

Abstract
Notch1 is an evolutionarily conserved receptor that regulates cell fate, including such events as differentiation, proliferation, and apoptosis. Myofibroblast differentiation is a key feature of lung fibrosis. Found in inflammatory zone 1 (FIZZ1) has direct fibrogenic properties because of its ability to induce myofibroblast differentiation. However, the downstream signaling pathway that mediates FIZZ1 induction of myofibroblast differentiation remains unknown. The objective of this study was to investigate the involvement of Notch signaling in FIZZ1 induction of lung myofibroblast differentiation and thus explore the potential role of Notch1 in pulmonary fibrosis. The results showed that FIZZ1 increased the expression levels of activated intracellular domain of Notch1 (NIC), its ligand Jagged1, and its target gene Hes1, which were associated with elevated α-smooth muscle actin expression levels. Fibroblast α-smooth muscle actin expression is induced by the overexpression of NIC but is suppressed by the inhibition of NIC. Moreover, lung fibroblasts that were isolated from mice lacking the GDP-4-keto-6-deoxymannose3,5-epimerase-4-reductase enzyme (FX knockout) exhibited significantly reduced responsiveness to FIZZ1, which was reversed by fucose supplementation. In the absence of exogenous fucose, these FX-deficient cells exhibited defective fucosylation, which is required for Notch signaling. These knockout mice also showed impaired lung fibrosis. These findings suggest that Notch1 signaling in response to FIZZ1 may play a significant role in myofibroblast differentiation during lung fibrosis.
Notch was initially identified as a transmembrane signaling protein that is located on the cell surface and is involved in Drosophila neurogenesis. Its large extracellular domain contains 36 tandem epidermal growth factor-like repeats and three cysteine-rich Notch/LIN-12 repeats. Six tandem ankyrin repeats, a glutamine-rich domain (opa), and a PEST sequence are found in the intracellular domain.1,2 On binding to Notch ligands (Jagged or Delta) expressed on adjacent cells, proteolytic cleavage involving γ-secretase releases the intracellular domain of Notch (NIC), which then translocates into the nucleus where it binds to its downstream transcription factor CSL [the mammalian homolog of Su(H), also known as CBF1 or RBP-Jk]. This results in the conversion of CSL from a repressor to an activator with consequent induction of the transcription of Notch-dependent target genes such as Hes-1 (hairy enhancer of split), HRT (hairy-related transcription), Deltes-1, pTα, Meltrin-β, and the Notch receptors themselves.3,4,5,6,7 Several studies implicate Notch signaling in CD4+ Th1 and Th2 cell differentiation.8,9,10 Notch ligands on antigen-presenting cells have distinct effects on T-cell cytokine production. Of the two Notch ligand families, Delta induces Th1, whereas Jagged induces Th2 response and immunity. Expression of these different ligands on antigen-presenting cells is induced by Th1- or Th2-promoting stimuli.11 Individual Notch receptors also induce different responses as Notch3 up-regulates interferon-γ, whereas Notch1 promoted GATA3 and interleukin (IL)-4 expression.12 There is evidence to suggest that Jagged1/Notch1 signaling directs Th2 differentiation by inducing GATA3 and by directly regulating IL-4 gene transcription through the RBP-Jk site.9,10 Th2 differentiation induced by antigen-presenting cells is abrogated in T cells lacking the Notch effector RBP-Jk.8,11,12 This evidence indicates that Jagged1/Notch1 signaling is involved in processes characterized by pathological Th2 cytokine responses such as asthma, tissue repair/remodeling, and pulmonary fibrosis.8,13
Progressive pulmonary fibrosis including idiopathic pulmonary fibrosis/usual interstitial pneumonitis usually has a fatal outcome, and its pathogenesis remains incompletely understood. It is postulated that cytokines and chemokines including transforming growth factor-β (TGF-β) produced by repeatedly injured and activated alveolar epithelial cells recruit and activate adjacent underlying fibroblasts to constitute fibroblast foci.14,15,16,17 Active pulmonary fibrosis is characterized by fibroblast proliferation, de novo emergence of myofibroblasts, extracellular matrix deposition, and tissue remodeling. Myofibroblasts are critical sources of extracellular matrix and fibrogenic cytokine production including TGF-β, which are key features of pulmonary fibrosis.18,19,20,21 Hence elucidating the mechanism of induction of myofibroblast differentiation may provide novel insight into pathogenesis