

Abstract
Cardiovascular abnormalities, especially structural congenital heart defects (CHDs), commonly occur in malformation syndromes and genetic disorders. Individuals with syndromes comprise a significant proportion of those affected with selected CHDs such as complete atrioventricular canal, interrupted arch type B, supravalvar aortic stenosis and pulmonary stenosis. As these individuals age, they contribute to the growing population of adults with special health care needs. Although most will require longterm cardiology followup, primary care providers, geneticists and other specialists should be aware of (1) the type and frequency of cardiovascular abnormalities, (2) the range of clinical outcomes, and (3) guidelines for prospective management and treatment of potential complications. This article reviews fundamental genetic, cardiac, medical and reproductive issues associated with common genetic syndromes which are frequently associated with a cardiovascular abnormality. New data are also provided about the cardiac status of adults with a 22q11.2 deletion and with Down syndrome.
Keywords: congenital heart defect, deletion 22q11, Down syndrome, Marfan syndrome, Noonan syndrome, Turner syndrome, Williams-Beuren syndrome
Cardiovascular abnormalities that occur in malformation syndromes and genetic conditions may affect the heart’s structure and great arteries (congenital heart defects, CHDs),1 muscle (cardiomyopathy), conduction system (arrhythmias, heart block), or connective tissue (aortic dilatation/dissection, mitral valve prolapse). Most clinical geneticists are familiar with the type, frequency and management of CHDs when they occur in children,2 but are less knowledgeable about the clinical history and treatment options for the adult. Depending on the specific syndrome, information about morbidity and mortality may or may not be available for those with a CHD.
Considerable attention has been devoted recently to the prevalence and management of CHDs in the adult.3–9 However, there has been little discussion about the subset of individuals whose CHD occurs in association with a syndrome or genetic disorder and who are now living longer and fuller lives which may include childbearing.10 Because of improved medical care, the number of adults with syndromes is a growing portion of the world’s population with special health care needs.11–12 As many patients transition from pediatric to adult caregivers, it becomes imperative to expand the knowledge base for both geneticists, cardiologists, other specialists and of course, primary care practitioners.
Estimates of the prevalence of adults with syndromes and genetic disorders who have a co-existing CHD do not currently exist. Fortunately, studies on the general population (e.g., non-syndromic) adult with a CHD provide frequency estimates from which we can extrapolate. The recent National Heart, Lung, and Blood Institute Working Group on Research in Adult Congenital Heart Disease estimated that the number of adults in the general population with “congenital heart disease” living in the year 2000 was 787,800, subdivided into 368,800 with “simple”, 302,500 with “moderate” and 117,000 with conditions of “great complexity”.5 Comparable data for children were calculated to be 623,000 total, 320,000 simple, 165,000 moderate and 138,000 highly complex conditions. Estimating the fraction with a syndrome or genetic condition is challenging, though it can be approximated from the population-based Baltimore-Washington Infant Study in which nearly 17% of infants with a CHD had an identifiable syndrome.13 Extrapolating this frequency provides an estimate that ~ 106,000 children are currently living with a syndrome and a CHD. Additional figures for gauging the birth prevalence of infants with a CHD and a syndrome (in contrast to the incidence in adults) are available (see Botto et al., Tables 25.3, 25.6).14 Thus, we believe there already are, or shortly will be, several hundred thousand adults with a genetic syndrome and a CHD in need of the specialized care as detailed below.
In addition to requiring medical care, adult women with genetic syndromes and coexisting cardiovascular abnormalities need answers to questions about fertility, childbearing and recurrence risk. For selected syndromes (i.e. Turner syndrome15, Marfan syndrome16) guidelines have addressed the care of the pregnant woman, but most of the syndromes in this review have not been studied to assess whether reproductive outcomes differ between adults with or without cardiovascular abnormalities.
This article reviews the spectrum of cardiovascular abnormalities, clinical history and general cardiac care guidelines for several common malformation syndromes and genetic conditions. Geneticists and pediatric cardiologists will encounter most of these conditions during their careers, but family practitioners, internists and adult cardiologists will meet them less frequently. Accordingly, we have provided a brief description of the clinical phenotype for the non-geneticist reader. A rich source of open access online information for selected disorders can be found at Online Mendelian Inheritance in Man (OMIM), and GeneTests/GeneReviews.org. Although the classic presentation of a syndrome such as Holt-Oram syndrome may be rare, the individual components such as an atrial septal defect, conduction heart block or hypoplastic thumb may not be uncommon in the practice of an adult cardiologist. It is essential to establish accurate diagnosis in order to direct immediate and long-term clinical management, including genetic counseling. With the elucidation of the underlying genetic cause of many of these syndromes, accurate diagnosis becomes more feasible.
The emphasis of this review is on structural CHDs, with some discussion about valve dysplasia, aortic enlargement, and hypertrophic cardiomyopathy. Excluded from this review are disorders which involve primarily the conduction system (e.g. Jervell-Nielsen syndrome) or myocardium (e.g. familial hypertrophic or dilated cardiomyopathy). Supplementing the text, Table 1 presents management issues derived in most cases from evidence quality that is “expert opinion, case reports and reasoning from first principles” rather than randomized control trials.17,18 It serves as a convenient single source and is not intended to be used as firm recommendations or a rigid care plan, since individual cardiologists and adult caregivers will customize the timing of follow-up and specific tests on a case by case basis. Certain guidelines are based on the specific CHD, irrespective of syndrome association. For example, the indications for antibiotic prophylaxis for bacterial endocarditis have decreased greatly, and should be prescribed in accordance with current American Heart Association guidelines for patients with CHDs.19 All adults should continue to maintain a heart healthy lifestyle. For the sake of completeness, Table 2 summarizes additional syndromes and conditions which are either (a) less common, (b) less likely to survive to adulthood or (c) associated with cardiovascular abnormalities which are less distinctive.
Table 1.
Common management issues for adults with geneticsyndromes and cardiovascular abnormalities: General information for pimary care providersa
| Syndrome | Type of heart problem (most common or distinctive) | Complication: Assume postoperative unless specified | Management issues |
|---|---|---|---|
| Chromosome abnormality | |||
| 22q11.2 deletion | Interrupted aortic arch type B | Left ventricular outflow tract obstruction. Residual coarctation. | Depending on degree of obstruction, balloon dilation or surgical correction may be needed. |
| Truncus arteriosus | Right ventricular outflow tract obstruction or insufficiency. Truncal valve regurgitation and stenosis. | Depending on degree of right ventricular, or truncal obstruction or regurgitation, surgery may be required, e.g. valve repair or replacement. | |
| Tetralogy of Fallot | Right ventricular outflow tract obstruction, including pulmonary artery obstruction, pulmonary regurgitation, right ventricular dilation | Depending on degree of obstruction or regurgitation, surgery may be required. | |
| Down syndrome | Complete AVCb | Common atrioventricular valve regurgitation or stenosis. | Depending on degree of regurgitation or stenosis, surgery may be required. |
| Residual shunt at the site of the septal defect closure. | Depending on size of shunt or residual lesion, surgery may be required. | ||
| Arrhythmia | Postoperative bradycardia, syncope | Monitor all cases. May need pacemaker | |
| Mitral valve prolapse, less frequently aortic and tricuspid prolapse | Usually asymptomatic. Mitral, aortic and tricuspid regurgitation may develop. | Auscultation in all individuals, obtain echocardiogram if abnormal. | |
| Coronary artery disease | Not known to have increased risk of angina or myocardial infarction. | Routine population screening. | |
| Turner syndrome | Bicuspid aortic valve | Can progress to aortic stenosis. As in general population, there is associated risk of aortic dilatation, aneurysm and rupture. | Monitor all cases with meticulous imaging of aortic root using echocardiography or MRI. |
| Coarctation | Residual obstruction at the site of coarctation repair may lead to hypertension. Some mild coarctation may go undetected until adulthood. Repaired or unrepaired, associated with risk of aortic dilatation, dissection, and rupture. | Depending on severity of obstruction, may require antihypertensive medication, interventional catheterization or surgical correction. Aorta requires meticulous imaging using echocardiograph, preferably MRI. | |
| Hypertension | Can be mild to severe. | Seek underlying cause, e.g. unrepaired coarctation. Treat aggressively when hypertension is essential since it is a risk factor for aortic dissection. | |
| Aortic root dilatation | Aortic dissection and rupture area associated with dilatation, although the natural history from dilatation to dissection has not been completely delineated. | Baseline imaging of aorta should be obtained in all individuals; at least one MRI examination may be needed to provide optimal imaging. Imaging should be repeated. Beta-blockade can be useful. | |
| ECG abnormalities | Aside from predisposition to resting tachycardia, not known to have pathologic arrhythmias. | Because of observed cardiac conduction and repolarization abnormalities, avoid drugs that increase risk of QT prolongation. | |
| Williams-Beuren syndrome | Supravalvar aortic stenosis, focal or diffuse. | Residual left ventricular outflow tract obstruction Re-stenosis |
Depending on degree of obstruction, surgical correction may be needed |
| Long-segment stenosis/hypoplasia of thoracic or descending aorta (“coarctation) | Re-coarctation | Depending on severity of obstruction, may require antihypertensive medication, interventional catheterization or surgical correction | |
| Coronary artery stenosis (due to medial hypertrophy or dysplastic aortic valve leaflet) | Coronary insufficiency | Screen coronary patency by echocardiogram (limited sensitivity), CTA, MRA or catheterization (if these procedures are indicated). Exercise stress testing is of limited utility due to poor exercise tolerance in most adults with WBS | |
| Hypertension | Can occur at any age | Screen for renovascular cause. Treat with antihypertensive medication. | |
| Pulmonary artery stenoses (PPS, main branch stenoses) | Balloon dilation or stenting for significant obstruction. Complications such as tears or aneurysms reported. | ||
| Gene abnormality | |||
| Holt-Oram syndrome | ASD, secundum-type, VSD, membranous, muscular | Residual shunt, rarely hemodynamic risk. Potential for stroke. | Depending on size of shunt, may need surgical closure. |
| ASD, secundum-type, VSD, membranous, muscular | Eisenmenger syndrome | Medication for pulmonary hypertension | |
| Conduction block | Can be progressive; when complete heart block is present, may be associated with atrial fibrillation | Need regular monitoring with ECG and 24 hour Holter monitoring in those with known conduction disease for development and/or progression. Severe cases may require permanent pacemaker; antiarrhythmics and anticoagulants (for atrial fibrillation) may be required | |
| Marfan syndrome | Aortic dilatation | Aortic dissection, rupture Aortic regurgitation | Prophylactic aortic root repair to prevent dissection (>45–50 mm); valve sparing technique option |
| Aortic dissection | Progressive dilatation, rupture | Prophylactic grafting of any portion of the aorta when the risks of surgery are less than the risks of medical management, or in the case of end-organ ischemia, persistent pain | |
| Mitral valve prolapse | Mitral regurgitation, heart failure | Mitral valve repair or replacement | |
| TGF-B receptor syndromes | Aortic dilatation | Aortic dissection, rupture | Prophylactic aortic root repair to prevention dissection, but using aortic diameter criteria less than what is used for Marfan syndrome (>40 mm instead of 50 mm in the adult) |
| Aneurysm of other major arteries | Arterial dissection, rupture | Prophylactic repair | |
| Hereditary Hemorrhagic Telangectasia | Pulmonary arterio-venous malformation | Cyanosis; paradoxic embolization leading to stroke or brain abscess | Therapeutic or prophylactic embolization of the AVM with coils when the diameter of the feeding artery is 1 mm or greater |
| Hepatic arteriovenous malformation | Cirrhosis; hepatic encephalopathy | Medical management of liver dysfunction | |
| Noonan syndrome | Pulmonic stenosis | Can present in adulthood for the first time; those with previous surgery or balloon angioplasty in childhood can have residual PS and/or pulmonary insufficiency | Options for severe pulmonary stenosis include balloon valvuloplasty or valve replacement; severe pulmonary insufficiency often requires valve replacement |
| ASD, secundum type | Residual shunt after repair in childhood; some adults have undetected large defect and fatigue symptoms; in combination with pulmonary stenosis may have risk for stroke | Depending on size of shunt may require surgical closure or closure by a septal occluder device | |
| Hypertrophic cardiomyopathy | Right ventricular outflow obstruction; can coexist with a CHD such as pulmonary stenosis; risk of arrhythmias | Treatment considerations include surgical myomectomy and beta blockers; careful monitoring for arrhythmias may also include treatment such as amiodarone, pacemaker or defibrillator | |
Guidelines provide general information for primary care providers, but individuals require specific management by cardiologists. In the interest of brevity, each cardiac abnormality for each disorder was not listed.
ASD, VSD and tetralogy of Fallot were not discussed Antibiotic prophylaxis for subacute bacterial endocarditis should be offered in appropriate doses per standard practice.19
ASD, atrial septal defect; AVC, atrioventricular canal defect; CAVC, complete atrioventricular canal; CHD, congenital heart defects; CTA, CT angiography; ECG, electrocardiographic; VSD, ventricular septal defect
Table 2.
Syndromes which are uncommon, have decreased survival to adulthood or infrequently associated with distinctive cardiovascular abnormalities
| Syndrome | Cardiovascular abnormality in adults |
|---|---|
| Chromosome abnormality | |
| Deletion 4p267 | Pulmonic stenosis, valvar |
| Mendelian syndrome | |
| Alagille syndrome267 | Pulmonic stenosis, valvar |
| Branch pulmonary stenosis | |
| Tetralogy of Fallot | |
| ASD, VSD | |
| Cardio-facio-cutaneous (CFC) syndrome269,270- | Pulmonic stenosis, valvar |
| HCM | |
| Costello syndrome274,272 | Pulmonic stenosis, valvar |
| HCM | |
| Fragile X syndrome273 | Mitral valve prolapse |
| Townes-Brocks syndrome274 | Conotruncal anomalies |
| ASD, VSD | |
| NKX2.5 deletion274 | ASD, VSD |
| Arrhythmias | |
| Tetralogy of Fallot | |
| Maternal exposure | |
| Diabetic embryopathy274 | HCM |
| Conotruncal anomalies | |
| ASD, VSD | |
| Maternal phenylketonuria274 | Conotruncal anomalies |
| Left ventricular outflow traction obstruction | |
| Isotretinoin embryopathy276,277 | Conotruncal anomalies |
ASD, atrial septal defect; ECG, electrocardiography; HCM, hypertrophic cardiomyopathy; VSD, ventricular septal defect
CHROMOSOME ABNORMALITY SYNDROMES
The 22q11.2 deletion
The 22q11.2 deletion (del 22q11.2) syndrome is the most commonly diagnosed chromosome deletion syndrome with an estimated prevalence between 1 in 4,000 and 1 in 7,000 livebirths.20–24 It is the most frequent genetic condition associated with a CHD after Down syndrome,21 and is calculated to be present in between 1.5% and 5% of children with a CHD22,24,25. Retrospectively, it has been identified in the majority of patients described clinically with DiGeorge syndrome,25 velocardiofacial syndrome,26 and conotruncal anomaly face syndrome,27 and in some patients with the autosomal dominant Opitz G/BBB syndrome28–30 and Cayler Cardiofacial syndrome (asymmetric crying facies syndrome).31 There is marked inter-and intrafamilial phenotypic variability though most patients have the same large (>3MB) deletion encompassing ~ 30 genes. A subset of patients have been identified with smaller distal deletions, an emerging phenotype which may differ from DiGeorge and velocardiofacial syndromes.32,33 Approximately 90% of deletions occur as a de novo event, but an affected individual has a 50% recurrence risk for each offspring, as in other contiguous deletion syndromes. Unlike the early reports of patients with DiGeorge syndrome, the current mortality rate is low (4%) primarily clustering in infancy (e.g., median age of death at 4 months).34 As a consequence of increased awareness and readily available diagnostic testing, as well as improved survival, the prevalence of diagnosed cases of this disorder is likely to increase. Studies will be needed to determine if life expectancy, notwithstanding significant multisystem morbidity in some patients, is similar to other adults with CHDs in the general population.
In addition to CHDs, features of this disorder are protean and can include: characteristic facial features in childhood (narrow palpebral fissures, “hooded” eyelids, prominent nasal root, full nasal tip, overfolded or squared off external ears, small mouth and chin), immunodeficiency, hypocalcemia, palate anomalies,34 gastrointestinal and feeding disorders, speech and learning disabilities, and behavioral as well as psychiatric disorders. Other findings include skeletal, renal and genitourinary anomalies, and endocrinologic issues. Infants often have significant feeding disorders with failure to thrive, while older children often experience substantial challenges in school with behavioral problems such as attention deficit/hyperactivity disorder or autism spectrum disorders. Adults can have recurrent respiratory tract infections, autoimmune disorders, and psychiatric disorders including schizophrenia (Figs. 1, 2).35,36 Hypocalcemia due to hypoparathyroidism often presents in the newborn, usually resolves in the first year of life, but may recur or occur later in childhood or adulthood.34
Fig. 1.
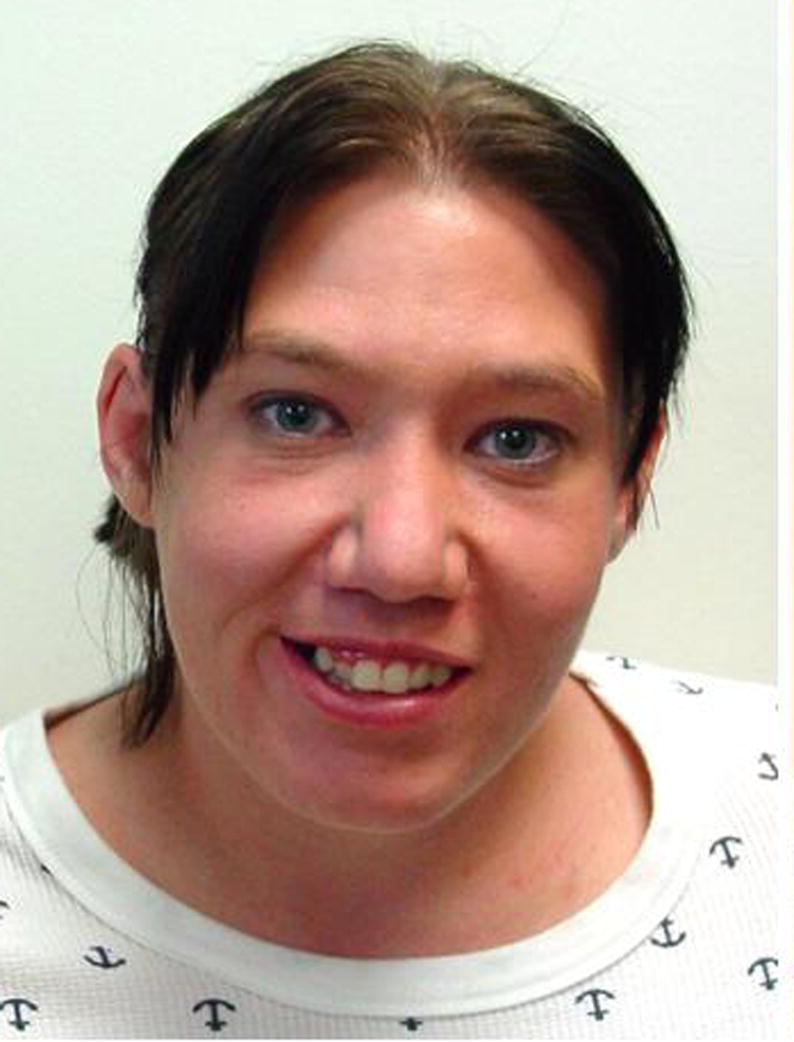
26-year-old woman with deletion 22q11.2 syndrome who had repair of a double aortic arch. Her ears are somewhat small and her palpebral fissures are relatively narrow compared to the prominence of her nose, she does not have the classic facies that can be seen in some young children with velocardiofacial syndrome. Her life has been challenged by an anxiety disorder with features of psychosis, and having a son with deletion 22q11.2. (courtesy of Alan F. Rope, M.D.).
Fig. 2.

48-year-old man with deletion 22q11.2 syndrome who had a stenotic, bicuspid aortic valve that required replacement in his mid-twenties. His facial appearance does not show the common dysmorphic features associate with velocardiofacial syndrome. He is receiving regular treatment for schizophrenia (courtesy of Alan F. Rope, M.D.).
