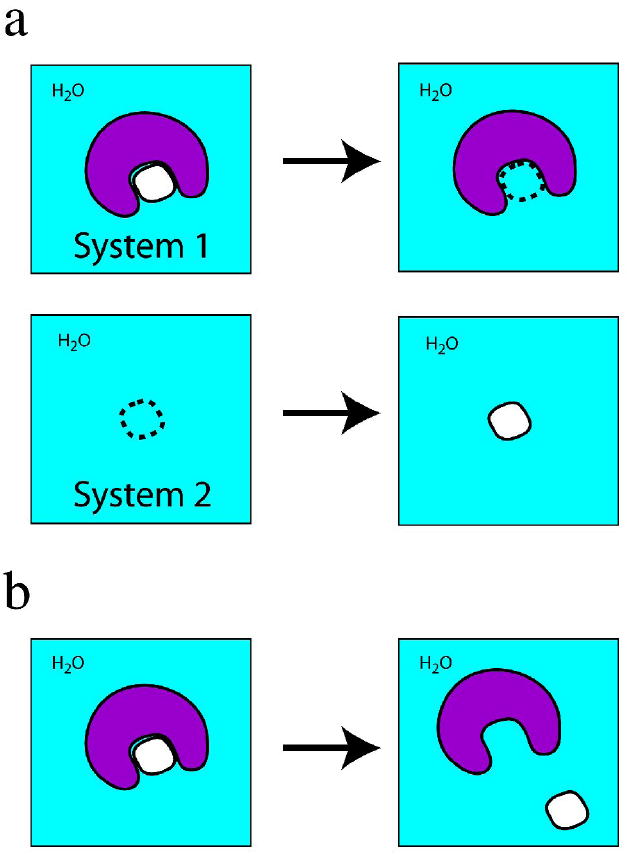
Schematic view of Class 1 (a) annihilation and (b) separation simulations. In both simulations, all atoms in the system, including water molecules (“H2O”) are explicitly represented. The protein is in purple and the small molecule in white. A dashed outline of the small molecule indicates interactions of the small molecule with the rest of the system (protein + water) have been decoupled, such that the small molecule is effectively “invisible” to the rest of the system.
The second Class 1 method is termed “separation.” A single system consisting of the protein – small molecule complex in water (i.e. System 1 in a Class 1 annihilation simulation) is simulated by MD. In contrast to annihilation, in which the small molecule is literally deleted from the binding pocket by uncoupling the small molecule interactions from the rest of the system, in a separation simulation the small molecule is gradually moved from the binding pocket to a distance representative of an unbound state while maintaining full interactions with the rest of the system. This separation is done in incremental stages, with each stage differing in the protein – small molecule separation distance. The effect of the biasing potential used to maintain the protein – small molecule separation distance is removed, and the free energies ΔGstage for the stages are summed to give the total free-enegy change ΔGtotal [9].
The annihilation approach has a long history in biomolecular simulations. Its initial incarnation as the “double annihilation” approach [10] did not properly incorporate standard states, a problem that was remedied in the “double decoupling” approach[8,11,12]. Two recent double-decoupling simulations demonstrating the utility of the method were those in which the small molecule was simply molecular water. In one instance, the free energy contributions of interfacial water molecules in an HIV-1 protease – small molecule complex were investigated, revealing only two of the four made significant binding free energy contributions [13]. In the other, the free energies for a set of five water molecules in a protein hydrophobic cavity were calculated to demonstrate that this cavity, which binds nonpolar small molecules, is completely dry in the apo protein [14]. With regard to drug-like small molecules, half in a series of 10 aromatic compounds with the T4 lysozyme L99A mutant showed agreement within 1 kcal/mol for absolute binding free energies, but larger errors were seen for the others, with one error as large as 4.4 kcal/mol [15]. More consistent agreement was seen for a set of polar small molecules binding to FKBP12, whose binding affinities were about twice those as the compounds in the T4 lysozyme study [16]. Very recently, quantitative agreement with experimental absolute binding free-energies was obtained for binding of the charged small molecules benzamidine and diazamidine to trypsin, in conjunction with a force field that included electronic polarizability [17], suggesting the introduction of polarizability into force fields [18] may help increase accuracy.