of pulmonary fibrosis. TGF-β and a recently identified novel mediator, found in inflammatory zone (FIZZ1), also known as resistin-like molecule-α, or RELM-α, are known inducers of myofibroblast differentiation and stimulate α-smooth muscle actin (α-SMA) expression in lung fibroblasts.22 Previous reports show that FIZZ1 is primarily expressed by airway epithelial cells and alveolar epithelial cells, and has direct fibrogenic properties by its ability to induce myofibroblast differentiation and α-SMA expression without affecting cell proliferation.22,23 The expression of FIZZ1 by alveolar epithelial cells is driven by Th2-type cytokines IL-4 and IL-13. The predominance of Th2-type responses in pulmonary fibrosis has been amply demonstrated.24,25,26,27 Moreover, the induction of FIZZ1 in epithelial cells by Th2-type cytokines could play an alternative or additional (to that of TGF-β for example) role in induction of myofibroblast differentiation in pulmonary fibrosis and remodeling.28,29 FIZZ1 is also highly up-regulated in a mouse model of hypoxia-induced pulmonary hypertension, where it is referred to as hypoxia-induced mitogenic factor (HIMF), FIZZ1/HIMF has mitogenic, angiogenic, and vasoconstrictive properties on pulmonary microvascular smooth muscle cells and endothelial cells via PI-3K/AKT signaling pathway.30,31,32,33 More recently, Bruton’s tyrosine kinase (BTK) was identified as a functional HIMF binding partner in primary cultured murine bone marrow cells through glutathione S-transferase-HIMF pull-down studies and mass spectrometry.34 However, the downstream signaling pathway mediating FIZZ1 induction of myofibroblast differentiation from lung fibroblasts, a key event in the propagation of fibrosis, remains essentially unexplored.
Recent reports show that Jagged1/Notch signaling induces epithelial-to mesenchymal transitions by interactions with the TGF-β/Smad pathway.35,36 Several studies also show that Notch signaling regulates α-SMA expression differentially via CSL or HRT in multiple cell types, including smooth muscle cells and mesenchymal fibroblasts, respectively.37,38,39,40,41 Because Notch1 is shown to mediate activation of Th2 differentiation,11 which is a predominant response in pulmonary fibrosis, the objective of this study was to clarify the activation of Jagged1/Notch1 signaling in response to FIZZ1 and analyze its role in myofibroblast differentiation in pulmonary fibrosis. These results for the first time showed the importance of Notch1 signaling in FIZZ1-induced myofibroblast differentiation and bleomycin (BLM)-induced pulmonary fibrosis.
Materials and Methods
Animals and Induction of Pulmonary Fibrosis
C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). FX knockout (KO) mice on C57BL/6 background were produced as previously described.42 These mice were propagated at the University of Michigan. To induce pulmonary fibrosis, BLM (Blenoxane, Mayne Pharma Inc, Paramus, NJ) was dissolved in sterile phosphate-buffered saline (PBS) and instilled endotracheally at a dose of 1.5 U/kg body weight. Control groups received PBS only (three to five mice per group). One half of the animals in all groups were fed either chow depleted of fucose (Notch-deficient) 1 month before BLM/saline treatment or chow replete with fucose (Notch-sufficient). At the indicated time points after BLM treatment, the mice were sacrificed and the lungs were harvested rapidly. For histological analysis, the lungs were formalin-fixed, and stained with routine hematoxylin and eosin (H&E). All animal studies have been reviewed and approved by the University of Michigan Committee on Use and Care of Animals at the University of Michigan.
Lung Fibroblast Isolation and Treatment
Fibroblasts were isolated from lung tissue by trypsinization as described previously.43 The cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% plasma-derived fetal bovine serum (Animal Technologie, Tyler, TX) 10 ng/ml of epidermal growth factor, and 5 ng/ml of platelet-derived growth factor (R&D Systems, Inc., Minneapolis, MN). Cells were used at passage 3 to 5 after primary culture. After plating as indicated, the cells were made quiescent by culturing in Dulbecco’s modified Eagle’s medium containing 0.5% plasma-derived fetal bovine serum for 24 hours when cells reached ∼85% confluent. Where indicated, recombinant mouse FIZZ1 (rmFIZZ1; Leinco Technologies, Inc., St. Louis, MO) were added at the indicated doses. In experiments to assess the role of γ-secretase inhibitor, 5 μmol/L of γ-secretase inhibitor X (Calbiochem, San Diego, CA) was added 24 hours before the addition of rmFIZZ1.