Cardiovascular abnormalities during childhood
Congenital heart defects occur in approximately 75% of individuals with the 22q11.2 deletion,23,34,37 reflecting what may be an ascertainment bias towards those who present early in life with CHDs.38,39 A subset of conotruncal defects are particularly common in the 22q11.2 deletion syndrome, such as tetralogy of Fallot (including the common form with valvar pulmonary stenosis and the less common varieties with absent pulmonary valve or pulmonary valve atresia with major aortopulmonary collateral arteries), interrupted aortic arch type B, truncus arteriosus, ventricular septal defect (VSD) (typically conventricular or membranous, rather than atrioventricular canal-type or muscular), and aortic arch anomalies. In contrast, very few patients with a 22q11.2 deletion have double outlet right ventricle or D-transposition of the great arteries. A variety of other CHDs have been seen in conjunction with a 22q11.2 deletion, including atrial septal defect, complete atrioventricular canal, patent ductus arteriosus, bicuspid aortic valve, hypoplastic left heart syndrome and heterotaxy.24,34,37
Conversely, a significant number of patients with particular types of conotruncal and related CHDs are found to have a 22q11.2 deletion. Several reports have estimated that 50–80% of patients with an interrupted aortic arch (IAA) type B, 40% of patients with truncus arteriosus, 15% of patients with tetralogy of Fallot (grouping all types together), 10% of patients with a conoventricular VSD and 24% of patients with an isolated aortic arch anomaly (with normal intracardiac anatomy) have a 22q11 deletion.40–42 Regardless of intracardiac anatomy, patients with an aortic arch anomaly (such as a cervical arch, right sided arch or abnormal origin of the subclavian artery) are at higher risk of having a 22q11 deletion than those with completely normal aortic arch sidedness and branching pattern.43
Most infants with a 22q11.2 deletion with a conotruncal defect or large VSD will undergo cardiac surgery within the first year of life. Post-operative complications following repair for IAA type B include re-coarctation of the aorta and/or recurrent obstruction to flow across the left ventricular outflow tract. Patients with a complete repair for truncus arteriosus can develop truncal valve stenosis or regurgitation, which may require surgical re-intervention. These patients also commonly require surgical revision of the conduit placed from the right ventricle to the branch pulmonary arteries, and may require interventional cardiac catheterization to reduce branch pulmonary artery stenosis. Patients with tetralogy of Fallot may have similar complications as those with truncus arteriosus due to recurrent right-sided obstruction, and those whose repair included a transannular patch will develop pulmonary insufficiency, leading to right ventricular dilation and dysfunction. This subset of patients with tetralogy of Fallot may require pulmonary valve replacement43. Surgical intervention is significantly more challenging when the pulmonary arteries are critically small, and/or when the blood supply to the lungs is derived in large part from aortopulmonary collaterals, which are both more likely to occur with the 22q11.2 deletion.44
Few studies have evaluated whether the immediate surgical or long-term clinical outcomes differ between those cardiac patients with and those without a 22q11.2 deletion. Mahle and colleagues45 reported that patients with tetralogy of Fallot/pulmonary atresia and a 22q11.2 deletion had a substantially lower rate of five-year survival (36%) than those without a 22q11.2 deletion (90%). Likewise, Anaclerio and colleagues46 reported a higher risk of surgical mortality in patients with a 22q11.2 deletion and either tetralogy of Fallot/pulmonary atresia or interrupted aortic arch type B as compared to non-syndromic patients, but also noted no difference in 10-year actuarial survival between those with and without a 22q11.2 deletion in a cohort with “classic” tetralogy of Fallot.47 Therefore, immediate and long-term surgical survival may differ between deleted and non-deleted patients in specific cardiac subsets, although further studies are required, particularly to address questions of morbidity rather than mortality alone.
Cardiovascular abnormalities in adulthood
Several studies have reported on adults with the 22q11.2 deletion syndrome34–38, 48–50 though some of these patients were identified only after the diagnosis of an affected child. This subset may represent the less severe end of CHD spectrum. With this in mind, Table 3 provides data on the presence, type and frequency of CHDs in 132 adults with the 22q11.2 deletion followed at The Children’s Hospital of Philadelphia. Nearly half of the cohort has either no overt cardiovascular involvement (41%) or minimal cardiovascular manifestations that have not required surgical intervention (9%), while the remainder has conotruncal, septal and aortic arch malformations typical of the 22q11.2 deletion syndrome.
Table 3.
Adults (>18years) with the deletion 22q11.2 followed at the Children’s Hospital of Philadelphia, Pennsylvania (n= 132)
| Type of CHD | Number pts (%) |
|---|---|
| All Patients | 132 |
| Patients with CHD | 78 (59%) |
| Patients with no CHD | 54 (41%) |
| Total with CHD Repair: | 67 (51% of total cohort) (86% of CHDs) |
| Conotruncal, total: | 33 |
| Tetralogy of Fallot | 19 |
| IAA Type B | 8 |
| Truncus arteriosus | 6 |
| Aortic Arch Anomaly: | 5 |
| Vascular Ring | 4 |
| Vascular Ring with aortic stenosis | 1 |
| ASD or VSD | 27 |
| VSD | 17 |
| ASD/VSD, type NS | 6 |
| ASD | 4 |
| Pulmonic Stenosis, NS if valvar | 2 |
| 11 | |
| Total with minor CHD, No intervention required | (7% of total cohort) (14% of CHDs) |
| Right Aortic Arch | 5 |
| Bicuspid aortic valve | 1 |
| Mitral valve prolapse | 3 |
| HCM and bicuspid aortic valve | 1 |
| Mild pulmonary artery stenosis | 1 |
| No CHD: | 54 (41% of total cohort) |
| Normal Echocardiogram | 22 |
| Normal by History | 27 |
| Normal with minor echocardiographic findings: | 5 |
| Mild Aortic Arch Dilatation | 1 |
| Mild MR, AR | 1 |
| Mild Tortuosity of the Thoracic Aorta | 1 |
| Enlarged pulmonary artery | 1 |
| Patent ductus arteriosus | 1 |
AR, aortic regurgitation; ASD, atrial septal defect; CHD, congenital heart defect; HCM, hypertrophic cardiomyopathy; IAA, interrupted aortic arch; MR, mitral regurgitation; VSD, ventricular septal defect
Adults with a conotruncal defect and the 22q11.2 deletion s1yndrome whose CHD was repaired in childhood are likely to have postoperative complications similar to their non-syndromic counterparts. The right ventricular to pulmonary artery conduits may require treatment for both pulmonary regurgitation and valvar stenosis. Individuals who had repair of an interrupted aortic arch may have recoarctation of the reconstructed aortic arch, but this is less likely in later years as an adult than in the first post-operative year. Residual or acquired left ventricular outflow obstruction at the level of the aortic valve, truncal valve or subaortic region may develop.41,51 In addition to structural and functional residua, tetralogy of Fallot is frequently associated with postoperative atrial tachyarrhythmias, ventricular tachyarrhythmias and heart block.48,52,53 These patients should have long term follow up with a cardiologist familiar with congenital heart disease. Given the lack of specific data, it is not clear whether patients with a 22q11.2 deletion are at greater risk than the general populations for essential hypertension, atherosclerosis, coronary artery disease, stroke, thromoboembolic events, or cardiomyopathy. Particularly in this population, counseling about appropriate and healthy levels of aerobic exercise and heart-healthy diets is clearly warranted.
Strong consideration should be given to testing an adult with a CHD typical of the 22q11.1 deletion syndrome given the implications for recurrence in offspring and the occurrence of non-cardiac features of the disorder in the adult. Testing the adult with a newly diagnosed CHD typical of the 22q11 deletion syndrome, such as an isolated arch anomaly, for a 22q11.2 deletion should also be considered. Those who have a newly diagnosed CHD that is not typical, such as a bicuspid aortic valve or atrial septal defect, should be tested for a 22q11 deletion only if additional syndromic features are observed. Special consideration should be given to the adult patient with previously diagnosed apparently nonsyndromic CHD, whose cardiac diagnosis was established prior to the advent of 22q11.2 deletion testing. Such patients are now candidates for deletion screening, particularly in the presence of a conotruncal CHD, or when CHD is associated with cleft palate, learning disability, or psychiatric disorder.32
Pregnancy-related cardiovascular issues
There are very little data on pregnancy in women with a 22q11.2 deletion. Their pregnancy-related personal medical risks are based on their own medical condition, with particular emphasis on the type of CHD and residual cardiovascular hemodynamic changes. Other factors which might affect pregnancy-related risks include asthma, autoimmune disease, and renal function. An affected parent has a 50% recurrence risk with each pregnancy, and 75% of affected fetuses are thought to be at risk for having a CHD. Prenatal ultrasonographic clues to the diagnosis of a 22q11.2 deletion include polyhydramnios, cleft palate, structural renal abnormalities; less commonly noted anomalies include, cleft lip and palate, polydactyly, myelomeningocele, and congenital diaphragmatic hernia.20, 42 54–55
Down syndrome
Approximately 95% of patients with Down syndrome have complete trisomy 21 in which there is an extra copy of chromosome 21. In rare cases, partial trisomy of chromosome 21 is present due to a chromosomal translocation or mosaicism. The latest national livebirth prevalence estimate for Down syndrome (1999–2001), adjusted for maternal age, is 1.36 per 1,000 (about 5,500 per year).56 Overall survival has improved, though prenatally diagnosed CHDs and/or intra-uterine growth retardation predict a worse outcome.57 Population-based data from the United States demonstrate that the median age at death increased from 25 to 49 years in the interval between 1983 to 1997.58 A recent analysis from Australia suggests that individuals with Down Syndrome may be expected to have a lifespan similar to the general population within the next generation,59 although additional data are needed to determine if this optimistic view is accurate worldwide.
The well-known facial appearance of Down syndrome varies with ethnic background and changes over time (Fig. 3). Frequent features include microbrachycephaly, sparse hair, midfacial hypoplasia with small nose, eyes, ears, and a protruding tongue. The eyes tend to slant upwards and have epicanthal folds, with Brushfield spots on the irides. Also common are single transverse palmar creases, fifth finger clinodactyly, brachydactyly, a gap between first and second toes, atlantoaxial instability, a hypoplastic pelvis, and joint laxity. Neurodevelopmental challenges include hypotonia, developmental delay, moderate mental retardation, visual impairment, and hearing loss.60 Structural and functional anomalies of the gastrointestinal and genitourinary systems are common, as are hematologic disorders.60
Fig. 3.

19-year-old woman with Down syndrome (47, XX,+21) and an unrepaired small membranous ventricular septal defect, which has remained hemodynamically insignificant since childhood (NHYA I). She has small, slightly upslanted palpebral fissures, and a small mouth. (courtesy of Catherine B. Nowak, M.D. and family)
Cardiovascular abnormalities during childhood
Depending on study design, approximately half of Down syndrome individuals are found to have a CHD,61,62 60% of whom have some type of an atrioventricular canal defect. Typically, there is a complete atrioventricular canal defect or primum type atrial septal defect (usually with a cleft mitral valve), and less commonly a canal-type VSD or transitional atrioventricular canal. Additional CHDs may include an atrial septal defect, VSD, patent ductus arteriosus, tetralogy of Fallot or double outlet right ventricle. Congenital heart defects with hemodynamically significant shunts are generally treated aggressively and repaired surgically in infancy.63
A slightly higher peri-operative and late cardiac mortality in patients with Down syndrome and complete atrioventricular canal defect has been observed, although the outcome for other CHDs was similar to children without Down syndrome.64,65 Currently, similar surgical options are offered to both Down syndrome and non-Down syndrome individuals, and reassuringly comparable post-operative complications have been observed. Post-operative issues include residual atrial or ventricular-level shunts, and more commonly, varying degrees of incompetence of the atrioventricular valve.65 Additional injury (or congenital dysfunction) to the atrioventricular node may be associated with variable heart block and associated dysrhythmias.
Cardiovascular abnormalities in adulthood
Considering it is such a common syndrome, there is a surprising lack of primary data about Down syndrome adults with a CHD. Depending on the underlying defect, some adults with Down syndrome may have a residual shunt at the atrial or ventricular level, atrioventricular valve regurgitation or, less commonly, stenosis of the surgically corrected valve. A characteristic complication of partial atrioventricular canal repair is left ventricular outflow tract obstruction in which the subaortic obstruction has the angiographic appearance of an elongated “gooseneck” deformity.63 In addition to these hemodynamic residua, there may be progressive heart block. In some individuals, bradycardia which may have been previously tolerated leads to exercise intolerance if the nodal heart rate decreases. In contrast to these postoperative cardiac issues, clinicians may infrequently encounter the Down syndrome individual with an unrepaired CHD, either diagnosed late or untreated for other reasons. These older individuals may have Eisenmenger syndrome with progressive cyanosis, pulmonary hypertension, subacute bacterial endocarditis and stroke.
Most reviews about Down syndrome have discussed cardiac issues in childhood without specifying the type and frequency of potential adult cardiac problems, or making specific recommendations regarding cardiology followup into adulthood.60,66,67 Several reports identified an increased risk of mitral valve prolapse (45%), increased echogenicity of the mitral valve and aortic regurgitation in Down syndrome adults without prior history of CHD.68–71 Less commonly noted are aortic regurgitation or tricuspid valve prolapse Previously, the occurrence of valve disease and consequent need for endocarditis prophylaxis was the reason to offer echocardiography in all adults,72 although others maintained that only abnormal auscultation should prompt referral.73
However, the current AHA Scientific Statement for typical mitral valve prolapse does not include a recommendation for routine antibiotics before dental procedures (http://circ.ahajournals.org).19 Thus, routine echocardiographic screening for adults with Down syndrome without pre-existing CHD is probably not necessary. When auscultation detects abnormal findings (i.e. systolic click, murmur of valve regurgitation), or if auscultation is limited by a large body size, then echocardiography is reasonable. It can be repeated at a prudent interval, e.g. every 5 years. In summary, current evidence does not prescribe a rigid schedule for routine echocardiography in adults with Down syndrome without known CHD, although it may be indicated in individual patients. A schedule of on-going cardiac surveillance in Down syndrome adults with CHD depends on the type of CHD and the potential for developing late complications.
Table 4 provides information on 48 non-institutionalized Down syndrome individuals aged 18 years or older followed at the National Birth Defects Center in Waltham, Massachusetts.74 Although this is a select non-population-based cohort, it provides new data reflecting individuals repaired within the last 37 years. Approximately 40% (19/48) of all adults had a CHD, which is similar to the occurrence in infants, and all but one had undergone surgical repair. The NYHA (New York Heart Association)18 functional cardiac status of those with a repaired CHD was judged to be Class I in 67% (12/18), Class II in 22% (4/18) and Class III in 11% (2/18). Among the 60% (29/48) of Down syndrome adults without a CHD only a small minority (3%) had a Class II cardiac status. Consistent with previous reports, mitral valve prolapse was common (12/48, 25%), but 75% of these adults had no prior CHD, suggesting that MVP is a late development. None of these Down syndrome adults had a murmur despite 42% (5/12) having some degree of regurgitation documented on echocardiogram. A click was heard by the cardiologist in only 33% (4/12) and never appreciated by a non-cardiologist. Among the three cases with mitral valve prolapse and a CHD, the nature of the CHD would not be expected to lead to mitral valve disease (i.e. two had spontaneous closure of a VSD and one had repaired double aortic arch.) Since over half of the adults in this series had no CHD, and two thirds of those with a repaired CHD were functioning as NHYA I, most of the adults with Down syndrome in this series, even those with a CHD, have no clinically or hemodynamically significant cardiac disease in adulthood.
Table 4.
Adults (>18 years) with Down syndrome followed at the National Birth Defects Center, Waltham, Massachusetts (n=48, ages 18–40 years, mean 27.8)
| Adult cardiac status (NYHA Classification) |
||||
|---|---|---|---|---|
| Total | Class I | Class II | Class III | |
| Type of CHD | ||||
| CHD repaired, total | (38%) | 12 | 4 | 2 |
| AV canal | 9 | 4 | 3 | 2 |
| Complete | 4 | 1 | 2 | 1 |
| ASD primum | 4 | 2 | 1 | 1 |
| VSD canal | 1 | 1 | 0 | 0 |
| ASD secundum | 1 | 1 | 0 | 0 |
| VSD membranous | 6 | 5 | 1 | 0 |
| Double Aortic Arch | 1 | 1 | 0 | 0 |
| PDA | 1 | 1 | 0 | 0 |
| CHD unrepaired, total | (2%) | 0 | 0 | 1 |
| AV canal | 1 | 0 | 0 | 1 |
| No CHD | (60%) | 27 | 1 | 1 |
| Total | (100%) | 39 | 5 | 4 |
ASD, atrial septal defect; AV canal, atrioventricular canal; CHD, congenital heart defect; NYHA, New York Heart Association classification of heart failure: Class I (No limitation of physical activity), Class II (mild) (Slight limitation of physical activity), Class III (moderate) (Marked limitation of physical activity); PDA, patent ductus arteriosus; VSD, ventricular septal defect
Down syndrome has been reported to be associated with a decrease in the frequency of atherosclerosis based on an autopsy series.75 Studies of lipid analyses have failed to detect any meaningful difference in cholesterol levels.76 There is also an intriguing observation that systemic hypertension is decreased. A decrease in PAI-1 has been noted, and a model for macroangiopathy protection has been hypothesized.77 The relative paucity of systemic hypertension contrasts to the increased risk of pulmonary vascular hypertension in childhood.63
Pregnancy-related cardiovascular issues
Pregnancy is rare in women with Down syndrome, with 31 pregnancies reported in 27 women.78–80 The cause of the reduced childbearing is thought to be due to social issues, but there are no reports which have studied the contribution of endocrine or fertility factors. Overall, the pregnancies are described as uneventful with rare maternal complications even with the presence of CHDs.78 In contrast, the labor and delivery were frequently complicated by fetal malpresentation, cephalopelvic disproportion and/or lack of maternal cooperation leading to interventions such as cesarean delivery, breech extraction or forceps delivery in nearly half of the deliveries.78 The greatest theoretical cardiovascular risk is for a woman with Down syndrome with an unrepaired complete atrioventricular canal or large VSD complicated by pulmonary hypertension since these defects could be associated with either a risk of cardiopulmonary failure due to the enormous changes in fluid balance associated with pregnancy, and to the possible risk of thromboembolism.
Turner syndrome
Turner syndrome refers to females with absence or structural abnormality of one of the two X chromosomes with the birth prevalence estimated as ~1/2000.81 A 30 year epidemiologic trend analysis to 2001 in Denmark observed no change in incidence, but a delay in diagnosis, i.e. approximately half of all Turner syndrome women were diagnosed after their fifteenth year. Mortality was increased compared with the background population of women, specifically for coronary artery disease, malformations, nutritional and metabolic disease.82 The heterogeneity of karyotypes results in a spectrum of phenotypes. 45, X occurs in almost half of Turner syndrome women; less frequently, there is an isochromosome X, short arm or long arm deletion, or ring chromosome (see reviews by Sybert and McCauley; Bondy et al.).83,15
The physical appearance of a Turner syndrome individual varies enormously from a massively hydropic spontaneously aborted fetus to that of a healthy woman with short stature and subtle dysmorphic facial features (Figure 4). Lymphatic malformations are common and contribute to neck webbing, protruding ears, loose nuchal skin, low hairline, puffy hands and feet, and deep-set nails.84,85 Often noted are renal anomalies, especially horseshoe kidney, nevi, widespaced nipples, short fourth metacarpals and metatarsals, cubitus valgus, Madelung deformity, knee anomalies, and osteoporosis. Chronic otitis media, conductive and sensorineural hearing loss, autoimmune diseases, type II diabetes, deficits in visual-spatial/perceptual abilities, attention deficit, and poor social skills are common (see reviews by Sybert and McCauley; Bondy et al.).83,15
Fig. 4.

22-year-old woman with Turner syndrome (45, X) who had a bicuspid aortic valve, mitral valve prolapse with mitral regurgitation and coarctation repair at age 2 ½ years, and balloon angioplasty and stent insertion at age 20 years. She received growth hormone for almost three years. She selected this photo to emphasize that the appearance of many adult women with Turner syndrome is not unusual, aside from short stature (height 5′ 1″) and bears little resemblance to the images portrayed in older medical textbooks (courtesy of Angela E. Lin, M.D. and family).
Cardiovascular abnormalities during childhood
The most dramatic hemodynamic alteration in Turner syndrome is severe hydrops and fetal demise.84 Throughout the lifespan, the most common CHDs are obstructive defects of the left heart which encompass bicuspid aortic valve (approximately 15%) with/without aortic stenosis, mitral valve anomalies (<5%), coarctation of the aorta (approximately 10%), and rarely, hypoplastic left heart syndrome.83,85–87 Additional CHDs include partial anomalous pulmonary venous connection (13%) and atrial septal defect or VSD (5% each). Magnetic resonance imaging has identified additional vascular anomalies, notably elongation of the transverse arch (almost 50%) which may be accompanied by pseudocoarctation.88,89 Recently, abnormalities on electrocardiogram, including prolongation of the QT interval, have been reported in girls with Turner syndrome, but the clinical implications are unclear aside from avoidance of certain drugs.90 Surgical treatment for a CHD is usually undertaken in an infant or a young girl with Turner syndrome, although milder cases of coarctation may escape detection in childhood, or may not become clinically significant until an older age. The main postoperative residua are recoarctation of the aorta, or re-stenosis of the aortic valve.