The separation approach has only recently been applied to protein – small molecule binding in a few studies that include proper accounting for standard states and the biasing potentials involved (separation simulations do have prior history in biomolecular simulation for other systems, including for the energetics of association in model peptide systems [19,20]). Studies include the binding of a short phosphotyrosine peptide to an SH2 domain [9], the binding of two small-molecules to FKBP [21], and the binding of the small molecule inhibitor pteroic acid to protein ricin toxin A-chain [22]. Agreement with the experimental binding free-energies to within 1 kcal/mol were obtained for SH2 – phosphotyrosine peptide and FKBP – FK506 binding; larger errors for the other systems suggest improvements in the force fields for these systems may be required, including possibly the introduction of electronic polarizability. The intuitive nature of separation simulations makes them appealing, and they provide information not only about the free energy difference between the bound and unbound states but also the free-energy profile along the separation path. However, separation simulations are not practically applicable in cases where large protein conformational rearrangement is required to create an exit path for the small molecule.
Despite the physical rigor and successes of Class 1 methods, they are computationally expensive because they use MD simulations that include a fully-mobile protein and explicit solvent molecules. Converged binding free-energy values depend on adequate sampling of the relevant protein, small molecule, and solvent conformations, which can take days to weeks for a single protein – small molecule complex on modern computer hardware. For a recent detailed review containing the background mathematics and physics of computing free energies from MD simulations, we refer readers to [23].
Class 2 methods: Fast but approximate
Class 2 methods are fast because they minimize the number of degrees of freedom in the system. In a protein + small molecule + explicit water system, most of the atoms belong to the protein and to the water molecules. Sampling the protein and water atoms in Class 1 methods takes most of the computational time, much more than is spent on the small molecule. Thus, Class 2 methods treat the protein as rigid and replace water molecules with a continuum description of solvation. With these two approximations, only the ligand degrees of freedom need to be sampled. By exploiting these assumptions commonly-used Class 2 methods [24,25] can computationally screen thousands to millions of compounds in a matter of days to weeks. These approaches are based on modeling the small molecule into the binding site, also referred to as docking or posing [26,27], followed by computation of the binding energy of the protein – small molecule complex.
Class 2 methods use a variety of expressions to compute the protein – small molecule binding energy. These include force-field based expressions, knowledge-based functions, and empirical functions [24,25,28]. These scoring functions are used both for docking the ligand to the protein and/or for ranking docked poses, with some protocols using a more expensive and presumably more accurate scoring function for the computationally less-expensive ranking phase. Unlike Class 1 methods, none of these binding scores are true free energies. The prevalence of the large variety of scoring functions is indicative of the fact that all are approximate and none offers the accuracy of Class 1 methods, which in turn has led to the use of “consensus” methods [29-32] in which multiple scoring functions are used to rank the ligands to arrive at a consensus rank.
In addition to the rigid protein and continuum solvent approximations, ligand entropy, which can make a large contribution to the binding free energy [33], needs to be accounted for by introducing an additional term into the scoring function or rescoring [34-36], and a score normalization based on molecular weight [37-39] is required in order to prevent over-favorable scoring of high molecular weight compounds.
Protein flexibility in Class 2 methods
The importance of protein flexibility for protein function is well-established [40], with the induced-fit and population-shift models used to rationalize [41] the binding of small molecules to protein conformations not seen in structural studies of apo proteins. The fact that these unobserved conformations are seen in complexes suggests that they are thermally accessible, though not as frequently sampled as the apo crystal structure in the absence of small molecule binding. Accordingly, one way of incorporating protein flexibility is the use of computational means to generate an ensemble of conformations and screen each small molecule against all protein conformations in the ensemble [42,43]. Since MD samples a thermodynamic ensemble, commonly-occurring thermally accessible non-crystallographic protein conformations will be present in snapshots from the simulation, and these conformations can be used in ensemble docking [44]. MD simulations have recently been used to generate ensembles that subsequently were used to successfully identify avian influenza neuraminidase [45], MDM2-p53 [46], DNA ligase [47], and selective SHP-2 phosphatase [48] inhibitors.