Cardiovascular abnormalities in adulthood
Some women with Turner syndrome enter adulthood with unsuspected or mild aortic root dilatation, bicuspid aortic valve, and even coarctation. As a result of childhood CHDs and possible corrective surgeries, they may have postoperative aortic valve re-stenosis, aortic re-coarctation or residual septal defect shunts. A new murmur can signify the appearance of mitral valve prolapse or aortic regurgitation. Since neck webbing has a strong correlation with the presence of coarctation and bicuspid aortic valve,85 an adult woman with Turner syndrome who has a characteristically severe appearance, but is not known to have a CHD, should be carefully re-evaluated to be certain that a bicuspid aortic valve and coarctation were not overlooked in childhood.
Patients with Turner syndrome and bicuspid aortic valve experience the same risks as non-Turner syndrome patients, i.e. risk of aortic dilation and dissection, progression to aortic stenosis and/or regurgitation.91 Natural history data for the rate and frequency of progression of bicuspid aortic valve to clinically significant aortic valve stenosis are not available, but are thought not to differ from the general population according to a recent consensus study group.15 Although individuals with bicuspid aortic valve, including women with Turner syndrome, had been advised to follow recommendations for subacute bacterial endocarditis, this policy has been changed recently.19
Aortic root dilatation, aneurysm, dissection and rupture in Turner syndrome is well-established.92–96 Early echocardiographic estimates of dilatation in ~10%92 have increased to ~30% using magnetic resonance angiography.95 In addition to aortic root disease, abnormalities of small bore arteries and the brachial and carotid vessels are sufficiently diffuse to suggest a vasculopathy.97 Most cases of dissection in Turner syndrome have been associated with an underlying risk factor, e.g. bicuspid aortic valve and/or coarctation (69%), or hypertension (54%) with or without a CHD. In an exhaustive literature review, the small number (11%) of reported cases with no associated risk factor were poorly documented,96 although a few may represent the intrinsic “aortopathy” of Turner syndrome. Despite the frequent association with bicuspid aortic valve, Turner syndrome itself is an independent risk factor for aortic dilation.98 Compared to the Danish Registry estimate of aortic dissection (78/100,000),99 a recent prospective study showed a much higher frequency of aortic dissection (618/100,000 Turner syndrome years).95 The authors proposed that prophylactic surgical aortic intervention for Turner syndrome should not follow the traditional Marfan syndrome guideline (50 mm), but use an actual dimension of 35 mm or body surface area-adjusted diameter ≥25 mm/m2. To monitor the occurrence of aortic dissection, the International Turner Syndrome Dissection Registry has been established in association with the Turner Syndrome Society of the United States.96
Whether growth hormone, which is used to treat many Turner syndrome girls for short stature, is a risk factor for cardiovascular disease will require ongoing clinical history studies. No reports have shown a clearcut deleterious impact. Several provide reassurance when left ventricular dimensions and/or aortic dimensions (among other measurements) have been analyzed.15,100–102
Half of Turner syndrome women have hypertension.92,103 Careful auscultation and lifelong blood pressure monitoring are essential, and for women who have had coarctation repair, the blood pressure cuff should be placed on the right arm. A variety of electrocardiographic and repolarization abnormalities have been reported which appear to expand the spectrum of cardiac involvement in Turner syndrome.104 Their clinical impact is not clear since these electrocardiographic abnormalities have not been correlated to clinical arrhythmias; prudence is advised when using drugs that have the potential to prolong the QTc interval. The lipid profile in women with Turner syndrome can be considered atherogenic since there is an elevation of low-density lipoprotein cholesterol and triglycerides, and a reduction in lipid particle size.105,106 Whether coronary artery disease is increased in Turner syndrome, independent of familial or nutritional factors, has not been determined. Two women younger than age 50 years have required coronary artery bypass grafting.91,107
The clinical phenotype of Turner syndrome may be mild and escape detection in childhood, so that undiagnosed women can present with reproductive endocrine symptoms such as menstrual “irregularity” or infertility. If the diagnosis is first established during adulthood, clinicians are reminded to obtain a comprehensive cardiology consultation with imaging of the aorta. Although this can be accomplished with high resolution echocardiography, recent guidelines encourage magnetic resonance imaging/angiography.15. The aorta should be imaged at 5–10 year intervals depending on aortic dimension, activity level, and other health issues, under the guidance of the primary care provider and/or cardiologist. Blood pressure should be monitored frequently and may require nocturnal monitoring to detect elevations which have been observed.103 Hypertension should be treated aggressively; recommendations for a specific antihypertensive medication based on results of treatment trials are not available, but beta-adrenergic receptor blockade is often selected if there is resting tachycardia, and because it is generally well-tolerated.15 The empiric use of beta-blockade for aortic dilatation in Turner syndrome has not been subjected to rigorous study. With the ongoing clinical trial using losartan to treat aortic disease in Marfan syndrome, there is optimism that the molecular insights might be applied to the study of aortic dissection in other syndromes, including Turner syndrome.95,108 Whether it is necessary to obtain periodic ECGs has not been determined.15,104
General lifestyle guidelines for older individuals with Turner syndrome have been modeled after the advice given to those with Marfan syndrome.15 These include an awareness that chest pain may have a significance which is not benign, avoidance of collision contact sports, promotion of regular moderate aerobic activity, discouraging highly competitive sports and very strenuous or isometric exercises. The latter are well-established risk factors for vulnerable aortas. Eligibility for competitive sports should be determined by a cardiologist after a comprehensive cardiac evaluation that includes recent magnetic resonance imaging of the aorta.15
Pregnancy-related cardiovascular issues
Spontaneous pregnancy is rare among Turner syndrome women, but with the availability of assisted reproductive techniques, pregnancy is increasingly pursued. Although pregnancy had not been reported to have serious cardiac risks, a recent survey of Turner syndrome women seeking assisted reproductive technology has alerted specialists about deaths associated with aortic dissection.95,109 Potential childbearing should be done in the context of promoting aortic “health”. Dissection is a rare event overall, but potentially lethal when it does occur. As in the non-gravid Turner syndrome woman, the risk of aortic dissection appears greater in women with bicuspid aortic valve, coarctation and hypertension. This risk may not be widely appreciated since reviews of pregnancy risks in non-syndromic women did not mention women with Turner syndrome as vulnerable patients.110–111 To identify Turner syndrome women with aortic dilatation who may have increased risk for dissection during the dramatic hemodynamic changes of pregnancy, high quality imaging of the aorta is advised using MRI to supplement echocardiography.15 A specific monitoring schedule is not available, but a prudent strategy would include consultation and imaging studies done before, during and after delivery with the support of a multidisciplinary team. In one series reporting pregnancy outcome of women with CHDs, a Turner syndrome woman status-post repair for coarctation of the aorta developed pre-eclampsia with hemolysis, elevated liver enzymes and low platelets (HELLP) syndrome.7
Williams-Beuren syndrome
Individuals with Williams-Beuren syndrome (WBS) display a unique pattern of physical, cognitive, and behavioral problems. The most characteristic non-cardiac findings include distinctive dysmorphic facial features (Figure 5), short stature, musculoskeletal abnormalities including spinal curvature and joint contractures, microdontia, and endocrine abnormalities such as hypercalcemia, hypothyroidism, and abnormal glucose metabolism.112–114 Other common medical problems are tremor, urinary frequency, and recurring abdominal pain variously related to gastroesophageal reflux, chronic constipation and diverticulitis (the result of infected colonic diverticuli). Intellectual disability is universal in WBS, and while the average IQ is 59 (mild mental retardation) there is a wide range which include individuals with low normal intelligence. Adults typically have better skills in socialization than in daily living.115 The WS behavioral phenotype is characterized by impulsivity, attention deficit disorder, overfriendliness, and anxiety that often includes specific phobias such as fear of medical procedures.116–117
Fig. 5.

Woman with Williams-Beurens syndrome at 5 and 20 and 1/2 years old illustrating the change in facial appearance. The adult has a prominenet supraorbital ridge and a narrow nasal root, but the facial asymmetry, full nasal tip, and wide mouth persist. She had repair of supravalvar aortic stenosis (hourglass type) at age 6 years (courtesy of Colleen A. Morris, M.D.)
As many as 1/10,000 individuals have WBS.118 It is caused by a micro-deletion of ~ 1.5 million DNA base pairs located on chromosome 7q [del (7)(q11.23]. This deletion is associated with loss of about two dozen genes, including the gene Elastin.119 It is loss of the elastin gene, resulting in decreased synthesis of the elastin protein, which causes the generalized arteriopathy spectrum of cardiovascular abnormalities seen in WBS. Other common problems due to elastin deficiency in WS include hoarse voice, inguinal hernia, bowel diverticuli, bladder diverticuli, and possibly some of the characteristic facial features. Deletion of additional genes, such as CYLN2, GTF2I, and GTF2IRD1, cause the remaining features of WBS though genotype-phenotype correlations are not well established. As with the other microdeletion syndromes already presented, i.e. chromosome 22q11.2 deletion, most deletions are de novo, but an affected individual has a 50% risk of transmitting the disorder to each of his/her offspring..113
Cardiovascular abnormalities during childhood
Supravalvar aortic stenosis is the most frequent cardiovascular abnormality, diagnosed in up to 75% of patients.120 Other vascular stenoses are also commonly observed, and any medium to large-sized artery may be narrowed due to medial hypertrophy. Narrowing is most commonly noted at arterial origins and can result in bruits, diminished peripheral pulses, hypertension (renal arteries), contribute to abdominal pain (mesenteric artery stenoses), or result in myocardial infarction (coronary arteries).121
Approximately half of infants with WBS have obstruction involving the pulmonary arterial tree (either peripheral, valvar or supravalvar pulmonary stenosis) that usually resolves within the first year of life without medical or surgical intervention, especially if mild. Severe supravalvar aortic stenosis leads to cardiac hypertrophy, heart failure and death if not treated surgically. Other potentially serious complications during childhood include idiopathic hypertension found in ~ 50%, diffuse aortic hypoplasia (10–20%), and renovascular stenosis (5–10%). Stroke and sudden death are rarely reported in children.121–125 Structural intracardiac defects, such as VSD and atrial septal defect, are reported in ~10%. Mitral valve prolapse also occurs in ~10% of children, although echocardiography found mitral valve anomalies in 41% (12/29) in one small series.126
Stenosis involving the ascending aorta, the descending aorta or its branches does not generally respond to balloon dilation, and, if clinically significant, requires surgical intervention. Branch pulmonary stenoses that do not resolve spontaneously or are sufficiently severe to cause right ventricular hypertrophy can be treated with balloon dilation and stenting, though complications such as aneurysm and rupture have been reported and the hemodynamic response can be modest.
Cardiovascular abnormalities during adulthood
The natural history of elastin arteriopathy requires that individuals with WBS receive regular and careful assessment of their cardiovascular system (Fig. 5). Vascular stenoses continue to be the most prevalent cardiovascular abnormalities among adults. Although the distribution and degree of cardiovascular involvement remains stable in the majority, stenosis can develop or progress to become clinically relevant in any artery. Across the lifespan, 25–30% of individuals with WBS will require surgical correction of their cardiovascular disease, most commonly in childhood.127–128 Surgery for restenosis of supravalvar aortic stenosis is more likely in individuals with concomitant descending aorta stenosis or diffuse aortic hypoplasia.127 Mitral valve prolapse can progress in adults, necessitating valve replacement.128,129 As in childhood, idiopathic hypertension is frequent and if new in onset, should prompt baseline noninvasive imaging to evaluate for a renovascular etiology. SVAS can result in elevated blood pressure readings on the right arm secondary to the Coanda effect;130 therefore the blood pressure should be measured bilaterally. Typically, the hypertension in WBS responds to standard antihypertensive therapy when it is not related to renal artery stenosis. No preferred anti-hypertensive treatment has been identified to date so that choice of medication is dictated by efficacy and tolerability. Stroke has been reported in adults with WBS, though it is fairly rare.131–132
Stenosis of the coronary ostia has been documented by angiography in children with WBS128 and can be caused by the typical medial hyperplasia, or obstruction secondary to aberrant dysplastic aortic valve leaflets. Coronary artery stenosis has been implicated as one of the causes of sudden death in both children and adults; the absolute risk of this potential complication appears low though the relative risk has been suggested to be 25–100 fold greater in individuals with WBS than the general population.133 Monitoring for coronary artery disease can be difficult as WBS adults fatigue easily and are often unable to perform standard stress exercise-based tests.134 Whether the adult with WBS is at especially high risk for coronary artery disease, given the potential for coronary artery involvement as part of the underlying elastin arteriopathy, plus development of age-related atherosclerosis, deserves further study.
Cases of sudden death in relation to exposure to anesthesia have also been reported, though it is unclear if increased risk is due to anesthesia itself or underlying cardiovascular disease. No systematic study has been performed in adults with WBS, but in a series of children undergoing procedures requiring anesthesia, the presence of pulmonary hypertension increased the relative risk of an adverse anesthesia-related event though the absolute risk was <2%.123 Careful studies in adults, including documentation of coronary artery patency, are required before risk estimates are available for adults.
Pregnancy-related cardiovascular issues
Pregnant women with WBS are high risk pregnancies, especially if they have pre-existing hemodynamically significant vascular stenoses. They should be monitored carefully for the development of pregnancy-induced hypertension, arrhythmias, or potentially even heart failure. Premature delivery and growth retardation of the fetus have been reported and since urinary tract infections may contribute to the former and are a known complication of WBS, regular urinalyses should be performed in late gestation. The number of carefully documented WBS pregnancy cases is small, but an increased frequency of non-WBS adverse outcomes is suggested. Thus, careful ultrasound and genetic monitoring of the fetus, above and beyond screening for inheritance of WBS, are indicated.135,136
MENDELIAN SYNDROMES
Marfan syndrome
The Marfan syndrome is a heritable disorder of connective tissue caused by mutations in the gene that encodes the large, extracellular matrix glycoprotein, fibrillin-1 (FBN1), on chromosome 15q21.137 People who are heterozygous for mutations in FBN1 usually have Marfan syndrome (more than 600 different mutations have been described), but they may also have one of the many conditions that phenotypically overlap with Marfan syndrome.137. However, Marfan syndrome remains a clinical diagnosis by using the “Ghent criteria” (or their modified form in preparation).138 Molecular analysis has a role, since the diagnostic criteria in an individual with borderline clinical features can be fulfilled when a FBN1 mutation is detected, or when linkage to a classically affected first degree relative is shown. Molecular testing is also useful when the differential diagnosis includes Loeys-Dietz syndrome and other disorders of TGF-beta receptors. For cases in which prenatal or presymptomatic testing of other family members is desired, molecular testing of the proband is necessary to determine if there is an identifiable FBN1 mutation. If molecular testing identifies a pathogenic FBN1 mutation, then prenatal testing, or presymptomatic diagnosis can be offered. The prevalence of Marfan syndrome is estimated at ~1 per 5,000 individuals, regardless of ethnicity. While intrafamilial variability is considerable, penetrance is very high, provided sufficient attention is directed to assessing the phenotype. Because Marfan syndrome is autosomal dominant, males and females are equally affected, and affected individuals have a 50% chance of having an affected child each time they conceive.
Multiple organ systems are affected to some degree in Marfan syndrome, in large part because fibrillin-1 is the major constituent of the extracellular microfibril found in most connective tissues. Organs with the greatest importance as diagnostic criteria and as medical problems are the musculoskeletal, ocular, cardiovascular, and pulmonary systems, and the abdominal viscera.138,139
Understanding the pathogenesis of Marfan syndrome has improved dramatically in the past few years. Rather than being due to “weak” connective tissue, most of the phenotypic features are now clearly caused by over expression of transforming growth factor-β (TGFβ). Mutant fibrillin has impaired ability to keep the latent TGFβ complex in the inactive form.140–142
Cardiovascular abnormalities during childhood
Most people with Marfan syndrome have cardiovascular involvement.137,143 The feature most likely to cause clinical problems in childhood is mitral regurgitation due to severe prolapse.144 Some infants with severe Marfan syndrome have congestive heart failure due to mitral regurgitation; the ventricular dysfunction may be out of proportion to the magnitude of the leakage, suggesting a primary cardiomyopathy. Mitral valve repair may be necessary in childhood. While the aortic root may be dilated in childhood, severe aortic regurgitation is uncommon. Likewise, aortic dissection is uncommon before adolescence. Indeed, dissection of the aorta or one of its branches in childhood should prompt consideration of an alternative diagnosis, such as Loeys-Dietz syndrome.145
In Marfan syndrome, the aortic root may be dilated at birth or exceed the upper limit of normal diameter later in childhood or adolescence.146 The rate of change of the diameter depends on both growth (which is predictable, based on body surface area)147 and pathologic dilatation. Treatment with beta-adrenergic blockade, at a dose sufficient to produce both a negative chronotropic and inotropic effect, reduces the rate of dilatation and reduces the risk of dissection.148,149
Cardiovascular abnormalities in adulthood
The best study of the natural history of Marfan syndrome showed that life expectancy is reduced, on average, by one-third.147 However, death can occur at any age. In the time before modern diagnostic and therapeutic advances, over 90 percent of deaths in Marfan syndrome were due to a cardiovascular complication.
Progressive dilatation of the main pulmonary artery rarely leads to clinically important pulmonic regurgitation and almost never leads to dissection.137 In contrast, progressive dilatation of the aortic sinuses of Valsalva leads to stretching of the commissures at the sinotubular junction and central aortic regurgitation; the larger the root, the greater the leakage. The relationship between the risk of a type A dissection (arising from the ascending aorta) and the aortic root diameter in Marfan syndrome is less predictable than the relationship with type B dissection (distal to the left subclavian). Clearly, the larger the diameter, the greater the risk.151 However, some patients suffer dissection at a modest degree of root dilatation (45–55 mm). This risk tends to track in families, which leads to the recommendation to perform prophylactic aortic surgery at a smaller diameter than usual when a relative with Marfan syndrome has suffered a type A dissection.
The surgical management of the aorta in Marfan syndrome has evolved tremendously over the past 30 years, beginning with the widespread adoption of the composite graft technique in the mid-1970’s.151 The implantation of an artificial valve in the end of a woven conduit, with reimplantation of the coronary arteries, remains the gold standard today. The combination of beta-adrenergic blockade, restriction from strenuous physical activity, replacement or repair of a severely leaking mitral valve, and prophylactic aortic repair has markedly improved life-expectancy in Marfan syndrome.152 By the mid-1990’s, average survival was approaching 70 years, and there has undoubtedly been further improvement over the past decade.
While the composite graft repair of the aortic root remains the standard, and is usually necessary in the face of an acute type A dissection, several techniques for repairing the aneurysm that preserve the native aortic valve have emerged. The preferred method is the ‘reimplantation’ technique.153,154 Valve-sparing can only be done when the leaflets are not stretched markedly or fenestrated. This necessitates performing the repair before the sinotubular junction dilates, so the indication for surgery has evolved to a maximal dimension of approximately 45–50 mm (at any location) in an adult. Because long-term results of valve-sparing operations are not yet available, the National Marfan Foundation has established a registry that is accumulating experience (www.marfan.org).
The adult with Marfan syndrome needs to be managed aggressively and regularly from the cardiovascular perspective. For a patient who has had prophylactic aortic root repair, imaging of the entire aorta by CT or magnetic resonance imaging should be performed every one to two years. A patient who has a dissection with a residual false lumen may require more frequent imaging. Indications for repair of a chronic dissection include extension, decreased blood flow to a branch artery, or progressive expansion of a dissected aneurysm. At the present time, endolumenal repair with a stent is not recommended. Adults should also have periodic echocardiography to follow the function of valves and myocardium. Chronic beta-blockade, in addition to whatever medications are needed to keep blood pressure well within the normal range, should be used. The angiotensin receptor-blocker, losartan, has the added effect of interfering with TGFβ activity. Treatment with losartan of mice with Marfan syndrome prevents (if the drug is started at birth) or reverses aortic wall pathology and dilatation.142 A human clinical trial comparing losartan with atenolol is now underway.155
The natural history of the arterial tree in adults with Marfan syndrome remains to be defined. Unanswered is whether older adults will be more prone to the inflammatory aneurysms that typically occur in the abdominal aorta in the general population. Certainly this risk can be reduced by controlling blood pressure, avoiding smoking, and monitoring other risk factors for atherosclerosis. Although the focus is typically the ascending aorta, Type B dissections represent about ten percent of all dissections in Marfan syndrome, and usually occur in the undilated postductal region. Ongoing research is needed to define the frequency of branch artery dilatation, aneurysm or dissection in Marfan syndrome. Currently, their presence in an individual should prompt review of the Marfan syndrome diagnosis, with consideration instead of one of the arteriopathies associated with mutations in TGFBR1 or TGFBR2.145,156
Pregnancy-related cardiovascular issues
Many case reports document that women with Marfan syndrome can suffer aortic dissection during the third trimester of pregnancy, during parturition, and during the six months post-partum. However, type A dissection usually occurs in the setting of pre-existing dilatation of the aortic root.137 The relative risk of dissection in Marfan syndrome women who have an aortic root diameter of < 40 mm is greater than in women who do not have Marfan syndrome, but fortunately the absolute risk of dissection is low so that pregnancy and delivery are usually uncomplicated from the cardiovascular perspective.157. Type B dissection can occur when the aortic root is not dilated. Women who have a maximal aortic root diameter > 40 mm should avoid pregnancy, or have a valve-sparing aortic root repair (if feasible) performed first as a mechanical aortic valve requires life-long anticoagulation with warfarin, which carries a risk of teratogenesis. In some women with Marfan syndrome, pregnancy will be complicated by mitral regurgitation or chronic dysrhythmia. Protection of the aortic root with a beta-adrenergic blocking drug, especially in the later stages of pregnancy and post-partum, is important. All pregnancies should be considered ‘high-risk’ and monitored appropriately. Delivery should be by whatever approach results in the least cardiovascular stress on the aorta. Because of rapid shifts in blood volume, cesarean section should be employed only when necessary for obstetric reasons; the route of delivery can be decided by the obstetrician and anesthesiologist as delivery approaches.
Disorders of the TGF-β Receptors (Loeys-Dietz syndrome and others)
Patients with a wide variety of phenotypes, but sharing a propensity to dilatation and dissection of larger arteries at any age, may have mutations in either of two genes encoding cell-surface receptors for TGF-β (TGFBR1 and TGFBR2). The most striking phenotype has been termed Loeys-Dietz syndrome (LDS).145 Patients have craniofacial abnormalities including cleft palate, bifid uvula and craniosynostosis, aneurysms of the aorta or its branches, arterial tortuosity, arachnodactyly, and dural ectasia. Importantly, they do not have ectopia lentis and typically do not have tall stature. At the other end of the spectrum are families that are prone to aneurysms, but have few other features.158 In between are patients who show partial overlap with the vascular form of Ehlers-Danlos syndrome and with Marfan syndrome, but have insufficient features to meet the Ghent criteria.159–161
Cardiovascular abnormalities during childhood
Anyone with a mutation in TGFBR1 or TGFBR2 should be evaluated and managed based on a propensity for arterial dilatation and dissection at an early age. Aortic dilation has been identified in fetal life in Loeys-Dietz syndrome.162 Echocardiography alone is insufficient to diagnose and follow arterial pathology, and thus, MRI/MRA should be used as needed.163
Cardiovascular abnormalities during adulthood
Any patient previously diagnosed with Marfan syndrome who has had particularly aggressive aneurysms in multiple sites, with or without dissection, should be considered for molecular testing and a revised diagnosis, especially if ectopia lentis is absent.164 Surgery of blood vessels does not appear to be complicated by the vascular fragility seen in vascular Ehlers-Danlos syndrome. As a result, prophylactic aortic root repair, and repair of other aneurysms, can be undertaken with confidence.165 However, the criteria used for recommending prophylactic repair in Marfan syndrome should not be used; rather, repair should be undertaken at a smaller diameter (40 mm, instead of 50 mm, in the adult).156 Whether medical therapy with β-adrenergic blocking drugs or angiotensin receptor blocking drugs will delay dilatation or prevent dissection has not been tested. In the absence of controlled trials, the use of either or both classes of medications seems warranted.
Pregnancy-related cardiovascular issues
Although prospective studies of pregnancy outcome have not yet been done, the cardiac risk for severe pregnancy complications appears to be greater for Loeys-Dietz syndrome than for Marfan syndrome, and also includes uterine rupture and bleeding. In the initial description of the disorder, half of the 12 women had either aortic dissection, uterine rupture or uterine hemorrhage during pregnancy or in the postpartum period.145 Women who carry the gene for a TGF-B receptor disorder should be advised of the pregnancy risk, and followed using the clinical care model for Marfan syndrome. As outlined above, this includes imaging the aorta preconceptually when possible, during pregnancy and after delivery.
Hereditary Hemorrhagic Telangiectasia (HHT)
This autosomal dominant disorder is due to defects in vascular development that result in abnormal connections between the arterial and venous circulations.162 Absence of capillaries between small arterioles and venules results in punctate telangiectases anywhere in the body, but typically on mucosal surfaces (nose, lips, gut) and skin (digits, face). In certain organs, arteriovenous malformations (AVM), or abnormal connections between larger arteries and veins, develop. Mutations in three disease genes (ENG, ALK1, SMAD4) have been identified to date for which clinical molecular testing is available.167,168 Studies point to at least two additional disease genes that have yet to be defined. Therefore, HHT is genetically heterogeneous. A correlation between genotype and phenotype is not well established at this time.
Cardiovascular abnormalities during childhood
All of the vascular abnormalities worsen with time. For example, the mucosal and skin telangiectases may not be present or detectable in children, whereas they will be apparent in all adults.169 The major concern is the development and/or progression of cerebral, spinal or pulmonary AVM. If a parent is affected, a child has a 50% risk of inheriting HHT. The initial evaluation of any patient with suspected HHT should include a complete physical examination and detailed family history. Contrast echocardiography should be done to look for late passage of contrast into the left side of the heart, which suggests a shunt at the level of the lungs. In addition, contrast magnetic resonance imaging of the brain should be done to identify occult cerebral abscess and large vascular malformations. If a person is known to have a pulmonary shunt, then high-resolution CT scanning with contrast should be performed to identify the number, size and location of AVMs. This scan can include the liver if evidence of hepatic AVMs is important to establish the diagnosis, or if the patient has a bruit over the right upper quadrant. Molecular testing of ENG, ALK1 and SMAD4 will detect a mutation in about 75–80% of patients who satisfy clinical criteria for HHT. Establishing the mutation is especially useful for screening relatives.
If a child has HHT, or continues to be suspected of having it, and the contrast echocardiogram is negative, it should be repeated in 5 years. If the CT scan does not reveal a pulmonary AVM, it should also be repeated in 5 years. Of note, the rate and timing of growth of pulmonary AVMs in children is not well documented. These recommendations serve as current guidelines for initial evaluation, but may need to be revised as more data become available.
Occasionally, a child with HHT will have one or more pulmonary AVMs that cause hypoxemia, pulmonary hypertension, or high-output cardiac failure. A pulmonary AVM can also be a conduit for a paradoxical embolism of either clot or bacteria to the systemic circulation. In all of these circumstances, occlusion of the pulmonary AVM(s) is an urgent matter. Most can be closed by an interventional radiologist using metal coils. Any pulmonary AVM with a feeding artery greater than 1 mm diameter should be occluded, if it can be reached by a catheter.170
Cardiovascular abnormalities during adulthood
Typically, pulmonary AVMs develop during adolescence and young adulthood. When one or more pulmonary AVMs are large, clinically important dyspnea and cyanosis can be the symptom and sign at the bedside. Unfortunately, a sudden cerebral event, such as an embolic stroke, due to paradoxical embolization can be the first indication of a pulmonary AVM. Thus, screening with a contrast echocardiogram is essential in any person suspected of having HHT. The interpretation, limitations, management, and follow-up are as described in the previous section.
Many patients with HHT develop intrahepatic vascular malformations. Fortunately, few of these lesions become symptomatic,171 though depending upon the nature of the aberrant connections, some patients develop hepatic encephalopathy from reduced effective portal flow through the liver, while others develop high-output cardiac failure.172 Embolization of hepatic vascular malformations is rarely an option because of the risk of fulminant hepatic infarction. If the consequences of liver involvement cannot be managed medically, transplantation is an option.173
In mid- to late-adulthood, gastrointestinal hemorrhage becomes a more prevalent and important problem in HHT. Intestinal bleeding from mucosal telangiectases or larger arteriovenous malformations can occur at any point, from the mouth to the anus, although lesions are most common in the stomach, duodenum and jejunum.174 If traditional upper and lower endoscopy cannot identify an important lesion, push-enteroscopy or capsule endoscopy can be attempted. Management by means of cautery can be effective, but is usually temporizing. Occasionally, when the transfusion requirement is exceptional, anti-fibrinolytics are necessary, but not always effective.
Pregnancy-related cardiovascular issues
Complications of pulmonary AVMs during pregnancy in women with HHT have been reported. Women may develop pulmonary hemorrhage, cardiac failure or profound dyspnea and cyanosis from increased shunting, or stroke. Any of these complications can pose a major risk to the fetus, but even more importantly constitute an emergency to the mother. With proper shielding of the gravid uterus, radiographic screening can be performed in a symptomatic pregnant woman, and any pulmonary AVM with a feeding artery > 1mm can be embolized. For the pregnant woman newly diagnosed with HHT and an unknown pulmonary circulation status, screening for pulmonary AVMs can be done by contrast echocardiography with minimal risk to the fetus.175
Holt-Oram syndrome
Holt-Oram syndrome (HOS) is the most common of the “heart-hand syndromes,” in which a CHD is associated with an upper limb deficiency. It is caused by mutations in the TBX5 gene on chromosome 12q24.1. Skeletal defects of the upper limb involve radial ray structures and may include hypoplasia or absence of the radius, absent thumb or triphalangeal fingerlike thumb, as well as anomalies of the scaphoid bone, humerus and clavicle. The most common CHD found in HOS is secundum atrial septal defect. Holt-Oram syndrome is not associated with dysmorphic facial features, extensive visceral malformations, lower limb anomalies or cognitive deficiencies.176 Most (~85%) cases of HOS are the result of new mutations in the TBX5 gene.177,178 Although classic HOS is rare, the adult cardiologist, and even the general internist or family practitioner, may encounter patients with an atrial septal defect whose family history indicates cardiac and/or skeletal features that may be part of the phenotypic spectrum of HOS. Establishing the diagnosis of HOS is important both because of the implications of this diagnosis on cardiac management and the potential for inheritance in at risk family members. The clinician can rely on a combination of clinical diagnostic criteria and molecular testing in assessing those patients where HOS is a potential diagnosis.
Cardiovascular abnormalities during childhood
Cardiovascular involvement including CHDs and/or conduction defects occurs in approximately 75% of individuals with Holt-Oram syndrome. The most common CHDs are ostium secundum atrial septal defect and VSDs, usually involving the muscular trabeculated septum, which may also be associated with cardiac chamber isomerism. Heterotaxy has been observed, i.e., a female with an atrial septal defect, left atrial isomerism, inferior vena cava interruption and bilateral superior vena cava,179 and “mirror image” Eustachian valve.180 Complex CHDs have been reported, including partial anomalous pulmonary venous return, but conotruncal malformations are uncommon in this syndrome.181–183
The vast majority of CHDs associated with Holt-Oram syndrome can be diagnosed by echocardiography, and in some instances, their detection on prenatal ultrasound combined with a radial deficiency skeletal defect suggests the diagnosis of HOS.184,185 Surgical repair of the CHDs can take place in infancy or later in childhood depending on the clinical significance, and as long as pulmonary hypertension or ventricular failure have not developed. Transcatheter atrial septal defect repair may be an option in some cases. However, many small secundum-type atrial septal defects, and membranous or muscular VSDs have a natural history similar to the non-HOS defects, and close spontaneously.186,187
Sinus bradycardia and first-degree atrioventricular block (i.e., PR interval prolongation) have been observed in affected neonates. Therefore ECG evaluation is warranted. At least an annual ECG should be performed in all individuals to capture those with new onset conduction disease. Since AV block is progressive and can evolve to be high grade associated with syncope, annual 24-hour Holter monitoring in those with known conduction disease should accompany the annual ECG.181–183 The ECG is usually not normal in HOS patients with a CHD, and the progression from first degree to more advanced heart block is highly variable.
Cardiovascular abnormalities during adulthood
Surgical repair of CHDs in adults with Holt-Oram syndrome needs to be assessed on a case-by-case basis. Repair of an atrial septal defect in an adult with HOS is determined using the same criteria as in a non-syndromic individual..3,186 Because of the phenotypic variability of Holt-Oram syndrome, it is conceivable that an adult with the disorder could escape diagnosis until the birth of a more severely affected child or other relative.181–183. A thorough cardiac assessment should be undertaken in any newly diagnosed individuals to assess for the presence of silent cardiac disease.
Adults with Holt-Oram syndrome need to be continually followed for the development of conduction disease, regardless of a known history of CHD or conduction disease.181–183. There is great intra- and interfamilial variability in progression of conduction block. Those individuals who progress to severe heart block may require a pacemaker. If complete heart block develops, it may be accompanied by atrial fibrillation, therefore consideration should be given to the potential need for cardioversion, antiarrhythmic drug therapy, and/or anticoagulation.
Serial imaging and assessment of cardiac function is recommended in those with CHDs. Individuals with unrepaired large atrial and/or ventricular septal defects are at risk to develop Eisenmenger syndrome, similar to non-syndromic individuals in the general population. Medication for pulmonary hypertension may be warranted in these individuals.
Pregnancy-related cardiovascular issues
Decreased fertility in affected males and females has not been reported in Holt-Oram syndrome. Published data on pregnancy outcome in affected females are essentially nonexistent, however in the experience of the Cardiovascular Genetics Center at Cornell University, numerous women with HOS have successfully carried pregnancies to term without complications.189 Report of a pregnant woman with classic Holt-Oram syndrome who required a pacemaker to treat bradycardic junctional escape rhythm highlights the point that repair of a CHD (in this case, an atrial septal defect) does not prevent progression of conduction abnormalities.190
Ultrasonographic prenatal diagnoses of HOS have been reported. However, the variable expressivity of HOS limits diagnostic specificity in fetuses with mild upper limb anomalies and small atrial septal defects and VSDs at 50% risk, and virtually precludes the ability to make a prenatal diagnosis of HOS in sporadic cases with mild upper limb involvement.181,182
Noonan syndrome
Noonan syndrome is a multi-organ disorder with autosomal dominant inheritance. Mutations in genes of the RAS-ERK signaling pathway, PTPN11, KRAS, SOS1 and most recently RAF1 have been found in approximately 50%,191–193 <5%,194–196 10–15%,197–198 and 3–5%199–200 of clinically diagnosed individuals with Noonan syndrome, respectively. Because of the existing genetic heterogeneity, and continued high percentage of mutation negative individuals, the discovery of additional genes in the future is likely. PTPN11 and RAF1 mutations also cause LEOPARD syndrome, which resembles Noonan syndrome but has lentigines and deafness.199,201,202 Noonan syndrome features include characteristic facial anomalies (hypertelorism, ptosis, bright irides, low-set ears), neck webbing due to a prominent trapezius muscle, chest deformity (pectus carinatum superior, excavatum inferior), short stature, undescended testes in the male, nevi, keloids and cardiac abnormalities.203–207 The face changes with time (Fig. 6).206 There can be lymphatic abnormalities, and a variety of bleeding problems including FactorXI deficiency, Von Willebrand disorder, platelet function defects and thrombocytopenia.203 Although mental retardation is not common in Noonan syndrome, learning disabilities are frequent.203,204
Fig. 6.

45-year-old man with classic Noonan syndrome facial appearance had surgical repair of pulmonary valvular stenosis at age 5 years (courtesy of Mary Ella Pierpont M.D. Ph.D. and patient). As an adult, his facial appearance is that of an inverted triangle with a high prominent forehead, small pointed twin, and downslanted palpebral fissures.
Cardiovascular abnormalities during childhood
The majority (>80%) of Noonan syndrome patients have some type of cardiac abnormality, typically pulmonary valve stenosis in infants and children. Other common CHDs include secundum-type atrial septal defect, branch pulmonary stenosis, VSD, tetralogy of Fallot and partial atrioventricular canal. Less common are aortic stenosis, subaortic stenosis, patent ductus arteriosus and coarctation.203,207–209. The presence of pulmonary stenosis with an atrial septal defect is most commonly associated with PTPN11 mutations.210 The characteristic electrocardiogram in Noonan syndrome shows left axis deviation or northwest axis and a dominant S wave over the precordial leads, even with minor or no CHD.
Some children with pulmonary valvar stenosis have dysplastic leaflets resulting in severe right ventricular outflow tract obstruction. Balloon valvuloplasty may be successful in reducing the gradient in mildly dysplastic pulmonary valves, but surgical treatment (either valvotomy or valve leaflet excisions) may still be necessary. However, pulmonary valvar stenosis is usually mild to moderate, and requires only periodic reevaluation in most individuals with Noonan syndrome. Other CHDs such as atrial septal defect, branch pulmonary stenosis, VSD or left ventricular outflow defects are treated by surgery if indicated.
Hypertrophic cardiomyopathy occurs in 20–30% of all children with Noonan syndrome, but in <10% of those with PTPN11 mutations. The recent discovery of RAF1 mutations as a cause of Noonan syndrome has provided new information about the causation of hypertrophic cardiomyopathy in this syndrome.199 Mutations in two hotspots of the RAF1 gene were highly correlated with the presence of hypertrophic cardiomyopathy. Individuals with LEOPARD syndrome also have a higher incidence of hypertrophic cardiomyopathy (>50%). The clinical course of hypertrophic cardiomyopathy is variable since it may develop in later childhood and gradually increase in severity, remain stable over many years, or be rapidly progressive in infancy.203,211 Histopathologic studies demonstrate myocyte fiber disarray resembling non-syndromic familial hypertrophic cardiomyopathy. Treatment is similar to that in familial hypertrophic cardiomyopathy including the use of beta blockers or surgery to reduce outflow tract obstruction. However, unlike classic familial hypertrophic cardiomyopathy, the right ventricle is often affected and hypertrophic cardiomyopathy may be accompanied by other cardiac defects especially pulmonary stenosis.
Cardiovascular Abnormalities in Adulthood
The phenotype of Noonan syndrome changes significantly over time. By adulthood, many individuals with milder features may blend into the normal population. It is not uncommon for a previously unrecognized mother with Noonan syndrome to be diagnosed after giving birth to an affected child. Even for those diagnosed in childhood, there is a scarcity of literature describing the cardiac status of adults with Noonan syndrome. In a recent review of 56 adults with Noonan syndrome, and cardiac outcome documented in 45, 93% of whom had a cardiac abnormality.204 About half had pulmonary valvar stenosis and had undergone surgery or balloon valvuloplasty. Some adults had not had cardiac re-evaluation in years. Potential problems for adults with pulmonary stenosis include severe pulmonary insufficiency, exercise intolerance, and right ventricular dysfunction. Appropriate follow-up is essential and may include pulmonary valve replacement. Three of the 45 adult patients had died of cardiac complications at 39, 48 and 49 years, respectively.204. An additional two had been told they would need a heart transplant, one other had a defibrillator, and two had pacemakers. 31% had ongoing cardiac problems. A number of adult patients demonstrate tachyarrhythmias such as atrial fibrillation (personal patients, J.N.). Several adults have developed mild aortic insufficiency or mild dilatation of the aorta. One of the original nine patients described by Noonan and Ehmke developed aortic obstruction at age 38 years including valvar, subvalvar and supravalvar aortic stenosis.208 He had previously undergone a pulmonary valvotomy at age 6 years for severe pulmonary valvar stenosis.
A recent natural history study of 107 Noonan syndrome patients (mean age 25.3 years) found pulmonary stenosis (65%) and hypertrophic cardiomyopathy (19%) as the most common cardiac abnormalities.213 Three of those with hypertrophic cardiomyopathy died from progressive heart failure. No cases of sudden death were documented. One patient underwent cardiac transplantation and one had surgical myomectomy. Individuals with hypertrophic cardiomyopathy have usually been given beta-blocker therapy or amiodarone. Studies assessing efficacy of beta-blockade have been infrequent and conflicting.214,215
Other adults have presented with marked right ventricular outflow tract obstruction due to subpulmonary or pulmonary valve stenosis.216–218 Balloon valvuloplasty has been successful in some adults leading to reduction in right ventricular pressure and alleviation of cardiac symptoms.216–218 One adult had balloon valvuloplasty as well as closure of a secundum atrial septal defect by a septal occluder device with resolution of symptoms.217
Some adults with Noonan syndrome have had cardiac abnormalities other than structural CHDs or hypertrophic cardiomyopathy. Aortic root dilation, aortic dissection, and giant aneurysms of the sinuses of Valsalva have been described in patients who were clinically diagnosed.219–221 and confirmed by PTPN11 mutation analysis.222 The giant aneurysms in two male patients caused compression of the right or left coronary artery ostia.220,222 Surgical composite graft replacement was performed to remove the giant aneurysms and histological evaluation revealed deficiency of medial elastin and non-specific myxoid degeneration. These findings show that some individuals with Noonan syndrome have a connective tissue disorder-like change in their aortas in adulthood, which lead to the suggestion that all adults with Noonan syndrome have periodic lifelong cardiac re-evaluation.