Explicitly sampling some subset of the protein degrees of freedom during docking is another way of including protein flexibility [42]. One advantage of this approach is that the protein and the small molecule can simultaneously relax to accommodate each other. However, this approach requires judicious choice of a subset of degrees of freedom to sample (e.g. sidechains in the binding pocket) in order to remain computationally tractable. A recent combination of simultaneous optimization of protein and small molecule degrees of freedom combined with ensemble docking looks to be promising [49], as does an automated approach in which the small molecule and the protein are iteratively optimized with the other molecule kept rigid [50].
Solvation in Class 2 methods
The most common approach to treating solvation effects in Class 2 methods is to disregard molecular water altogether and treat all the solvent as a continuum. The physics of solvation in a continuum scheme is often broken down into three components: solvent screening of electrostatic interactions, changes in solvent polarization energy upon binding, and the hydrophobic effect. Commonly-used methods addressing the first two phenomena are the Poisson-Boltzmann (PB) [51] and generalized Born (GB) [52] models, with the latter being a fast approximation of the former. Much of the research on GB models centers on protein stability and folding [53], though advances in the field are transferable to the treatment of small molecules. By way of an example, depending on the GB method used, quantitative agreement or gross errors are seen for the absolute binding free energy for a protein – small molecule complex [22]. The hydrophobic effect can be approximated using surface area (SA) models, and like the PB and GB models, research focusing on proteins [54,55] will hopefully lead to advances in modeling protein – small molecule interactions.
Despite advances in continuum solvation methods, the molecular nature of water continues to be difficult to capture in continuum representation, and inclusion of select water molecules explicitly Class 2 methods is a way of including molecular solvation effects [56,57]. Water molecules are often seen at binding interfaces and are important to binding [58], and “special” water molecules can make large contributions to the binding free energy of small molecules [59]. Explicit-solvent MD and Monte Carlo simulations can be used to inform the placement of water molecules that are conserved at interfaces, as they have longer residence times relative to other water molecules on the protein surface [60] and larger association free energies [61]. Such results are in turn being used to construct advanced continuum models [62].
Conclusion
Computational methods for the evaluation of protein – small molecule binding affinities have advanced to the stage where quantitative agreement can be achieved using Class 1 methods, and the approximate Class 2 methods are of adequate speed and sufficient accuracy to routinely inform the drug discovery process. Clearly, both approaches will see significant advances based on improved technologies combined with careful validation [63], as well as simply based on increased computer power. While this review has focused on Class 1 methods (“accurate but slow”) and Class 2 methods (“fast but approximate”), these two classes in reality represent ends of a spectrum. There exist a number of “intermediate” methods, ones that are not-too-slow and not-too-approximate. While not fast enough for computational high throughput screening, these intermediate methods are fast enough to be used routinely, and they are in principle more accurate than Class 2 methods. Notable among these intermediate methods are MM-PBSA and LIE [64]. Both use MD simulations of the protein – small molecule complex, and therefore incorporate protein flexibility, yet require significantly less computer time than a Class 1 simulation. This is because the stages between the fully bound and unbound stages in Class 1 methods are skipped, giving MM-PBSA and LIE the designation of “end point” free-energy simulations [23,64,65]. While both approaches have an extensive literature regarding their theoretical basis, they use implicit-solvent calculations on snapshots stripped of water molecules from the MD simulations, and are therefore by definition approximate [64]. They nonetheless benefit from being both more computationally feasible and more straightforward to execute than Class 1 methods as well as from having a more physically detailed basis than Class 2 methods.
Acknowledgments
This work was supported in part by the University of Maryland Computer-Aided Drug Design Center, the Waxman Foundation, NIH GM51501, CA107331, CA120215, HL082670 (ADM) and F32CA1197712 (OG).
Recent articles of special interest
*[13][Lu YP, Yang CY, Wang SM; 2006; Class 1 annihilation simulations were used to determine which interfacial water molecules are important contributors to binding free-energy in the tightly-bound HIV-1 – Kynostatin 272 complex, including an interesting analysis on the influence of protonation state on binding free-energy results.]
**[14][Qvist J, Davidovic M, Hamelberg D, Halle B; 2008; Class 1 annihilation simulations were used to confirm experimental data suggesting a hydrophobic binding cavity in bovine P-lactoglobulin is empty, i.e. contains no water molecules, in the apo state.]