Hypertrophic cardiomyopathy is the most common type of cardiomyopathy seen in adults with Noonan syndrome. Hypertrophic cardiomyopathy can progress in the adult with Noonan syndrome to right ventricular outflow tract obstruction due to septal hypertrophy, can be associated with severe pulmonary valve stenosis, or can evolve into a dilated form of cardiomyopathy.223–226 Restrictive cardiomyopathy and constrictive pericarditis have been rarely reported.226–228. Idiopathic pulmonary hypertension has been reported in 2 adults and one female teenager with Noonan syndrome.229–231 In the teenager, progression of the hypertension was relentless and she died awaiting heart lung transplantation.231
Pregnancy-related cardiovascular issues
Although pregnancy is successful in the majority of women with Noonan syndrome, the occurrence of polyhydramnios is relatively high and expert prenatal care is often needed. Women with mild pulmonary stenosis and no other cardiac abnormalities can have normal pregnancies. If unsuspected cardiac disease such as coarctation of the aorta is found, maternal hypertension and/or heart failure symptoms must be managed.232 Hypertrophic cardiomyopathy in the mother does not preclude successful pregnancy.233
Hydrops fetalis is uncommon and despite the fact that it signifies a serious problem in the fetus, successful subsequent pregnancy has been reported.234 With the increasing use of fetal ultrasound, a transient cystic hygroma may be noted as common feature in the fetus with Noonan syndrome. Presumably, when a nuchal hygroma is found in a pregnancy involving a mother or father with Noonan syndrome, it is likely, though not a certainty, that the fetus has inherited Noonan syndrome, since nuchal hygroma can be associated with many other conditions. Management of fetal hydrops is problematic, with early delivery a consideration for survival of the fetus. Fetal echocardiography should be considered for identification of fetal heart disease in the offspring of men or women with Noonan syndrome. If severe disease such as critical pulmonary stenosis or severe hypertrophic cardiomyopathy is found, appropriate plans can be made for care of the cardiac condition of the infant after birth.
Another maternal issue to consider with pregnancy, labor and delivery is maternal clotting and bleeding disorders, frequently found in Noonan syndrome. If one of these is present, this can have a significant impact on choices of anesthesia and post-partum care.235 Epidural anesthesia may not be the most appropriate choice due to the risk of epidural hematoma in females with platelet function abnormalities or factor deficiencies.
REPRODUCTION AND GENETIC COUNSELING
When a young adult with a syndrome and a cardiovascular abnormality is physically and emotionally mature, the primary caregiver or geneticist should begin the discussion about reproductive options. Apart from recurrence risk which apply to both men and women, several clinical issues relate to the impact of pregnancy on a mother’s reproductive health and cardiovascular system. Pregnancy outcome data for Turner syndrome and Marfan syndrome are emerging, but prospective studies are lacking for most conditions.
Menarche in a female with a CHD tends to occur at a slightly older age compared to population controls.236 Contraception counseling should be offered under the supervision of the gynecologist and cardiologist. Individualized guidelines will be based on the underlying CHD, surgical interventions, and post-operative residua.237 In general, women with residual shunts and/or obstruction, prosthetic valves, or pulmonary and/or systemic hypertension are advised against taking combined oral contraceptives (agents with low dose estrogen combined with varying doses of progestin) because of an increased risk for thromboembolitic events and fluid retention.238 For those approaching menopause, special consideration to the risks and benefits of hormone replacement therapy should also be addressed.
Since pregnancy data are not available for many genetic syndromes, it may be useful to extrapolate from two large studies of general population (e.g., non-syndromic) pregnant women with a cardiovascular abnormality. A prospective multicenter study of broadly defined “heart disease”,6 and a retrospective single center study of CHDs7 noted maternal cardiac and neonatal complications in the range of 13–19%. Siu et al. included 10 mothers with Marfan syndrome in their review of 599 pregnancies, but 22% of the total series had acquired heart disease. The more relevant study is that of Khairy et al. who excluded Marfan syndrome, and identified one woman with Turner syndrome in their cohort of 90 pregnancies born to 53 women with a CHD (discussed previously in the section on Turner syndrome). The identification of CHDs in 7% of the live offspring in this series was significantly greater than the general population but similar to other studies of children born to women with CHDs.7 Adverse maternal complications included pulmonary edema and arrhythmia. Risk factors included prior history of heart failure, NYHA functional class ≥2, decreased subpulmonary ventricular function and/or pulmonary regurgitation, and smoking.7
Whether an individual with a syndrome and cardiovascular abnormality is able to become pregnant depends on the underlying condition and the impact of the heart disease. Genetic counseling should emphasize the risk of recurrence, potential variable expression in affected offspring, and effects of pregnancy on maternal and fetal health.239 Like adults with a nonsyndromic CHD, they may require psychosocial support to deal with their own disease-related concerns as well as those of their potential offspring.240 The use of prenatal folic acid which is advised for all pregnancies to prevent neural tube defects has not been studied in women with syndromes and a CHD; there is no a priori reason to assume it is contraindicated but there are also no data that demonstrate its efficacy.
Recurrence risk counseling depends on the underlying genetic etiology. Theoretically, an individual with a chromosomal disorder such as Turner syndrome or Down syndrome has an increased risk to have a child with an aneuploid condition as well. However, it is extremely difficult to estimate the exact recurrence risk because other factors contribute to fertility and pregnancy loss. Because most individuals with Turner syndrome are infertile, childbearing may occur with assisted reproductive technology using a donor egg. Some females with partial monosomy X or low-level mosaicism for 45, X may be able to conceive naturally.15,241 One series indicated that the risk for an abnormal pregnancy outcome in women with nonmosaic Turner syndrome could be as high as 50%, when taking into account miscarriage and stillbirth.242 An earlier study had reported both on both fetal wastage (22/46), as well as chromosome abnormalities among liveborns (8/26), including 3 with trisomy 21.243 Amniocentesis should be offered to women with Turner syndrome who conceive. Since the vast majority of individuals with Down syndrome do not have children of their own, this discussion is mostly theoretical.
The individual with a syndrome and cardiovascular abnormality and their partner should meet with a geneticist or genetic counselor, an experienced obstetrician/gynecologist, and, as needed, an infertility specialist, e.g. woman with Turner syndrome, man with Klinefelter syndrome. Establishing a genetic diagnosis in the individual is as important as defining the cardiovascular abnormality.239 In addition to a high quality clinical genetic evaluation, genetic testing may be needed to confirm the specific syndrome and it should be performed in a CLIA-approved clinical laboratory prior to conception whenever possible. If a gene mutation or chromosome abnormality cannot be identified in the affected individual, the individual may be informed that ongoing follow-up may provide the opportunity for genetic confirmation of the diagnosis in the future. In the meantime, specific prenatal diagnosis of the condition would not be possible. Genetic counseling should also include a discussion of possible maternal risk factors (reviewed by Botto et al.)14 including maternal diseases such as diabetes, medications which are known teratogens, and exposures which are evolving risks. The most notable example is warfarin, a pregnancy category X medication, which may be given to someone with a mechanical valve or atrial fibrillation. Individuals who are treated with known teratogens must have a thorough discussion with their obstetrician and cardiologist whether there are appropriate pharmaceutical alternatives that are nonteratogenic and that can achieve the required effect during pregnancy.
Ideally the discussion between care providers and patients about reproduction and genetics, especially medication use, should occur preconception, given that teratogens can act early in human development. Likewise, prenatal testing options, as well as their risks, benefits and limitations should be discussed prior to conception or early in pregnancy with all at-risk couples. Non-invasive screening (i.e. high-level ultrasound, fetal echocardiography and first or second trimester maternal serum screening) can be routinely offered to all couples at risk for syndromic CHD regardless of the ability to offer genetic testing for the specific syndrome.244–247 Diagnostic prenatal testing (i.e. chorionic villi sampling or amniocentesis) can be offered to those couples where prenatal genetic testing for the known syndrome is available. Screening based advanced maternal age, and ethnicity for common conditions, such as Down syndrome, cystic fibrosis and Tay-Sachs disease, should be made available in accordance with ACOG guidelines.248,249
The potential to offer pre-implantation genetic diagnosis (PGD) also exists when genetic testing is available for the syndrome of interest and the specific disease causing mutation or chromosome anomaly can be identified. This should not be viewed as the standard of care for all genetic diseases and syndromes, but a specific technology with ongoing promise. When the affected patient is female, discussion with her obstetrician, cardiologist, and a fertility specialist can determine whether her cardiovascular system can tolerate the hormonal stimulation required for PGD.250,251 For those individuals with decreased fertility associated with their specific genetic condition, other reproductive options may include sperm/egg donation, or adoption. Surrogacy may be an option for those women who are fertile, but who are counseled against a full term pregnancy and delivery, yet could withstand hormonal stimulation and oocyte retrieval.251 Even when technically feasible, there are significant financial and psychosocial implications associated with these options which must be addressed as part of any couple’s decision-making process.253–255
TRANSITIONING FOR YOUNG ADULTS WITH SYNDROMES AND CARDIOVASCULAR ABNORMALITIES
Individuals with a cardiovascular abnormality as part of a syndrome require increased services to help in the transition from pediatric to adult care. Many have multiple chronic health conditions, including developmental disabilities. The American Academy of Pediatrics and American Academy of Family Physicians published a consensus statement on health care transitions for young adults with special healthcare needs.256 They recommend identifying a specific health care professional to work with each adolescent and family to co-ordinate the transition, maintaining an up-to-date, accessible, portable medical summary and transition plan. Other articles have addressed the transfer of management from pediatric to adult careproviders in individuals with Turner syndrome,15,257 Williams syndrome258 and Noonan syndrome.206
Studies of the status of nonsyndromic adults with a CHD in the United States,9,259 Canada,260 and Europe261 demonstrate significant progress in creating a standard of care, especially with respect to the development of regional centers. Many young adults with a CHD still are not receiving appropriate care because of the lack of appropriately trained adult-level caregivers for CHDs, attachment of patients and families to pediatric caregivers, lack of patient and family understanding of the chronic nature of the illness, emotional issues of independence, risk-taking behaviors and desire for peer acceptance, lack of transition planning, and problems with continuity of health insurance. Interestingly, adolescents with a CHD rate their quality of life and health status favorably262,263, but usually do not recognize the probability of a shortened life expectancy.264 More than 25% of adult patients with a CHD had no follow-up after age18 years.9. This can be attributed to many factors including denial of disability status or need for services, the misperception that individuals with a CHD have been cured by surgery and no longer require specialist care, and lack of insurance or other resources. Overall, fewer than half of the patients with a CHD in Canada were judged to have made a successful transition from pediatric to adult cardiology. Some continued to be followed in pediatrics where they may not receive appropriate care for adult cardiac issues such as atherosclerosis. Many seemed unaware of the need or lacked the resources to obtain follow-up care by an adult cardiologist interested in CHDs.9 Furthermore, two-thirds of adults with a CHD have experienced problems obtaining health insurance.265
It is a reasonable extrapolation that if these problems exist for normally intelligent young adults with a CHD without a syndrome, they are presumably more severe for patients with multiple chronic conditions and disabilities who need more services, and often are less capable of understanding their own needs. The need for cardiology follow-up may seem less urgent than the need for guardianship for those with developmental disabilities. Adults with a genetic syndrome require more urgent healthcare visits than the general population266 which may be due in part because the healthcare system is not meeting, and possibly not even recognizing all of their special healthcare needs. Individuals with complex healthcare needs often require help in establishing appropriate adult healthcare when transitioning out of the parents’ home, especially if cognition is compromised. Medical conditions including a CHD may affect employability which in turn may limit insurance coverage. Many young adults may not be aware of the resources available and/or may not want to be labeled as having a “disability”. For individuals age 18–21, the federally mandated Children & Youth With Special Health Care Needs is a useful resource. Some young adults with disabilities may qualify for help from the Social Security Administration (federal), Department of Health and Family Services (county social services), Aging and Disability resource centers, Department of Vocational Rehabilitation, Independent Living Centers, and disease specific programs in some states. Many hospitals provide free care. Those who require expensive medications may be eligible for drug programs to nonpaying (“indigent”, “charity”) patients through pharmaceutical companies. Since many of these resources are not advertised and may not be very user friendly to young adults, most individuals with a CHD will require help from a caseworker to determine if they qualify for any type of assistance.
FUTURE DIRECTIONS
The strength of this review is the authorship by experts in their respective fields, referenced with both seminal papers and current literature, and enriched with anecdotal experience. However, much of the clinical care information was derived from consensus-based practice guidelines published from similar “expert” task forces.15,138 In the absence of evidence-based practice guidelines, it is necessary to approach each disorder by defining the scope of the problem, describing existing practices, and evaluating future needs. Ideally, each disorder, whether isolated or syndromic, would be the subject of a natural history study covering the entire lifespan which then would serve as a starting point from which therapy could be tested and clinical care practice guidelines could be derived. As a long term goal, physicians caring for individuals with a cardiovascular abnormality should work together to develop this type of natural history information and guidelines for clinical care. In the short term, the authors hope that this paper will serve to alert and encourage more healthcare providers to address the immediate needs of young people with a genetic syndromes and cardiovascular abnormality now making the transition to adulthood.
Acknowledgments
The authors thank Dr. Leah Burke for sponsoring the session at the 2003 American College of Medical Genetics Annual Meeting which inspired this paper. A.E.L. acknowledges the support of the Massachusetts Center for Birth Defects Research and Prevention. Meaghan Muir and Cindy Marcus provided research assistance. Dr. Suzanne Cassidy, Dr. Claus Gravholt, Susan M. Fernandes, MHP, PA-C, and Dr. Robert Geggel provided helpful comments.
Angela E. Lin is supported in part by the Massachusetts Department of Public Health, Massachusetts Centers for Birth Defects Research and Prevention (#U50/CCU 1132247-03). Craig T. Basson and Deborah A. McDermott are supported by NIH HL80663 and the Snart Cardiovascular Fund. Craig T. Basson is an Established Investigator of the American Heart Association. The research of Colleen A. Morris is funded by grant N535102 from the National Institute of Neurological Disorders and Stroke.
Footnotes
Adults with syndromes and cardiovascular abnormality
Conflict of interest statement
The authors have no commercial association that might pose or create a conflict of interest with the information presented.
Note to editor and publisher: For an article of this length, with well defined sections, please allow us to use the following section headings for the references, too.
References
- 1.Pierpont ME, Basson CT, Benson DW, Gelb BD, et al. The genetic basis for congenital heart defects: Current knowledge. A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–3058. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- 2.Goldmuntz E, Lin AE. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, including the Fetus and Young Adult. 7. Philadelphia: Lippincott Williams & Wilkins; 2008. Genetics of congenital heart defects; pp. 545–572. [Google Scholar]
- 3.Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. First of two parts. N Engl J Med. 2000;342:256–263. doi: 10.1056/NEJM200001273420407. [DOI] [PubMed] [Google Scholar]
- 4.Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med. 2000;342:334–342. doi: 10.1056/NEJM200002033420507. [DOI] [PubMed] [Google Scholar]
- 5.Foster E, Graham TP, Driscoll DJ, Reid GJ, et al. Task Force 2: Special health care needs of adults with congenital heart disease. J Am Coll Cardiol. 2001;37:1161–1198. doi: 10.1016/s0735-1097(01)01277-3. [DOI] [PubMed] [Google Scholar]
- 6.Siu SC, Sermer M, Colman JM, Alvarez N, et al. Prospective multicenter study of pregnancy outcomes in women with heart disease. Circulation. 2001;104:515–521. doi: 10.1161/hc3001.093437. [DOI] [PubMed] [Google Scholar]
- 7.Khairy P, Ouyang DW, Fernandes SM, Lee-Parritz A, et al. Pregnancy outcomes in women with congenital heart disease. Circulation. 2006;113:517–24. doi: 10.1161/CIRCULATIONAHA.105.589655. [DOI] [PubMed] [Google Scholar]