*[22][Lee MS, Olson MA; 2008; In addition to being one of the few cases of a Class 1 separation simulation, this study vividly illustrates the failure of two different continuum solvent methods.]
**[23][Gilson MK, Zhou HX; 2007; Excellent recent review on computational protein – small molecule binding including an accessible overview on the statistical thermodynamics of binding and a brief but thorough discussion of the MM-PBSA and LIE mehods.]
*[28][Rajamani R, Good AC; 2007; Recent review of Class 2 methodology, including discussion of scoring functions.]
*[30][Feher M; 2006; Good review of consensus scoring.]
*[32][Konstantinou-Kirtay C, Mitchell JBO, Lumley JA; 2007; Recent case study on heat shock protein 90 using a variety of scoring functions and consensus scoring, showing that though the top single scoring function was different in two different docking protocols, consensus scoring did as well as the top single scoring function in both cases.]
**[33][Chang CEA, Chen W, Gilson MK; 25 kcal/mol loss in small molecule entropy upon binding was calculated, with the vast majority due to loss of vibrational entropy, while only 10% was from a drop in the number of stable conformers. Scoring terms to account for small molecule entropy used in Class 2 scoring functions are typically a function of the number of rotatable bonds, and therefore take into account conformational entropy and not vibrational entropy. The results therefore suggest a reconsideration of how to account for small molecule entropy in scoring functions.]
*[41][Okazaki KI, Takada S; 2008; Good discussion of induced fit and population-shift, with simulation results suggesting flexible protein – small molecule binding falls into the population-shift class.]
*[42][Wong CF; 2008; Good review of incorporating protein flexibility into Class 2 methods, with a focus on kinases as case studies.]
*[59][Young T, Abel R, Kim B, Berne BJ, Friesner RA; 2007; Molecular dynamics simulations were used to show how water molecules confined in a hydrophobic binding pocket can exhibit significantly different entropic and enthalpic behavior than bulk water.]
*[60][Samsonov S, Teyra J, Pisabarro MT; 2008; Comprehensive study of “wet spots,” i.e. interfacial residues interacting through only one water molecule.]
**[62][Abel R, Young T, Farid R, Berne BJ, Friesner RA; 2008; New continuum model that takes into account molecular nature of water.]
*[63][Kirchmair J, Markt P, Distinto S, Wolber G, Langer T; 2008; Recent virtual screening review with critical assessment of enrichment studies.]
*[64][Foloppe N, Hubbard R; Review with focus on MM-PBSA and LIE.]
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nature Structural Biology. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 2.Adcock SA, McCammon JA. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chemical Reviews. 2006;106:1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacKerell AD., Jr Empirical force fields for biological macromolecules: overview and issues. Journal Of Computational Chemistry. 2004;25:1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 4.Guvench O, MacKerell AD. Comparison of protein force fields for molecular dynamics simulations. In: Kukol A, editor. Molecular Modeling of Proteins. Humana Press, Inc.; 2008. pp. 63–88. Methods in Molecular Biology. [DOI] [PubMed] [Google Scholar]
- 5.Tuckerman ME, Martyna GJ. Understanding modern molecular dynamics: Techniques and applications. Journal of Physical Chemistry B. 2000;104:159–178. [Google Scholar]
- 6.Tuckerman ME, Liu Y, Ciccotti G, Martyna GJ. Non-Hamiltonian molecular dynamics: Generalizing Hamiltonian phase space principles to non-Hamiltonian systems. Journal of Chemical Physics. 2001;115:1678–1702. [Google Scholar]
- 7.Simonson T. Free energy calculations. In: Becker OM, MacKerell AD, Roux B, Watanabe M, editors. Computational biochemistry and biophysics. Marcel Dekker, Inc.; 2001. pp. 169–197. [Google Scholar]
- 8.Gilson MK, Given JA, Bush BL, McCammon JA. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophysical Journal. 1997;72:1047–1069. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woo HJ, Roux B. Calculation of absolute protein-ligand binding free energy from computer simulations. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:6825–6830. doi: 10.1073/pnas.0409005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jorgensen WL, Buckner JK, Boudon S, Tiradorives J. Efficient computation of absolute free-energies of binding by computer-simulations: application to the methane dimer in water. Journal of Chemical Physics. 1988;89:3742–3746. [Google Scholar]
- 11.Roux B, Nina M, Pomes R, Smith JC. Thermodynamic stability of water molecules in the bacteriorhodopsin proton channel: A molecular dynamics free energy perturbation study. Biophysical Journal. 1996;71:670–681. doi: 10.1016/S0006-3495(96)79267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boresch S, Tettinger F, Leitgeb M, Karplus M. Absolute binding free energies: A quantitative approach for their calculation. Journal of Physical Chemistry B. 2003;107:9535–9551. [Google Scholar]
- 13.Lu YP, Yang CY, Wang SM. Binding free energy contributions of interfacial waters in HIV-1 protease/inhibitor complexes. Journal of the American Chemical Society. 2006;128:11830–11839. doi: 10.1021/ja058042g. [DOI] [PubMed] [Google Scholar]
- 14.Qvist J, Davidovic M, Hamelberg D, Halle B. A dry ligand-binding cavity in a solvated protein. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6296–6301. doi: 10.1073/pnas.0709844105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng YQ, Roux B. Calculation of standard binding free energies: Aromatic molecules in the T4 lysozyme L99A mutant. Journal of Chemical Theory and Computation. 2006;2:1255–1273. doi: 10.1021/ct060037v. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Deng Y, Roux B. Absolute binding free energy calculations using molecular dynamics simulations with restraining potentials. Biophysical Journal. 2006;91:2798–2814. doi: 10.1529/biophysj.106.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiao D, Golubkov PA, Darden TA, Ren P. Calculation of protein-ligand binding free energy by using a polarizable potential. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6290–6295. doi: 10.1073/pnas.0711686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rick SW, Stuart SJ. Reviews in Computational Chemistry. Vol. 18. Wiley-Vch, Inc; 2002. Potentials and algorithms for incorporating polarizability in computer simulations; pp. 89–146. Reviews in Computational Chemistry. [Google Scholar]
- 19.Sneddon SF, Tobias DJ, Brooks CL., III Thermodynamics of amide hydrogen bond formation in polar and apolar solvents. Journal of Molecular Biology. 1989;209:817–820. doi: 10.1016/0022-2836(89)90609-8. [DOI] [PubMed] [Google Scholar]
- 20.Tobias DJ, Sneddon SF, Brooks CL. Stability of a model beta-sheet in water. Journal of Molecular Biology. 1992;227:1244–1252. doi: 10.1016/0022-2836(92)90534-q. [DOI] [PubMed] [Google Scholar]
- 21.Lee MS, Olson MA. Calculation of absolute protein-ligand binding affinity using path and endpoint approaches. Biophysical Journal. 2006;90:864–877. doi: 10.1529/biophysj.105.071589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee MS, Olson MA. Calculation of absolute ligand binding free energy to a ribosome-targeting protein as a function of solvent model. Journal of Physical Chemistry B. 2008;112:13411–13417. doi: 10.1021/jp802460p. [DOI] [PubMed] [Google Scholar]
- 23.Gilson MK, Zhou HX. Calculation of protein-ligand binding affinities. Annual Review of Biophysics and Biomolecular Structure. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 24.Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annual Review of Biophysics and Biomolecular Structure. 2003;32:335–373. doi: 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- 25.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: Current status and future challenges. Proteins-Structure Function and Bioinformatics. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 26.Alonso H, Bliznyuk AA, Gready JE. Combining docking and molecular dynamic simulations in drug design. Medicinal Research Reviews. 2006;26:531–568. doi: 10.1002/med.20067. [DOI] [PubMed] [Google Scholar]
- 27.Zhong SJ, Macias AT, MacKerell AD. Computational identification of inhibitors of protein-protein interactions. Current Topics in Medicinal Chemistry. 2007;7:63–82. doi: 10.2174/156802607779318334. [DOI] [PubMed] [Google Scholar]