- 8.Avila WS, Rossi EG, Ramires JA, Grinberg M, et al. Pregnancy in patients with heart disease: experience with 1,000 cases. Clin Cardiol. 2003;26:135–142. doi: 10.1002/clc.4960260308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams RG, Pearson GD, Barst RJ, Child JS, et al. Report of the National Heart, Lung, and Blood Institute Working Group on Research in Adult Congenital Heart Disease. J Am Coll Cardiol. 2006;47:701–707. doi: 10.1016/j.jacc.2005.08.074. [DOI] [PubMed] [Google Scholar]
- 10.Rimoin DL, Connor JM, Pyeritz RE, Korf BR. Nature and frequency of genetic disease. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Emery and Rimoin’s principle and practice of medical genetics. 4. Vol. 1. New York: Churchill Livingstone; 2002. pp. 55–59. [Google Scholar]
- 11.Williams MS. Introduction: Adult dysmorphology: Perspectives on approach to diagnosis and care. Am J Med Genet Part C Semin Med Genet. 2007;145C:227–229. doi: 10.1002/ajmg.c.30134. [DOI] [PubMed] [Google Scholar]
- 12.Schrander-Strumpel CTRM, Sinnema M, Van Den Hout L, Maaskant MA. Healthcare transition in persons with intellectual disabilities: General issues, the Masstricht model, and Prader-Willi model. Am J Med Genet Part C Semin Med Genet. 2007;145C:241–247. doi: 10.1002/ajmg.c.30136. [DOI] [PubMed] [Google Scholar]
- 13.Ferencz C, Rubin JD, Loffredo CA, Magee CA. Epidemiology of congenital heart disease: The Baltimore-Washington Infant Study: 1981–1989. Armonk, NY: Futura Publishing Company, Inc; 1993. [Google Scholar]
- 14.Botto L, Goldmuntz E, Lin AE. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, including the Fetus and Young Adult. 7. Philadelphia: Lippincott Williams & Wilkins; 2008. Epidemiology and prevention of congenital heart defects; pp. 524–545. [Google Scholar]
- 15.Bondy CA writing for the Turner Syndrome Consensus Group. Clinical Practice Guideline. Care of Girls and Women with Turner Syndrome: A Guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab. 2007;92:10–25. doi: 10.1210/jc.2006-1374. [DOI] [PubMed] [Google Scholar]
- 16.Pyeritz RE. Maternal and fetal complications of pregnancy in the Marfan syndrome. Am J Med. 1991;71:784–790. doi: 10.1016/0002-9343(81)90365-x. [DOI] [PubMed] [Google Scholar]
- 17.Classifying recommendations for clinical practice guidelines. Pediatrics. 2004;114:874–877. doi: 10.1542/peds.2004-1260. [DOI] [PubMed] [Google Scholar]
- 18.The Criteria Committee of the New York Heart Association. Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels. 9. Boston, Mass: Little, Brown & Co; 1994. pp. 253–256. [Google Scholar]
- 19.Wilson W, Taubert KA, Gewitz M, Lockhart PB, et al. Prevention of Infective Endocarditis. Guidelines From the American Heart Association. A Guideline From the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116:1736–1754. doi: 10.1161/CIRCULATIONAHA.106.183095. [DOI] [PubMed] [Google Scholar]
Deletion 22q11.2
- 20.Devriendt K, Fryns JP, Mortier G, van Thienen MN, et al. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet. 1998;35:789–790. doi: 10.1136/jmg.35.9.789-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79:348–351. doi: 10.1136/adc.79.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Botto LD, May K, Fernhoff PM, Correa A, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- 23.Wilson DI, Burn J, Scambler P, Goodship J. DiGeorge syndrome: part of CATCH 22. J Med Genet. 1993;30:852–856. doi: 10.1136/jmg.30.10.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oskarsdottir S. Itellecta DocusysAB. Vastra Frolunda; Goteborg, Sweden: 2005. The 22q11 deletion syndrome, A clinical and epidemiological study; pp. 1–183. [Google Scholar]
- 25.Driscoll DA, Spinner NB, Budarf ML, McDonald-McGinn DM, et al. Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome. Am J Med Genet. 1992;44:261–268. doi: 10.1002/ajmg.1320440237. [DOI] [PubMed] [Google Scholar]
- 26.Scambler PJ, Kelly D, Lindsay E, Williamson R, et al. Velo-cardio-facial syndrome associated with chromosome 22 deletions encompassing the DiGeorge locus. Lancet. 1992;339:1138–1139. doi: 10.1016/0140-6736(92)90734-k. [DOI] [PubMed] [Google Scholar]
- 27.Burn J, Takao A, Wilson D, Cross I, et al. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J Med Genet. 1993;30:822–824. doi: 10.1136/jmg.30.10.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, et al. Autosomal dominant “Opitz” GBBB syndrome due to a 22q11.2 deletion. Am J Med Genet. 1995;59:103–113. doi: 10.1002/ajmg.1320590122. [DOI] [PubMed] [Google Scholar]
- 29.Lacassie Y, Arriaza MI. Opitz GBBB syndrome and the 22q11.2 deletion. Am J Med Genet. 1996;62:318. doi: 10.1002/(SICI)1096-8628(19960329)62:3<318::AID-AJMG21>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 30.Fryburg JS, Lin KY, Golden WL. Chromosome 22q11.2 deletion in a boy with Opitz (G/BBB) syndrome. Am J Med Genet. 1996;62:274–275. doi: 10.1002/(SICI)1096-8628(19960329)62:3<274::AID-AJMG13>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 31.Giannotti A, Digilio MC, Marino B, Mingarelli R, et al. Cayler cardiofacial syndrome and del 22q11: part of the CATCH22 phenotype. Am J Med Genet. 1994;53:303–304. doi: 10.1002/ajmg.1320530320. [DOI] [PubMed] [Google Scholar]
- 32.McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, et al. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 2001;3:23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- 33.Ben-Shachar S, Ou Z, Shaw CA, Belmont JW, et al. 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am J Hum Genet. 2008;82:214–221. doi: 10.1016/j.ajhg.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, et al. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genet Couns. 1999;10:11–24. [PubMed] [Google Scholar]
- 35.Bassett AS, Hodgkinson K, Chow EW, Correia S, et al. 22q11 deletion syndrome in adults with schizophrenia. Am J Med Genet. 1998;81:328–337. [PMC free article] [PubMed] [Google Scholar]
- 36.Bassett AS, Chow EW, Husted J, Weksberg R, et al. Clinical features of 78 adults with 22q11 Deletion Syndrome. Am J Med Genet A. 2005;138:307–313. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryan AK, Goodship JA, Wilson DI, Philip N, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen E, Chow EW, Weksberg R, Bassett AS. Phenotype of adults with the 22q11 deletion syndrome: A review. Am J Med Genet. 1999;86:359–365. doi: 10.1002/(sici)1096-8628(19991008)86:4<359::aid-ajmg10>3.0.co;2-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shooner KA, Rope AF, Hopkin RJ, Andelfinger GU, et al. Genetic analyses in two extended families with deletion 22q11 syndrome: importance of extracardiac manifestations. J Pediatr. 2005;146:382–387. doi: 10.1016/j.jpeds.2004.10.038. [DOI] [PubMed] [Google Scholar]
- 40.Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998;32:492–498. doi: 10.1016/s0735-1097(98)00259-9. [DOI] [PubMed] [Google Scholar]
- 41.Menahem S, Rahayoe AU, Brawn WJ, Mee RB. Interrupted aortic arch in infancy: a 10-year experience. Pediatr Cardiol. 1992;13:214–221. doi: 10.1007/BF00838779. [DOI] [PubMed] [Google Scholar]
- 42.Marino B, Digilio MC, Toscano A, Anaclerio S, et al. Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet Med. 2001;3:45–48. doi: 10.1097/00125817-200101000-00010. [DOI] [PubMed] [Google Scholar]
- 43.McElhinney DB, Clark BJ, III, Weinberg PM, Kenton ML, McDonald-McGinn D, Driscoll DA, Zackai EH, Goldmuntz E. Association of chromosome 22q11 deletion with isolated anomalies of aortic arch laterality and branching. J Am Coll Cardiol. 2001;37:2114–2119. doi: 10.1016/s0735-1097(01)01286-4. [DOI] [PubMed] [Google Scholar]
- 44.Carotti A, Marino B, Di Donato RM. Influence of chromosome 22q11.2 microdeletion on surgical outcome after treatment of tetralogy of fallot with pulmonary atresia. J Thorac Cardiovasc Surg. 2003;126:1666–1667. doi: 10.1016/s0022-5223(03)01196-6. [DOI] [PubMed] [Google Scholar]
- 45.Mahle WT, Crisalli J, Coleman K, Campbell RM, et al. Deletion of chromosome 22q11.2 and outcome in patients with pulmonary atresia and ventricular septal defect. Ann Thorac Surg. 2003;76:567–571. doi: 10.1016/s0003-4975(03)00516-2. [DOI] [PubMed] [Google Scholar]
- 46.Anaclerio S, Di Ciommo V, Michielon G, Digilio MC, et al. Conotruncal heart defects: impact of genetic syndromes on immediate operative mortality. Ital Heart J. 2004;5:624–628. [PubMed] [Google Scholar]
- 47.Michielon G, Marino B, Formigani R, Gargiulo G, et al. Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Ann Thorac Surg. 2006;81:968–975. doi: 10.1016/j.athoracsur.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 48.Gatzoulis MA, Balaji S, Webber SA, Siu SC, et al. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: a multicentre study. Lancet. 2000;356:975–981. doi: 10.1016/S0140-6736(00)02714-8. [DOI] [PubMed] [Google Scholar]
- 49.Hokanson JS, Pierpont ME, Hirsch B, Moller JH. 22q11.2 microdeletions in adults with familial tetralogy of Fallot. Genet Med. 2001;3:61–64. doi: 10.1097/00125817-200101000-00013. [DOI] [PubMed] [Google Scholar]
- 50.Beauchesne LM, Warnes CA, Connolly HM, Ammash NM, et al. Prevalence and clinical manifestations of 22q11.2 microdeletion in adults with selected conotruncal anomalies. J Am Coll Cardiol. 2005;45:595–598. doi: 10.1016/j.jacc.2004.10.056. [DOI] [PubMed] [Google Scholar]
- 51.Jacobs MLCA, Rychik J, Steven JM, Nicolson SC, et al. Interrupted aortic arch. Impact of subaortic stenosis on management and outcome. Circulation. 1995;92:128–131. doi: 10.1161/01.cir.92.9.128. [DOI] [PubMed] [Google Scholar]
- 52.Dittrich S, Vogel M, Dahnert I, Berger F, et al. Surgical repair of tetralogy of Fallot in adults today. Clin Cardiol. 1999;22:460–464. doi: 10.1002/clc.4960220705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harrison DA, Siu SC, Hussain F, MacLoghlin CJ, et al. Sustained atrial arrhythmias in adults late after repair of tetralogy of fallot. Am J Cardiol. 2001;87:584–588. doi: 10.1016/s0002-9149(00)01435-1. [DOI] [PubMed] [Google Scholar]
- 54.McDonald-McGinn DM, Driscoll DA, Saitta S, Jawad A, et al. Guidelines for prenatal detection of the 22q11.2 deletion. Am J Hum Genet. 2002;71:198. (A173) [Google Scholar]
- 55.McDonald-McGinn DM, Driscoll DA, Zackai EH. Guidelines for the prenatal detection of the 22q11.2 deletion syndrome. Proc Greenwood Genet Center. 2007;26:155. [Google Scholar]
Down syndrome
- 56.Centers for Disease Control and Prevention. Improved national prevalence estimates for 18 selected major birth defects- United States, 1999–2001. MMWR. 2005;54:1301–1305. [PubMed] [Google Scholar]
- 57.Wessels MW, Los FJ, Frohn-Mulder IME, Niermeijer MF, et al. Poor outcome in Down syndrome fetuses with cardiac anomalies or growth retardation. Am J Med Genet. 2003;116A:147–151. doi: 10.1002/ajmg.a.10823. [DOI] [PubMed] [Google Scholar]
- 58.Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down’s syndrome in the USA from 1983 to 1997: a population-based study. Lancet. 2002;359:1019–1025. doi: 10.1016/s0140-6736(02)08092-3. [DOI] [PubMed] [Google Scholar]
- 59.Bittles AH, Glasson EJ. Clinical, social and ethical implications of changing life expectancy in Down syndrome. Dev Med Child Neurol. 2004;46:282–286. doi: 10.1017/s0012162204000441. [DOI] [PubMed] [Google Scholar]
- 60.Hunter AGW. Down syndrome. In: Cassidy SB, Allanson JE, editors. Management of Genetic Syndromes. 2. John Wiley and Sons; Hoboken: 2004. pp. 191–210. [Google Scholar]
- 61.Freeman SB, Taft LF, Dooley KJ, Allran K, et al. Population-based study of congenital heart defects in Down syndrome. Am J Med Genet. 1998;80:213–217. [PubMed] [Google Scholar]
- 62.McElhinney DB, Straka M, Goldmuntz E, Zackai EH. Correlation between abnormal cardiac physical examination and echocardiographic findings in neonates with Down syndrome. Am J Med Genet. 2002;113:238–241. doi: 10.1002/ajmg.10803. [DOI] [PubMed] [Google Scholar]
- 63.Cetta F, Minich LL, Edwards WD, Dearani JA, et al. Moss and Adams Heart Disease in Infants, Children, and Adolescents, including the Fetus and Young Adult. 7. Philadelphia: Lippincott Williams & Wilkins; 2008. Atrioventricular septal defects; pp. 646–667. [Google Scholar]
- 64.Reller MD, Morris CD. Is Down syndrome a risk factor for poor outcome after repair of congenital heart defects? J Pediatr. 1998;132:738–741. doi: 10.1016/s0022-3476(98)70372-5. [DOI] [PubMed] [Google Scholar]
- 65.Marino B, Diociaiuti L, Calcagni G, Mastroiacovo P. Outcome in Down syndrome fetuses with cardiac anomalies. Am J Med Genet. 2004;128A:101–102. doi: 10.1002/ajmg.a.20690. [DOI] [PubMed] [Google Scholar]
- 66.American Academy of Pediatrics, Committee on Genetic. Health supervision for children with Down syndrome. Pediatrics. 2001;107:442–449. doi: 10.1542/peds.107.2.442. [DOI] [PubMed] [Google Scholar]
- 67.Roizen NJ, Patterson D. Down’s syndrome. Lancet. 2003;361:1281–1289. doi: 10.1016/S0140-6736(03)12987-X. [DOI] [PubMed] [Google Scholar]
- 68.Goldhaber SZ, Rubin IL, Brown W, Robertson N, et al. Valvular heart disease (Aortic regurgitation and mitral valve prolapse) among institutionalized adults with Down’s syndrome. Am J Cardiol. 1986;57:278–281. doi: 10.1016/0002-9149(86)90905-7. [DOI] [PubMed] [Google Scholar]
- 69.Geggel RL, O’Brien JE, Feingold M. Development of valve dysfunction in adolescents and young adults with Down syndrome and no known congenital heart disease. J Pediatr. 1993;122:821–823. doi: 10.1016/s0022-3476(06)80036-3. [DOI] [PubMed] [Google Scholar]
- 70.Hamada T, Gejyo F, Koshino Y, Murata T, et al. Echocardiographic evaluation of cardiac valvular abnormalities in adults with Down’s syndrome. Tohoku J Exp Med. 1998;185:31–35. doi: 10.1620/tjem.185.31. [DOI] [PubMed] [Google Scholar]
- 71.Marino B, Digilio MC. Health supervision for Children with Down Syndrome (letter) Pediatrics. 2001;108:1384–1385. [PubMed] [Google Scholar]
- 72.Feingold M, Geggel RL, Marino B, Digilio MC, et al. Health supervision for children with Down syndrome. Letters and reply. Pediatrics. 2001;108:1384–1385. [PubMed] [Google Scholar]
- 73.Smith DS. Health care management of adults with Down syndrome. Am Fam Pract. 2001;64:1031–1038. [PubMed] [Google Scholar]
- 74.Nowak CB, Feingold M. Cardiac status in Down syndrome adults. Poster presentation at the Am Coll Med Genet; March 22, 2007; Nashville, TN. [Google Scholar]
- 75.Murdoch JC, Rodger JC, Rao SS, Fletcher CD, et al. Down’s syndrome: an atheroma-free model? Brit Med J. 1971;2:223–228. doi: 10.1136/bmj.2.6081.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pueschel SM, Craig WY, Haddow JE. Lipids and lipoproteins in persons with Down’s syndrome. J Intell Disabil Res. 1992;36:365–369. doi: 10.1111/j.1365-2788.1992.tb00535.x. [DOI] [PubMed] [Google Scholar]
- 77.Hopkins WE, Fukagawa NK, Sobel BE, Schneider DJ. Plasminogen activator inhibitor type 1 in adults with Down syndrome and protection against macrovascular disease. Am J Cardiol. 2000;85:784–786. doi: 10.1016/s0002-9149(99)00864-4. [DOI] [PubMed] [Google Scholar]
- 78.Bovicelli L, Orsini LF, Rizzo N, Montacuti V, et al. Reproduction in Down syndrome. Obstet Gynecol. 1982;59:13S–17S. [PubMed] [Google Scholar]
- 79.Shobha Rani A, Jyothi A, Reddy PP, Reddy OS. Reproduction in Down’s syndrome. Int J Gynaecol Obstet. 1990;31:81–86. doi: 10.1016/0020-7292(90)90187-p. [DOI] [PubMed] [Google Scholar]
- 80.Mullins DH, Estrada WR, Gready TG. Pregnancy in an adult mongoloid female. Obstet Gynecol. 1960;15:781. [PubMed] [Google Scholar]
Turner syndrome
- 81.Nielsen J, Wohlert M. Sex chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Birth Defects Orig Artic Ser. 1990;26:209–223. [PubMed] [Google Scholar]
- 82.Stochholm K, Juul S, Juel K, Naera RW, Gravholt CH. Prevalence, incidence, diagnostic delay and mortality in Turner syndrome. J Clin Endocrinol Metab. 2006;91:3897–3902. doi: 10.1210/jc.2006-0558. [DOI] [PubMed] [Google Scholar]
- 83.Sybert VP, McCauley E. Turner’s syndrome. N Engl J Med. 2004;351:1227–1238. doi: 10.1056/NEJMra030360. [DOI] [PubMed] [Google Scholar]
- 84.Lacro RV, Jones KL, Benirschke K. Coarctation of the aorta in Turner syndrome: a pathologic study of fetuses with nuchal cystic hygromas, hydrops fetalis, and female genitalia. Pediatrics. 81:445–451. [PubMed] [Google Scholar]
- 85.Loscalzo ML, Van PL, Ho VB, Bakalov VK, et al. Association between fetal lymphedema and congenital cardiovascular defects in Turner syndrome. Pediatrics. 2005:115732–735. doi: 10.1542/peds.2004-1369. [DOI] [PubMed] [Google Scholar]
- 86.Mazzanti L, Cacciari E. Congenital heart disease in patients with Turner’s syndrome. Italian Study Group for Turner Syndrome (ISGTS) J Pediatr. 1998;133:688–692. doi: 10.1016/s0022-3476(98)70119-2. [DOI] [PubMed] [Google Scholar]
- 87.Sybert VP. Cardiovascular malformations and complications in Turner syndrome. Pediatrics. 1998;101:e11. doi: 10.1542/peds.101.1.e11. [DOI] [PubMed] [Google Scholar]
- 88.Ho VB, Bakalov VK, Cooley M, Van PL, et al. Major vascular anomalies in Turner syndrome. Prevalence and magnetic resonance angiographic features. Circulation. 2004;110:1694–1700. doi: 10.1161/01.CIR.0000142290.35842.B0. [DOI] [PubMed] [Google Scholar]
- 89.Bechtold SM, Dalla Pozza R, Becker A, Meidert A, et al. Partial anomalous pulmonary vein connection: an underestimated cardiovascular defect in Ullrich-Turner syndrome. Eur J Pediatr. 2004;163:158–162. doi: 10.1007/s00431-003-1384-7. [DOI] [PubMed] [Google Scholar]
- 90.Bondy CA, Ceniceros I, Van PL, Bakalov VK, et al. Prolonged rate-corrected QT interval and other electrocardiogram abnormalities in girls with Turner syndrome. Pediatrics. 2006;118:e1220–1225. doi: 10.1542/peds.2006-0776. [DOI] [PubMed] [Google Scholar]
- 91.Cecconi M, Nistri S, Quarti A, Manfrin M, et al. Aortic dilatation in patients with bicuspid aortic valve. J Cardiovasc Med. 2006;7:11–20. doi: 10.2459/01.JCM.0000199777.85343.ec. [DOI] [PubMed] [Google Scholar]
- 92.Lin AE, Lippe B, Rosenthal R. Further delineation of aortic dilation, dissection and rupture in patients with Turner syndrome. Pediatrics. 1998;102(1) doi: 10.1542/peds.102.1.e12. URL: http://www.pediatrics.org.cgi/content/full/102/1/e12. [DOI] [PubMed]
- 93.Elsheikh M, Casadei B, Conway GS, Wass JA. Hypertension is a major risk factor for aortic root dilatation in women with Turner’s syndrome. Clin Endocrinol (Oxf) 2001;54:69–73. doi: 10.1046/j.1365-2265.2001.01154.x. [DOI] [PubMed] [Google Scholar]
- 94.Ostberg JE, Brookes JAS, McCarthy C, Halcox J, et al. A comparison of echocardiography and magnetic resonance imaging in cardiovascular screening of adults with Turner syndrome. J Clin Endocrinol Metab. 2004;80:5966–5971. doi: 10.1210/jc.2004-1090. [DOI] [PubMed] [Google Scholar]
- 95.Matura LA, Ho VB, Rosing DR, Bondy CA. Aortic dilatation and dissection in Turner syndrome. Circulation. 2007;116:1663–1670. doi: 10.1161/CIRCULATIONAHA.106.685487. [DOI] [PubMed] [Google Scholar]
- 96.Carlson M, Silberbach M. Dissection of the aorta in Turner syndrome: Two cases and review of 85 cases in the literature. J Med Genet. 2007;44:745–749. doi: 10.1136/jmg.2007.052019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ostberg JE, Donald AE, Halcox JP, Storry C, et al. Vasculopathy in Turner syndrome: arterial dilatation and intimal thickening without endothelial dysfunction. J Clin Endocrinol Metab. 2005;90:5161–5166. doi: 10.1210/jc.2005-0677. [DOI] [PubMed] [Google Scholar]
- 98.Lopez L, Arheart KL, Colan SD, Stein NS, et al. Turner syndrome is an independent risk factor for aortic dilation in the young. Pediatrics. 2008;121:e1622–e1627. doi: 10.1542/peds.2007-2807. [DOI] [PubMed] [Google Scholar]