- 28.Rajamani R, Good AC. Ranking poses in structure-based lead discovery and optimization: Current trends in scoring function development. Current Opinion in Drug Discovery & Development. 2007;10:308–315. [PubMed] [Google Scholar]
- 29.Clark RD, Strizhev A, Leonard JM, Blake JF, Matthew JB. Consensus scoring for ligand/protein interactions. Journal of Molecular Graphics & Modelling. 2002;20:281–295. doi: 10.1016/s1093-3263(01)00125-5. [DOI] [PubMed] [Google Scholar]
- 30.Feher M. Consensus scoring for protein-ligand interactions. Drug Discovery Today. 2006;11:421–428. doi: 10.1016/j.drudis.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 31.Oda A, Tsuchida K, Takakura T, Yamaotsu N, Hirono S. Comparison of consensus scoring strategies for evaluating computational models of protein-ligand complexes. Journal of Chemical Information and Modeling. 2006;46:380–391. doi: 10.1021/ci050283k. [DOI] [PubMed] [Google Scholar]
- 32.Konstantinou-Kirtay C, Mitchell JBO, Lumley JA. Scoring functions and enrichment: a case study on Hsp90. BMC Bioinformatics. 2007;8:9. doi: 10.1186/1471-2105-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang CEA, Chen W, Gilson MK. Ligand configurational entropy and protein binding. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1534–1539. doi: 10.1073/pnas.0610494104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salaniwal S, Manas ES, Alvarez JC, Unwalla RJ. Critical evaluation of methods to incorporate entropy loss upon binding in high-throughput docking. Proteins-Structure Function and Bioinformatics. 2007;66:422–435. doi: 10.1002/prot.21180. [DOI] [PubMed] [Google Scholar]
- 35.Ruvinsky AM. Role of binding entropy in the refinement of protein-ligand docking predictions: Analysis based on the use of 11 scoring functions. Journal of Computational Chemistry. 2007;28:1364–1372. doi: 10.1002/jcc.20580. [DOI] [PubMed] [Google Scholar]
- 36.Lee J, Seok C. A statistical rescoring scheme for protein-ligand docking: Consideration of entropic effect. Proteins-Structure Function and Bioinformatics. 2008;70:1074–1083. doi: 10.1002/prot.21844. [DOI] [PubMed] [Google Scholar]
- 37.Pan Y, Huang N, Cho S, MacKerell AD. Consideration of molecular weight during compound selection in virtual target-based database screening. Journal of Chemical Information and Computer Sciences. 2003;43:267–272. doi: 10.1021/ci020055f. [DOI] [PubMed] [Google Scholar]
- 38.Jacobsson M, Karlen A. Ligand bias of scoring functions in structure-based virtual screening. Journal of Chemical Information and Modeling. 2006;46:1334–1343. doi: 10.1021/ci050407t. [DOI] [PubMed] [Google Scholar]
- 39.Carta G, Knox AJS, Lloyd DG. Unbiasing scoring functions: A new normalization and rescoring strategy. Journal of Chemical Information and Modeling. 2007;47:1564–1571. doi: 10.1021/ci600471m. [DOI] [PubMed] [Google Scholar]
- 40.Dodson GG, Lane DP, Verma CS. Molecular simulations of protein dynamics: New windows on mechanisms in biology. EMBO Reports. 2008;9:144–150. doi: 10.1038/sj.embor.7401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okazaki KI, Takada S. Dynamic energy landscape view of coupled binding and protein conformational change: Induced-fit versus population-shift mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11182–11187. doi: 10.1073/pnas.0802524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong CF. Flexible ligand-flexible protein docking in protein kinase systems. Biochimica et Biophysica Acta-Proteins and Proteomics. 2008;1784:244–251. doi: 10.1016/j.bbapap.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Current Opinion in Structural Biology. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amaro RE, Baron R, McCammon JA. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. Journal of Computer-Aided Molecular Design. 2008;22:693–705. doi: 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng LS, Amaro RE, Xu D, Li WW, Arzberger PW, McCammon JA. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. Journal of Medicinal Chemistry. 2008;51:3878–3894. doi: 10.1021/jm8001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowman AL, Nikolovska-Coleska Z, Zhong H, Wang S, Carlson HA. Small molecule inhibitors of the MDM2-p53 interaction discovered by ensemble-based receptor models. Journal of the American Chemical Society. 2007;129:12809–12814. doi: 10.1021/ja073687x. [DOI] [PubMed] [Google Scholar]