- 99.Gravholt CH, Landin-Wilhelmsen K, Stochholm K, Hjerrild BE, et al. Clinical and epidemiological description of aortic dissection in Turner syndrome. Cardiol Young. 2006;16:1–7. doi: 10.1017/S1047951106000928. [DOI] [PubMed] [Google Scholar]
- 100.Bondy CA, van PL, Bakalov VK, Ho VB. Growth hormone treatment and aortic dimensions in Turner syndrome. J Clin Endocrinol Metab. 2006;91:1785–1788. doi: 10.1210/jc.2005-2625. [DOI] [PubMed] [Google Scholar]
- 101.van den Berg J, Bannink EM, Wielopolski PA, Pattynama PM, de Muinck Keizer-Schrama SM, Helbing WA. Aortic distensibility and dimensions and the effects of growth hormone treatment in the Turner syndrome. Am J Cardiol. 2006;97:1644–1649. doi: 10.1016/j.amjcard.2005.12.058. [DOI] [PubMed] [Google Scholar]
- 102.Matura LA, Sachdev V, Bakalov V, Rosing DR, Bondy CA. Growth hormone treatment and left ventricular dimensions in Turner syndrome. J Pediatr. 2007;150:587–591. doi: 10.1016/j.jpeds.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gravholt CH, Hansen KW, Erlandsen M, Ebbehoj E, et al. Nocturnal hypertension and impaired sympathovagal tone in Turner syndrome. J Hypertens. 2006;24:353–360. doi: 10.1097/01.hjh.0000200509.17947.0f. [DOI] [PubMed] [Google Scholar]
- 104.Bondy CA, Van PK, Bakalov VK, Sachdev V, et al. Prolongation of the cardiac QTc interval in Turner syndrome. Medicine. 2006;85:1–7. doi: 10.1097/01.md.0000205629.16302.bc. [DOI] [PubMed] [Google Scholar]
- 105.Cooley M, Bakalov V, Bondy CA. Lipid profiles in women with 45, X vs 46, XX primary ovarian failure. JAMA. 2003;290:2127–2128. doi: 10.1001/jama.290.16.2127. [DOI] [PubMed] [Google Scholar]
- 106.Van PL, Bakalov VK, Bondy CA. Monosomy for the X chromosome is associated with an atherogenic lipid profile. J Clin Endocrinol Metab. 2006;91:2867–2870. doi: 10.1210/jc.2006-0503. [DOI] [PubMed] [Google Scholar]
- 107.Aligeti C, Horn HR. Turner’s syndrome and coronary artery disease. Am J Cardiol. 2007;99:741–742. doi: 10.1016/j.amjcard.2006.10.027. [DOI] [PubMed] [Google Scholar]
- 108.Lin AE, Silberbach M. Focus on the heart and aorta in Turner syndrome. J Pediatr. 2007;150:572–574. doi: 10.1016/j.jpeds.2007.03.038. [DOI] [PubMed] [Google Scholar]
- 109.Karnis MF, Zimon AE, Lalwani SI, Timmreck LS, et al. Risk of death in pregnancy achieved through oocyte donation in patients with Turner syndrome: a national survey. Fertil Steril. 2001;80:498–501. doi: 10.1016/s0015-0282(03)00974-9. [DOI] [PubMed] [Google Scholar]
- 110.Mendelson MA. Pregnancy in patients with obstructive lesions: aortic stenosis, coarctation of the aorta and mitral stenosis. Progr Pediatr Cardiol. 2004:61–70. [Google Scholar]
- 111.Van Voorhis BJ. In vitro fertilization. N Engl J Med. 2007;356:379–86. doi: 10.1056/NEJMcp065743. [DOI] [PubMed] [Google Scholar]
Williams-Beuren syndrome
- 112.Cherniske EM, Carpenter TO, Klaiman C, Young E, et al. Multisystem study of 20 older adults with Williams syndrome. Am J Med Genet A. 2004;131:255–264. doi: 10.1002/ajmg.a.30400. [DOI] [PubMed] [Google Scholar]
- 113.Morris CA, Thomas IT, Greenberg F. Williams syndrome: autosomal dominant inheritance. Am J Med Genet. 1993;47:478–481. doi: 10.1002/ajmg.1320470409. [DOI] [PubMed] [Google Scholar]
- 114.Morris CA, Demsey SA, Leonard CO, Dilts C, et al. Natural history of Williams syndrome: physical characteristics. J Pediatr. 1988;113:318–326. doi: 10.1016/s0022-3476(88)80272-5. [DOI] [PubMed] [Google Scholar]
- 115.Mervis CB, Robinson BF, Bertrand J, Morris CA, et al. The Williams syndrome cognitive profile. Brain Cogn. 2000;44:604–628. doi: 10.1006/brcg.2000.1232. [DOI] [PubMed] [Google Scholar]
- 116.Klein-Tasman BP, Risi S, Lord CE. Socio-communicative deficits in young children with Williams syndrome: performance on the autism diagnostic observation schedule. Child Neuropsychol. 2007;13:444–67. doi: 10.1080/09297040601033680. [DOI] [PubMed] [Google Scholar]
- 117.Mervis CB. Williams syndrome: 15 years of psychological research. Dev Neuropsychol. 2003;23:1–12. doi: 10.1080/87565641.2003.9651884. [DOI] [PubMed] [Google Scholar]
- 118.Stromme P, Bjornstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17:269–271. doi: 10.1177/088307380201700406. [DOI] [PubMed] [Google Scholar]
- 119.Osborne LR. The Molecular Basis of a Multisystem Disorder. In: Morris CA, Lenhoff HM, Wang PP, editors. Williams-Beuren syndrome: Research, Evaluation, and Treatment. 1. Baltimore: Johns Hopkins University Press; 2006. pp. 18–58. [Google Scholar]
- 120.Eronen M, Peippo M, Hiippala A, Raatikka M, et al. Cardiovascular manifestations in 75 patients with Williams syndrome. J Med Genet. 2002;39:554–558. doi: 10.1136/jmg.39.8.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bird LM, Billman GF, Lacro RV, Spicer RL, et al. Sudden death in Williams syndrome: report of ten cases. J Pediatr. 1996;129:926–931. doi: 10.1016/s0022-3476(96)70042-2. [DOI] [PubMed] [Google Scholar]
- 122.Ardinger RH, Jr, Goertz KK, Mattioli LF. Cerebrovascular stenoses with cerebral infarction in a child with Williams syndrome. Am J Med Genet. 1994;51:200–202. doi: 10.1002/ajmg.1320510305. [DOI] [PubMed] [Google Scholar]
- 123.Kaplan P. The Medical Management of Children with Williams-Beuren syndrome. In: Morris CA, Lenhoff HM, Wang PP, editors. Williams-Beuren syndrome: Research, Evaluation, and Treatment. 1. Baltimore: Johns Hopkins University Press; 2006. pp. 83–106. [Google Scholar]
- 124.Kaplan P, Levinson M, Kaplan BS. Cerebral artery stenoses in Williams syndrome cause strokes in childhood. J Pediatr. 1995;126:943–945. doi: 10.1016/s0022-3476(95)70216-4. [DOI] [PubMed] [Google Scholar]
- 125.Soper R, Chaloupka JC, Fayad PB, Greally JM, et al. Ischemic stroke and intracranial multifocal cerebral arteriopathy in Williams syndrome. J Pediatr. 1995;126:945–948. doi: 10.1016/s0022-3476(95)70217-2. [DOI] [PubMed] [Google Scholar]
- 126.Scheiber D, Fekete G, Urban Z, Tarjan I, et al. Echocardiographic findings in patients with Williams-Beuren syndrome. Wien Klin Wochenschr. 2006;118:538–554. doi: 10.1007/s00508-006-0658-2. [DOI] [PubMed] [Google Scholar]
- 127.Kececioglu D, Kotthoff S, Vogt J. Williams-Beuren syndrome: a 30-year follow-up of natural and postoperative course. Eur Heart J. 1993;14:1458–1464. doi: 10.1093/eurheartj/14.11.1458. [DOI] [PubMed] [Google Scholar]
- 128.Stamm C, Friehs I, Ho SY, Moran AM, et al. Congenital supravalvar aortic stenosis: a simple lesion? Eur J Cardiothorac Surg. 2001;19:195–202. doi: 10.1016/s1010-7940(00)00647-3. [DOI] [PubMed] [Google Scholar]
- 129.Morris CA, Leonard CO, Dilts C, Demsey SA. Adults with Williams syndrome. Am J Med Genet Suppl. 1990;6:102–107. doi: 10.1002/ajmg.1320370619. [DOI] [PubMed] [Google Scholar]
- 130.French JW, Guntheroth WG. An explanation of asymmetric upper extremity blood pressures in supravalvular aortic stenosis: the Coanda effect. Circulation. 1970;42:31–36. doi: 10.1161/01.cir.42.1.31. [DOI] [PubMed] [Google Scholar]
- 131.Kawai M, Nishikawa T, Tanaka M, Ando A, et al. An autopsied case of Williams syndrome complicated by moyamoya disease. Acta Paediatr Jpn. 1993;35:63–67. doi: 10.1111/j.1442-200x.1993.tb03009.x. [DOI] [PubMed] [Google Scholar]
- 132.Wollack JB, Kaifer M, LaMonte MP, Rothman M. Stroke in Williams syndrome. Stroke. 1996;27:143–146. doi: 10.1161/01.str.27.1.143. [DOI] [PubMed] [Google Scholar]
- 133.Wessel A, Gravenhorst V, Buchhorn R, Gosch A, et al. Risk of sudden death in the Williams-Beuren syndrome. Am J Med Genet A. 2004;127:234–237. doi: 10.1002/ajmg.a.30012. [DOI] [PubMed] [Google Scholar]
- 134.Giordano U, Turchetta A, Giannotti A, Digilio MC, et al. Exercise testing and 24-hour ambulatory blood pressure monitoring in children with Williams syndrome. Pediatr Cardiol. 2001;22:509–511. doi: 10.1007/s002460010285. [DOI] [PubMed] [Google Scholar]
- 135.Mulik VV, Templeston KI, Howe DT. Two pregnancies in a woman with Williams syndrome. Brit J Obstet Gynecol. 2004;111:511–512. doi: 10.1111/j.1471-0528.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 136.van der Tuuk KV, Drenthen W, Moons P, Budts W. Three live-birth pregnancies in a woman with Williams syndrome. Congenit Heart Dis. 2007;2:139–142. doi: 10.1111/j.1747-0803.2007.00088.x. [DOI] [PubMed] [Google Scholar]
Marfan syndrome
- 137.Pyeritz RE. Marfan syndrome and related disorders. In: Rimoin DL, Conner JM, Pyeritz RE, Korf BR, editors. Principles and Practice of Medical Genetics. 5. Philadelphia: Churchill Livingstone; 2007. pp. 3579–3624. [Google Scholar]
- 138.DePaepe A, Deitz HC, Devereux RB, Hennekem R, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–426. doi: 10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 139.Chow K, Litt HI, Pyeritz RE. Abdominal visceral findings in the Marfan syndrome. Genet Med. 2007;9:208–212. doi: 10.1097/gim.0b013e3180423cb3. [DOI] [PubMed] [Google Scholar]
- 140.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nature Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 141.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, et al. TGF-beta-dependent pathogenesis of mitral valve}prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Habashi JP, Judge DP, Holm TM, Cohn RD, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–21. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.McKusick VA. The cardiovascular aspects of Marfan’s syndrome: A heritable disorder of connective tissue. Circulation. 1955;11:321–341. doi: 10.1161/01.cir.11.3.321. [DOI] [PubMed] [Google Scholar]
- 144.Morse RP, Rockenmacher S, Pyeritz RE, Sanders SP, et al. Diagnosis and management of Marfan syndrome in infants. Pediatrics. 1990;86:888–895. [PubMed] [Google Scholar]
- 145.Loeys BL, Chen J, Neptune ER, Judge DP, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 146.Sisk HE, Zahka KG, Pyeritz RE. The Marfan syndrome in early childhood: Analysis of 15 patients diagnosed at less than four years of age. Am J Cardiol. 1983;52:353–358. doi: 10.1016/0002-9149(83)90138-8. [DOI] [PubMed] [Google Scholar]
- 147.Roman MJ, Devereux RB, Kramer-Fox R, O’Loughlin J. Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am J Cardiol. 1989;64:507–512. doi: 10.1016/0002-9149(89)90430-x. [DOI] [PubMed] [Google Scholar]
- 148.Salim MA, Alpert BS, Ward JC, Pyeritz RE. Effect of beta-adrenergic blockade on aortic root rate of dilation in the Marfan syndrome. Am J Cardiol. 1994;74:629–633. doi: 10.1016/0002-9149(94)90762-5. [DOI] [PubMed] [Google Scholar]
- 149.Shores J, Berger KR, Murphy EA, Pyeritz RE. Chronic β-adrenergic blockade protects the aorta in the Marfan syndrome: a prospective, randomized trial of propranolol. N Engl J Med. 1994;330:1335–1341. doi: 10.1056/NEJM199405123301902. [DOI] [PubMed] [Google Scholar]
- 150.Murdoch JL, Walker BA, Halpern BL, Kuzma JW, et al. Life expectancy and causes of death in the Marfan syndrome. N Engl J Med. 1972;286:804–808. doi: 10.1056/NEJM197204132861502. [DOI] [PubMed] [Google Scholar]
- 151.Gott VL, Greene PS, Alejo DE, Cameron DE, et al. Surgery for ascending aortic disease in Marfan patients: a multi-center study. N Engl J Med. 1999;340:1307–1313. doi: 10.1056/NEJM199904293401702. [DOI] [PubMed] [Google Scholar]
- 152.Silverman DI, Burton KJ, Gray J, Bosner MS, et al. Life expectancy in the Marfan syndrome. Am J Cardiol. 1995;75:157–160. doi: 10.1016/s0002-9149(00)80066-1. [DOI] [PubMed] [Google Scholar]
- 153.Miller DC. Valve-sparing aortic root replacement in patients with the Marfan syndrome. J Thorac Cardiovasc Surg. 2003;125:773–778. doi: 10.1067/mtc.2003.162. [DOI] [PubMed] [Google Scholar]
- 154.David TE, Feindel CM, Webb GD, Coleman JM, et al. Aortic valve preservation in patients with aortic root aneurysm: results of the reimplantation technique. Ann Thoracic Surg. 2007;83:S732–5. doi: 10.1016/j.athoracsur.2006.10.080. [DOI] [PubMed] [Google Scholar]
- 155.Lacro RV, Dietz H, Wruck L, Bradley T, et al. Rationale and design of a randomized clinical trial of β-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome. Am Heart J. 2007;1544:624–31. doi: 10.1016/j.ahj.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Loeys BL, Schwarze U, Holm T, Callewaert BL, et al. Aneurysm syndromes caused by mutations in TGF-β receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 157.Rossiter JP, Repke JT, Morales AJ, Murphy EA, et al. A prospective longitudinal evaluation of pregnancy in the Marfan syndrome. Am J Obstet Gynecol. 1995;173:1599–1606. doi: 10.1016/0002-9378(95)90655-x. [DOI] [PubMed] [Google Scholar]
Disorders of TGF-β Receptors
- 158.Pannu H, Fadulu VT, Chang J, Lafont A, et al. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 159.Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sakai H, Visser R, Ikegawa S, Ito E, et al. Comprehensive genetic analysis of relevant four genes in 49 patients with Marfan syndrome and Marfan-related phenotypes. Am J Med Genet A. 2006;140:1719–1725. doi: 10.1002/ajmg.a.31353. [DOI] [PubMed] [Google Scholar]
- 161.Adès LC, Sullivan K, Biggin A, Haan EA, Brett M, Holman KJ, Dixon J, Robertson S, Holmes AD, Rogers J, Bennetts B. FBN1, TGFBR1, and the Marfan-craniosynostosis/mental retardation disorders revisited. Am J Med Genet A. 2006;140:1047–1058. doi: 10.1002/ajmg.a.31202. [DOI] [PubMed] [Google Scholar]
- 162.Viassolo V, Lituania M, Marasini M, Dietz H, et al. Fetal aortic root dilation: a prenatal feature of the Loeys-Dietz syndrome. Prenat Diagn. 2006;26:1081–1083. doi: 10.1002/pd.1565. [DOI] [PubMed] [Google Scholar]
- 163.Johnson PT, Chen JK, Loeys BL, Dietz HC, et al. Loeys-Dietz syndrome: MDCT angiography findings. AJR. 2007;189:W29–35. doi: 10.2214/AJR.06.1316. [DOI] [PubMed] [Google Scholar]
- 164.LeMaire SA, Pannu H, Tran-Fadulu V, Carter SA, et al. Severe aortic and arterial aneurysms associated with a TGFBR2 mutation. Nat Clin Pract Cardiovasc Med. 2007;4:167–171. doi: 10.1038/ncpcardio0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Williams JA, Loeys BL, Nwakanma LU, Dietz HC, Spevak PJ, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg. 2007;83:S757–763. doi: 10.1016/j.athoracsur.2006.10.091. [DOI] [PubMed] [Google Scholar]
Hereditary Hemorrhagic Telangectasia
- 166.Guttmacher A, Marchuck D, Pyeritz RE. Hereditary hemorrhagic telangiectasia. In: Rimoin DL, Conner JM, Pyeritz RE, Korf BR, editors. Principles and Practice of Medical Genetics. 5. Philadelphia: Churchill Livingstone; 2007. pp. 1200–1213. [Google Scholar]
- 167.Bossler AD, Richards J, George C, Godmilow L, et al. A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): correlation of genotype with phenotype. Hum Mutat. 2006;27:667–75. doi: 10.1002/humu.20342. [DOI] [PubMed] [Google Scholar]
- 168.Gallione CJ, Richards JA, Letteboer TGW, Rushlow D, et al. SMAD4 mutations found in unselected HHT patients. Eur J Hum Genet. 2006;43:793–797. doi: 10.1136/jmg.2006.041517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Curie A, Lesca G, Cottin V, Edery P, et al. Long-term follow-up in 12 children with pulmonary arteriovenous malformations: confirmation of hereditary hemorrhagic telangiectasia in all cases. J Pediatr. 2007;151:299–306. doi: 10.1016/j.jpeds.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 170.Pollack JS, Saluja S, Thabet A, Henderson KJ, et al. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radiol. 2006;17:35–45. doi: 10.1097/01.RVI.0000191410.13974.B6. [DOI] [PubMed] [Google Scholar]
- 171.Wu JS, Saluja S, Garcia-Tsao G, Chong A, et al. Liver involvement in hereditary hemorrhagic telangiectasia: CT and clinical findings do not correlate in symptomatic patients. Am J Radiol. 2006;187:W399–405. doi: 10.2214/AJR.05.1068. [DOI] [PubMed] [Google Scholar]
- 172.Garcia-Tsao G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT) J Hepatol. 2007;46:499–507. doi: 10.1016/j.jhep.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 173.Lerut J, Orlando G, Adam R, Sabba C, et al. Liver transplantation or hereditary hemorrhagic telangiectasia. Ann Surg. 2006;244:854–62. doi: 10.1097/01.sla.0000247258.35406.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Longacre AV, Gross CP, Gallitelli M, Henderson KJ, et al. Diagnosis and management of gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterol. 2003;98:59–65. doi: 10.1111/j.1572-0241.2003.07185.x. [DOI] [PubMed] [Google Scholar]
- 175.Gershon AS, Faughnan ME, Chon KS, Pugash RA, et al. Transcatheter embolotherapy of maternal pulmonary arteriovenous malformations during pregnancy. Chest. 2001;119:470–77. doi: 10.1378/chest.119.2.470. [DOI] [PubMed] [Google Scholar]
Holt-Oram syndrome
- 176.McDermott DA, Bressan MC, He J, Lee JS, et al. TBX5 genetic testing validates strict clinical criteria for Holt-Oram syndrome. Pediatr Res. 2005;58:981–6. doi: 10.1203/01.PDR.0000182593.95441.64. [DOI] [PubMed] [Google Scholar]
- 177.Csaba E, Marta V, Endre C. Holt-Oram syndrome. Orv Hetil. 1991;132:73–78. [PubMed] [Google Scholar]
- 178.Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet. 1997;15:21–29. doi: 10.1038/ng0197-21. [DOI] [PubMed] [Google Scholar]
- 179.Varma PK, Padmakumar R, Harikrishnan s, Koshy T, et al. Holt-Oram syndrome with hemiazygous continuation of the inferior vena cava. Asian Cardiovasc Thorac Ann. 2002;14:161–163. doi: 10.1177/021849230601400218. [DOI] [PubMed] [Google Scholar]
- 180.Vaughan CJ, Basson CT. Molecular determinants of atrial and ventricular septal defects and patent ductus arteriosus. Am J Med Genet. 2000;97:304–309. doi: 10.1002/1096-8628(200024)97:4<304::aid-ajmg1281>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 181.Basson CT, Cowley GS, Solomon SD, Weissman B, et al. The clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndrome) N Engl J Med. 1994;330:885–891. doi: 10.1056/NEJM199403313301302. [DOI] [PubMed] [Google Scholar]
- 182.Basson CT, Bachinsky DR, Lin RC, Levi T, et al. Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet. 1997;15:30–35. doi: 10.1038/ng0197-30. [DOI] [PubMed] [Google Scholar]
- 183.Basson CT, Huang T, Lin RC, Bachinsky DR, et al. Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc Natl Acad Sci USA. 1999;96:2919–2924. doi: 10.1073/pnas.96.6.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Muller LM, De Jong G, Van Heerden KM. The antenatal ultrasonographic detection of the Holt-Oram syndrome. S Afr Med J. 1985;68:313–5. [PubMed] [Google Scholar]
- 185.Tongsong T, Chanprapaph P. Prenatal sonographic diagnosis of Holt-Oram syndrome. J Clin Ultrasound. 2000;28:98–100. doi: 10.1002/(sici)1097-0096(200002)28:2<98::aid-jcu8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 186.Porter CJ, Feldt RH, Edwards WD. Atrial septal defects. In: Allen HD, Driscoll DJ, Shaddy RE, Feltes TF, editors. Moss and Adams Heart Disease in Infants, Children, and Adolescents, including the Fetus and Young Adult. 7. Philadelphia: Lippincott Williams & Wilkins; 2008. pp. 632–646. [Google Scholar]
- 187.McDaniel NL, Gutgesell HP. Ventricular septal defects. In: Allen HD, Driscoll DJ, Shaddy RE, Feltes TF, editors. Moss and Adams Heart Disease in Infants, Children, and Adolescents Including the Fetus and Young Adult. 7. Philadelphia: Lippincott Williams & Wilkins; 2008. pp. 667–683. [Google Scholar]
- 188.Rigby ML. Atrial septal defects. In: Gatzoulis MA, Webb GP, Daubeney PEP, editors. Diagnosis and Management of Adult Congenital Heart Disease. Edinburgh, Scotland: Churchill Livingston; 2003. pp. 163–178. [Google Scholar]
- 189.Hatcher CJ, Basson CT. Holt-Oram syndrome and the TBX5 transcription factor in cardiogenesis. In: Berul CI, Towbin JA, editors. Molecular genetics of cardiac electrophysiology. Kluwer Academic Publisher; Boston: 2000. pp. 297–315. [Google Scholar]
- 190.Tucker KJ, Murphy J, Conti JB, Curtis AB. Syncope and sinus arrest associated with upper limb-cardiovascular (Holt-Oram) syndrome. Pace. 1994;17:1678–1680. doi: 10.1111/j.1540-8159.1994.tb02364.x. [DOI] [PubMed] [Google Scholar]
Noonan syndrome
- 191.Tartaglia M, Mehler EL, Goldberg R, Zampino G, et al. Mutations in PTPN11, encoding the protein tyrosine, phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 192.Zenker M, Buheitel G, Rauch R, Koenig R, et al. Genotype-phenotype correlations in Noonan syndrome. J Pediatr. 2004;144:368–374. doi: 10.1016/j.jpeds.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 193.Jongmans M, Sistermans EA, Rikken A, Nillesen WM, et al. Genotypic and phenotypic characterization of Noonan syndrome: new data and review of the literature. Am J Med Genet A. 2005;134:165–70. doi: 10.1002/ajmg.a.30598. [DOI] [PubMed] [Google Scholar]
- 194.Schubbert S, Zenker M, Rowe SL, Boll S, et al. KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–336. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 195.Carta C, Pantaleoni F, Bocchinfuso G, Stella L, et al. Germline missense mutations affecting KRAS Isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet. 2006;9:129–135. doi: 10.1086/504394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zenker M, Lehmann K, Schulz AL, Barth H, et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet. 2007;44:131–135. doi: 10.1136/jmg.2006.046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Roberts AE, Araki T, Swanson KD, Montgomery KT, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70–74. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- 198.Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 199.Pandit B, Sarkozy A, Pennacchio LA, Carta C, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–1012. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- 200.Razzaque MA, Nishizawa T, Komoike Y, Yagi H, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39:1013–1017. doi: 10.1038/ng2078. [DOI] [PubMed] [Google Scholar]
- 201.Sarkozy A, Conti E, Seripa D, Digilio MC, et al. Correlation between PTPN11 gene mutations and congenital heart defects in Noonan and LEOPARD syndromes. J Med Genet. 2003;40:704–708. doi: 10.1136/jmg.40.9.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Keren B, Hadchouel A, Saba S, Sznajer Y, et al. for the French Collaborative Noonan Study Group. PTPN11 mutations in patients with LEOPARD syndrome:A French multicentric experience. J Med Genet. 2004;41:e117. doi: 10.1136/jmg.2004.021451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Noonan JA. Noonan syndrome and related disorders. Prog Pediatr Cardiol. 2005a;20:177–185. [Google Scholar]
- 204.Noonan JA. Noonan Syndrome. In: Goldstein-Reynolds, editor. Handbook of Neurodevelopmental and Genetic Disorders in Adults. The Gilford Press; New York-London: 2005b. pp. 308–319. Chapter 15. [Google Scholar]
- 205.Digilio MC, Marino B. Clinical Manifestations of Noonan syndrome. Images Paediatr Cardiol. 2001;7:19–30. [PMC free article] [PubMed] [Google Scholar]
- 206.Allanson JE. Noonan syndrome. Am J Med Genet Part C Semin Med Genet. 2007;145C:274–279. doi: 10.1002/ajmg.c.30138. [DOI] [PubMed] [Google Scholar]
- 207.Sharland M, Burch M, McKenna WM, Patton MA. A clinical study of Noonan syndrome. Arch Dis Child. 1992;67:178–183. doi: 10.1136/adc.67.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Burch M, Sharland M, Shinebourne E, Smith G, et al. Cardiologic abnormalities in Noonan syndrome: Phenotypic diagnosis and echocardiographic assessment of 118 patients. J Am Coll Cardiol. 1993;22:1189–1192. doi: 10.1016/0735-1097(93)90436-5. [DOI] [PubMed] [Google Scholar]
- 209.Marino B, Digilio MC, Toscano A, Gionnotti A, et al. Congenital heart disease in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr. 1999;135:703–706. doi: 10.1016/s0022-3476(99)70088-0. [DOI] [PubMed] [Google Scholar]
- 210.Sznajer Y, Keren B, Baumann C, Pereira S, et al. The spectrum of cardiac anomalies in Noonan syndrome as a result of mutations in the PTPN11 gene. Pediatrics. 2007;119:e1325–31. doi: 10.1542/peds.2006-0211. [DOI] [PubMed] [Google Scholar]
- 211.Hirsch HD, Gelband H, Garcia O, Gottlieb S, et al. Rapidly progressive obstructive cardiomyopathy in infants with Noonan’s syndrome. Circulation. 1975;52:1161–1165. doi: 10.1161/01.cir.52.6.1161. [DOI] [PubMed] [Google Scholar]
- 212.Danetz JS, Donofrio MT, Embrey RP. Multiple left-sided cardiac lesions in one of Noonan’s original patients. Cardiol Young. 1999;9:610–612. doi: 10.1017/s1047951100005679. [DOI] [PubMed] [Google Scholar]
- 213.Shaw AC, Kalidas K, Crosby AH, Jeffery S, et al. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. 2006;92:128–132. doi: 10.1136/adc.2006.104547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. New Engl J Med. 1997;336:775–785. doi: 10.1056/NEJM199703133361107. [DOI] [PubMed] [Google Scholar]
- 215.Ostman-Smith I, Wettrell G, Riesenfeld R. A cohort study of childhood hypertrophic cardiomyopathy. Improved survival following high-dose beta-adrenoreceptor antagonist treatment. J Am Coll Cardiol. 1999;34:1813–1833. doi: 10.1016/s0735-1097(99)00421-0. [DOI] [PubMed] [Google Scholar]
- 216.Carter JE, Feldman T, Carroll JD. Sustained reversal of right-to-left atrial septal defect flow after pulmonic valvuloplasty in an adult. Eur Heart J. 1994;15:575–576. doi: 10.1093/oxfordjournals.eurheartj.a060545. [DOI] [PubMed] [Google Scholar]
- 217.Zanchetta M, Colonna S, Rigatelli G, Pedon L, et al. Combined catheter-based pulmonary valvuloplasty and atrial septal defect closure in Noonan syndrome. Minerva Cardioangiol. 2002;50:383–388. [PubMed] [Google Scholar]
- 218.Loukas M, Dabrowski M, Kantoch M, Ruzyllo W, et al. A case report of Noonan’s syndrome with pulmonary valvar stenosis and coronary aneurysms. Med Sci Monit. 2004;10:CS80–83. [PubMed] [Google Scholar]
- 219.Schacter N, Perloff JK, Mulder DG. Aortic dissection in Noonan’s syndrome (46 XY) Am J Cardiol. 1984;54:464–465. doi: 10.1016/0002-9149(84)90228-5. [DOI] [PubMed] [Google Scholar]
- 220.Morgan JM, Coupe MO, Honey M, Miller GAH. Aneurysms of the sinuses of Valsalva in Noonan’s syndrome. Eur Heart J. 1989;10:190–193. doi: 10.1093/oxfordjournals.eurheartj.a059462. [DOI] [PubMed] [Google Scholar]
- 221.Purnell R, Williams I, Von Oppell U, Wood A. Giant aneurysms of the sinuses of Valsalva and aortic regurgitation in a patient with Noonan’s syndrome. Eur J Cardiothorac Surg. 2005;28:346–348. doi: 10.1016/j.ejcts.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 222.Power PD, Lewin MB, Hannibal MA, Glass IA. Aortic dilatation is a rare complication of Noonan Syndrome. Pediatr Cardiol. 2006;27:478–480. doi: 10.1007/s00246-006-1210-x. [DOI] [PubMed] [Google Scholar]
- 223.Doyama K, Hirose K, Fujiwara H. Asymmetric septal hypertrophy in a 41-year-old woman with Noonan’s syndrome. Chest. 1990;97:1480–1481. doi: 10.1378/chest.97.6.1480. [DOI] [PubMed] [Google Scholar]
- 224.Shimizu A, Oku Y, Matsuo K, Hashiba K. Hypertrophic cardiomyopathy progressing to a dilated cardiomyopathy-like feature in Noonan’s syndrome. Am Heart J. 1992;123:814–816. doi: 10.1016/0002-8703(92)90534-3. [DOI] [PubMed] [Google Scholar]
- 225.Hayashi S, Tojyo K, Uchikawa S, Momose T, et al. Biventricular hypertrophic cardiomyopathy with right ventricular outflow tract obstruction associated with Noonan syndrome in an adult. Jpn Circ J. 2001;65:132–135. doi: 10.1253/jcj.65.132. [DOI] [PubMed] [Google Scholar]
- 226.Wilmshurst PT, Katritsis D. Restrictive and hypertrophic cardiomyopathies in Noonan syndrome: the overlap syndromes. Heart. 1996;75:94–97. doi: 10.1136/hrt.75.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Cooke RA, Chambers JB, Curry PVL. Noonan’s cardiomyopathy: a non-hypertrophic variant. Br Heart J. 1994;71:561–565. doi: 10.1136/hrt.71.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.O’Sullivan T, Hally M, Cronin CC, Bashyam M, et al. Constrictive pericarditis in a patient with the Noonan syndrome. Ir J Med Sci. 1993;162:180–181. doi: 10.1007/BF02945180. [DOI] [PubMed] [Google Scholar]
- 229.Okahata H, Nishi Y, Yoshimitsu H, Tanimoto T, Usui T. A case of Noonan’s with primary hypertension. Hiroshima J Med Science. 1982;31:27–29. [PubMed] [Google Scholar]
- 230.Tinker A, Uren N, Schofield J. Severe pulmonary hypertension in Ullrich-Noonan syndrome. Brit Heart J. 1989;62:74–77. doi: 10.1136/hrt.62.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Huff DS, Zackai EH. Fatal pulmonary hypertension in a 17 year old girl with Noonan’s syndrome: a third case. Proc Greenwood Gen Cr. 2004;23:153. [Google Scholar]
- 232.Cullimore AJ, Smedstad KG, Brennan BG. Pregnancy in women with Noonan syndrome:report of two cases. Obstet Gynecol. 1999;93:813–816. doi: 10.1016/s0029-7844(98)00413-x. [DOI] [PubMed] [Google Scholar]
- 233.Yadav BS, Indurkar M, Bisarya BN. Normal pregnancy in a case of Noonan syndrome with hypertrophic cardiomyopathy. J Assoc Physicians India. 1996;44:494–495. [PubMed] [Google Scholar]
- 234.Joo JG, Beke A, Toth-Pal E, Csaba A, et al. Successful pregnancy in a Noonan syndrome patient after 3 unsuccessful pregnancies from severe fetal hydrops:a case report. J Reprod Med. 2005;50:373–376. [PubMed] [Google Scholar]
- 235.Grange CS, Heide R, Lucas SB, Ross PL, et al. Anaesthesia in a patient with Noonan’s syndrome. Can J Anaesth. 1998;45:332–336. doi: 10.1007/BF03012024. [DOI] [PubMed] [Google Scholar]
Reproduction and Genetic Counseling
- 236.Canobbio MM, Rapkin AJ, Perloff JK, Lin A, Child JS. Menstrual patterns in women with congenital heart disease. Pediatr Cardiol. 1995;16:12–15. doi: 10.1007/BF02310327. [DOI] [PubMed] [Google Scholar]
- 237.Canobbio MM, Perloff JK, Rapkin AJ. Gynecological health of females with congenital heart disease. Inter J Cardiol. 2005;98:379–387. doi: 10.1016/j.ijcard.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 238.Vessey M, Mant J, Smith A, Yeates D. Oral contraceptive and venous thromboembolism: indices in a large prospective study. BMJ. 1986;292:526. doi: 10.1136/bmj.292.6519.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Hoess K, Goldmuntz E, Pyeritz RE. Genetic counseling for congenital heart disease: new approaches for a new decade. Curr Cardiol Rep. 2002;4:68–75. doi: 10.1007/s11886-002-0129-y. [DOI] [PubMed] [Google Scholar]
- 240.Van Rijen EH, Utens EM, Roos-Hesselink JW, Merijboom FJ, van Domdurg RT, Roelandt JR, Bogers AJ, Verhulst FC. Current subjective state of health, and longitudinal psychological well-being over a period of 10 years, in a cohort of adults with congenital cardiac disease. Cardiol Young. 2005;15:168–175. doi: 10.1017/S1047951105000351. [DOI] [PubMed] [Google Scholar]
- 241.Rizk DE, Deb P. A spontaneous and uneventful pregnancy in a Turner mosaic with previous recurrent miscarriages. J Pediatr Adolesc Gynecol. 2003;16:87–88. doi: 10.1016/s1083-3188(03)00009-3. [DOI] [PubMed] [Google Scholar]
- 242.Kaneko N, Kawagoe S, Hiroi M. Turner’s syndrome – review of the literature with reference to a successful pregnancy outcome. Gynecol Obstet Invest. 1990;29:81–87. doi: 10.1159/000293307. [DOI] [PubMed] [Google Scholar]
- 243.King CR, Magenis E, Bennett S. Pregnancy and the Turner syndrome. Obstet Gynecol. 1978;52:617–624. [PubMed] [Google Scholar]
- 244.Allan L. Prentatal diagnosis of structural cardiac defects. Am J Med Genet Part C Semin Med Genet. 2007;145C:73–76. doi: 10.1002/ajmg.c.30123. [DOI] [PubMed] [Google Scholar]
- 245.Sonek J. First trimester ultrasonography in screening and detection of fetal anomalies. Am J Med Genet Part C Semin Med Genet. 2007;145C:45–61. doi: 10.1002/ajmg.c.30120. [DOI] [PubMed] [Google Scholar]
- 246.Breathnach FM, Fleming A, Malone FD. The second trimester genetic sonogram. Am J Med Genet Part C Semin Med Genet. 2007;145C:62–72. doi: 10.1002/ajmg.c.30116. [DOI] [PubMed] [Google Scholar]
- 247.Spencer K. Aneuploidy screening in the first trimester. Am J Med Genet Part C Semin Med Genet. 2007;145C:18–32. doi: 10.1002/ajmg.c.30119. [DOI] [PubMed] [Google Scholar]
- 248.ACOG Committee on Genetics (2) ACOG committee opinion. Number 298, August 2004. Prenatal and preconceptional carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstet Gynecol. 2004;104:425–8. doi: 10.1097/00006250-200408000-00050. [DOI] [PubMed] [Google Scholar]
- 249.Committee on Genetics, American College of Obstestricians and Gynecologists (1) ACOG Committee Opinion. Number 325, December 2005. Update on carrier screening for cystic fibrosis. Obstet Gynecol. 2005;106:1465–1468. doi: 10.1097/00006250-200512000-00055. [DOI] [PubMed] [Google Scholar]
- 250.Kuliev A, Verlinsky Y. Place of preimplantation diagnosis in genetic practice. Am J Med Genet. 2005;134:105–110. doi: 10.1002/ajmg.a.30635. [DOI] [PubMed] [Google Scholar]
- 251.Thornhill AR, deDie-Smudlers CE, Geraedts JO, Harper JC, Harton GL, Lavery SA, Moutou C, Robinson MD, Schmutzler AG, Scriven PN, Sermon KD, Wilton L ESHRE PGD Consortium. ESHRE PGD Consortium “Best practice guidelines for clinical preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS)”. Hum Reprod. 2005;20:35–48. doi: 10.1093/humrep/deh579. [DOI] [PubMed] [Google Scholar]
- 252.Press F, Shapiro HM, Cowell CA, Oliver GD. Outcome of ovum donation in Turner’s syndrome patients. Fertil Steril. 1995;64:995–998. [PubMed] [Google Scholar]
- 253.Bui TH, Harper JC. Preimplantation genetic diagnosis. Clin Obstet Gynecol. 2002;45:640–648. doi: 10.1097/00003081-200209000-00007. [DOI] [PubMed] [Google Scholar]
- 254.Jain T, Harlow BL, Hornstein MD. Insurance coverage and outcomes of in vitro fertilization. N Eng J Med. 2002;347:661–666. doi: 10.1056/NEJMsa013491. [DOI] [PubMed] [Google Scholar]
- 255.Lavery SA, Aurell R, Turner C, Castello C, Veiga A, Barri PN, Winston RM. Preimplantation genetic diagnosis: Patients’ experiences and attitudes. Hum Reprod. 2002;17:2464–2467. doi: 10.1093/humrep/17.9.2464. [DOI] [PubMed] [Google Scholar]
Transitioning care
- 256.American Academy of Pediatrics; American Academy of Family Physicians; American College of Physicians-American Society of Internal Medicine. A consensus statement on health care transitions for young adults with special health care needs. Pediatrics. 2002;110:1304–6. [PubMed] [Google Scholar]
- 257.Donaldson MDC, Gault EJ, Tan KW, Dunger DB. Optimising management in Turner syndrome: from infancy to adult transfer. Arch Dis Child. 2006;91:513–520. doi: 10.1136/adc.2003.035907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Pober BR, Morris CA. Diagnosis and management of medical problems in adults with Williams-Beuren syndrome. 2007. Am J Med Genet Part C Semin Med Genet. 2007;145C:280–290. doi: 10.1002/ajmg.c.30139. [DOI] [PubMed] [Google Scholar]
- 259.Fernandes SM, Landsberg MJ. Transitioning the young adult with congenital heart disease for life-long medical care. Pediatr Clin N Am. 2004;51:1739–1748. doi: 10.1016/j.pcl.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 260.Reid GJ, Irvine MJ, McCrindle BW, Sananes R, et al. Prevalence and correlates of successful transfer from pediatric to adult health care among a cohort of young adults with complex congenital heart defects. Pediatrics. 2004;113:e97–e205. doi: 10.1542/peds.113.3.e197. [DOI] [PubMed] [Google Scholar]
- 261.Chessa M, Arciprete P, Bossone E, Daliento L, et al. A multicentre approach for the management of adults with congenital heart disease. J Cardiovasc Med. 2006;7:701–705. doi: 10.2459/01.JCM.0000243005.13041.2e. [DOI] [PubMed] [Google Scholar]
- 262.Moons P, Van Deyk K, De Bleser L, Marquet K, et al. Quality of life and health status in adults with congenital heart disease: a direct comparison with healthy counterparts. Eur J Cardiovasc Prevent and Rehab. 2006;13:407–413. doi: 10.1097/01.hjr.0000221864.19415.a0. [DOI] [PubMed] [Google Scholar]
- 263.Moons P, Engelfriet P, Kaemmerer H, Meijboom FJ, et al. on behalf of the Expert Committee of Euro Heart Survey on Adult Congenital Heart Disease. Delivery of care for adult patients with congenital heart disease in Europe: results from the Euro Heart Survey. Eur Heart J. 2006;27:1324–1330. doi: 10.1093/eurheartj/ehi858. [DOI] [PubMed] [Google Scholar]
- 264.Reid GJ, Webb GD, Barzel M, McCrindle B, et al. Estimates of life expectancy by adolescents and young adults with congenital heart disease. J Am Coll Cardiol. 2006;48:349–355. doi: 10.1016/j.jacc.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 265.Skorton D, Garson A, Allen HD, Fox JM, et al. Task force 5: Adults With congenital Heart Disease: Access to Care. J Am Coll Cardiol. 2001;37:1161–1198. doi: 10.1016/s0735-1097(01)01274-8. [DOI] [PubMed] [Google Scholar]
- 266.Giampietro P, Greenlee R, McPherson E, Benetti L, et al. Acute health events in adult patients with genetic disorders: The Marshfield Epidemiologic Study Area. Genet Med. 2006;8:474–490. doi: 10.1097/01.gim.0000232479.90268.40. [DOI] [PubMed] [Google Scholar]
Tables
- 267.Battaglia A, Carey JC, Wright TJ. Wolf-Hirschhorn (4p-) syndrome. Adv Pediatr. 2001;48:75–113. [PubMed] [Google Scholar]
- 268.Spinner NB. Genetics of Alagille syndrome. Progr Pediatr Cardiol. 2005;20:169–176. [Google Scholar]
- 269.Roberts A, Allanson J, Jadico SK, Kavamura MI, et al. The cardiofaciocutaneous syndrome. J Med Genet. 2006;43:833–842. doi: 10.1136/jmg.2006.042796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Narumi Y, Aoki Y, Niihori T, Neri G, et al. Molecular and clinical characterization of cardio-facio-cutaneous (CFC) syndrome. Am J Med Genet. 143A:799–807. doi: 10.1002/ajmg.a.31658. Part A. [DOI] [PubMed] [Google Scholar]
- 271.Lin AE, Grossfeld PD, Hamilton R, Smoot L, Proud V, Weksberg R, Gripp K, Wheeler P, Picker J, Irons M, Zackai E, Scott CI, Nicholson L. Further delineation of cardiac anomalies in Costello syndrome. Am J Med Genet. 2002;111:115–129. doi: 10.1002/ajmg.10558. [DOI] [PubMed] [Google Scholar]
- 272.White S, Graham JM, Jr, Kerr B, Gripp K. The adult phenotype in Costello syndrome. Am J Med Genet Part A. 2005;136:128–135. doi: 10.1002/ajmg.a.30747. [DOI] [PubMed] [Google Scholar]
- 273.Hagerman RJ. Fragile X syndrome. In: Cassidy SB, Allanson JE, editors. Management of Genetic Syndromes. 2. New Jersey: John Wiley and Sons; 2005. p. 258. [Google Scholar]
- 274.Surka WS, Kohlhase J, Neunert CE, Schneider DS, et al. Unique family with Townes-Brocks syndrome, SALL1 mutation, and cardiac defects. Am J Med Genet. 2001;102:250–257. doi: 10.1002/1096-8628(20010815)102:3<250::aid-ajmg1479>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 275.Benson DW, Silberbach GM, Kavanaugh-McHugh A, et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Lofreddo CA, Wilson PD, Ferencz C. Maternal diabetes: An independent risk factor for major cardiovascular malformations with increased mortality of affected infants. Teratology. 2001;64:98–106. doi: 10.1002/tera.1051. [DOI] [PubMed] [Google Scholar]
- 277.Levy HL, Guldberg P, Guttler F, et al. Congenital heart disease in maternal phenylketonuria: report from the Maternal PKU Collaborative Study. Pediatr Res. 2001;49:636–42. doi: 10.1203/00006450-200105000-00005. [DOI] [PubMed] [Google Scholar]
- 278.Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, Curry CJ, Fernhoff PM, Grix AW, Jr, Lott IT, et al. Retinoic acid embryopathy. N Engl J Med. 1985;313:837–41. doi: 10.1056/NEJM198510033131401. [DOI] [PubMed] [Google Scholar]
- 279.Honein MA, Paulozzi LJ, Erickson JD. Continued occurrence of Accutane-exposed pregnancies. Teratology. 2001;64:142–7. doi: 10.1002/tera.1057. [DOI] [PubMed] [Google Scholar]