- 47.Zhong S, Chen X, Zhu X, Dziegielewska B, Bachman KE, Ellenberger T, Ballin JD, Wilson GM, Tomkinson AE, MacKerell AD. Identification and validation of human DNA ligase inhibitors using computer-aided drug design. Journal of Medicinal Chemistry. 2008;51:4553–4562. doi: 10.1021/jm8001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu W-M, Guvench O, Qu C-K, MacKerell AD., Jr Identification of small molecular weight inhibitors of SHP-2 tyrosine phosphatase via in silico screening combined with experimental assay. Journal of Medicinal Chemistry. doi: 10.1021/jm800229d. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nabuurs SB, Wagener M, De Vlieg J. A flexible approach to induced fit docking. Journal of Medicinal Chemistry. 2007;50:6507–6518. doi: 10.1021/jm070593p. [DOI] [PubMed] [Google Scholar]
- 50.Koska J, Spassov VZ, Maynard AJ, Yan L, Austin N, Flook PK, Venkatachalam CM. Fully automated molecular mechanics based induced fit protein-ligand docking method. Journal of Chemical Information and Modeling. 2008 doi: 10.1021/ci800081s. [DOI] [PubMed] [Google Scholar]
- 51.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 52.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. Journal of the American Chemical Society. 1990;112:6127–6129. [Google Scholar]
- 53.Chen JH, Brooks CL, Khandogin J. Recent advances in implicit solvent-based methods for biomolecular simulations. Current Opinion in Structural Biology. 2008;18:140–148. doi: 10.1016/j.sbi.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wagoner JA, Baker NA. Assessing implicit models for nonpolar mean solvation forces: The importance of dispersion and volume terms. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8331–8336. doi: 10.1073/pnas.0600118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin MS, Fawzi NL, Head-Gordon T. Hydrophobic potential of mean force as a solvation function for protein structure prediction. Structure. 2007;15:727–740. doi: 10.1016/j.str.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 56.Rarey M, Kramer B, Lengauer T. The particle concept: Placing discrete water molecules during protein-ligand docking predictions. Proteins-Structure Function and Genetics. 1999;34:17–28. [PubMed] [Google Scholar]
- 57.Mancera RL. Molecular modeling of hydration in drug design. Current Opinion in Drug Discovery & Development. 2007;10:275–280. [PubMed] [Google Scholar]
- 58.Li Z, Lazaridis T. Water at biomolecular binding interfaces. Phys Chem Chem Phys. 2007;9:573–581. doi: 10.1039/b612449f. [DOI] [PubMed] [Google Scholar]
- 59.Young T, Abel R, Kim B, Berne BJ, Friesner RA. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proceedings of the National Academy of Sciences. 2007;104:808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Samsonov S, Teyra J, Pisabarro MT. A molecular dynamics approach to study the importance of solvent in protein interactions. Proteins-Structure Function and Bioinformatics. 2008;73:515–525. doi: 10.1002/prot.22076. [DOI] [PubMed] [Google Scholar]
- 61.Barillari C, Taylor J, Viner R, Essex JW. Classification of water molecules in protein binding sites. Journal of the American Chemical Society. 2007;129:2577–2587. doi: 10.1021/ja066980q. [DOI] [PubMed] [Google Scholar]
- 62.Abel R, Young T, Farid R, Berne BJ, Friesner RA. Role of the active-site solvent in the thermodynamics of Factor Xa ligand binding. Journal of the American Chemical Society. 2008;130:2817–2831. doi: 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirchmair J, Markt P, Distinto S, Wolber G, Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection - What can we learn from earlier mistakes? Journal of Computer-Aided Molecular Design. 2008;22:213–228. doi: 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]
- 64.Foloppe N, Hubbard R. Towards predictive ligand design with free-energy based computational methods? Current Medicinal Chemistry. 2006;13:3583–3608. doi: 10.2174/092986706779026165. [DOI] [PubMed] [Google Scholar]
- 65.Huang N, Jacobson MP. Physics-based methods for studying protein-ligand interactions. Current Opinion in Drug Discovery & Development. 2007;10:325–331. [PubMed] [Google Scholar]