Plasmid Constructs and Cell Transfection
Human NIC and Jagged1 constructs pcDNA3.0-NIC and pcDNA3.1-Jagged1 expression plasmids were kind gifts from Dr. Lucio Miele (Cardinal Bernardin Cancer Center, Loyola University Medical Center, Maywood, IL) and Dr. Igor Prudovsky (Center for Molecular Medicine, Maine Medical Center Research Institute, Scarborough, ME). Notch1 siRNA (5′-UGGCUUGCAGUAGCAAGGAAGCUAA-3′) and control siRNA (5′-UGGACGUGAUGGAACGAAGCUCUAA-3′) were self-designed and purchased from Invitrogen (Carlsbad, CA). Predesigned Jagged1 siRNA and its negative control siRNA were purchased from Ambion, Inc. (Austin, TX). Primary cultured lung fibroblasts were transfected with 2 μg of either pcDNA3.0-NIC or pcDNA3.1-Jagged1 by electroporation Nucleofector II (Amaxa Biosystem, Gaithersburg, MD). After 20 hours, rmFIZZ1 was added and incubated for another 48 hours. Plasmid pcDNA3.0 or pcDNA3.1 was used as negative controls. Transfection with 100 nmol/L of Notch1, Jagged1 siRNAs (Ambion, Inc.) and their control siRNAs were conducted as described above with siRNA test kit (Amaxa Biosystem).
α-SMA Promoter Construct and Activity Assay
The α-SMA promoter was cloned by polymerase chain reaction (PCR) from rat genomic DNA as previously described.44 The promoter activity was evaluated by a dual-luciferase reporter assay kit (Promega, Madison, WI). Briefly, normal rat lung fibroblasts were co-transfected with 2 μg of α-SMA promoter construct and pcDNA-NIC or 100 nmol/L of Notch 1 siRNA along with 50 ng of control vector pRL-CMV in transfect reagent Fugene6 (Roche, Indianapolis, IN). FIZZ1 was added as indicated, and the cells were harvested 48 hours after transfection. Firefly luciferase (Luc) and Renilla reniformis luciferase (RlLuc) activities were determined with microplate luminometer (Turner Designs, Sunnyvale, CA). Luc activity was normalized to RlLuc activity and shown as relative luciferase activity.
mRNA Analysis by Quantitative Reverse Transcriptase (RT)-PCR
For mRNA analysis, total RNA was isolated from lung tissue or fibroblasts. Primer Express 2.0 software (Applied Biosystems, Foster City, CA) was used to design TaqMan primers and MGB probes. The primer sequences were as follows: α-SMA: forward primer, 5′-CAACAGGATGAAGACTGCAACCT-3′; reverse primer, 5′-GGGACCATCAGCTAAAGAAG-3′; probe, 5′-6FAM-CCCTTCTCATCTGCGTCT-3′. Type I procollagen (collagen I): forward primer, 5′-CAACCGTGCTTCTCAGAACAT-3′; reverse primer, 5′-TGCCCGTCTCCTCATCCA-3′; probe, 5′-6FAM-ACCACTGCAAGAAC-3′. FIZZ1: forward primer, 5′-TCCAGCTAACTATCCCTCCACTGT-3′; reverse primer, 5′-GGCCCATCTGTTCATAGTCTTGA-3′; probe, 5′-6FAM-CGAAGACTCTCTCTTGC-3′. Jagged1: forward primer, 5′-GTCTTCCCCTTGTGCCTTTG-3′; reverse primer, 5′-ATGTCCTGGAGGGCAGATACA-3′; probe, 5′-6FAM-CACCTGTGTGGATGAGA-3′. Primers and probe for murine tumor necrosis factor (TNF)-α, TGF-β, MCP-1, IL-4, Delta4, and GAPDH were purchased from Applied Biosystems. GAPDH mRNA was used as internal control. One-step real time qPCR was undertaken by using a GeneAmp 7500 sequence detection system (Applied Biosystems). Results were expressed as 2−ΔΔCT as previously described.45
Western Blotting Analysis and Immunofluorescence Staining
To detect protein expressions, 20 μg (or 5 μg for α-SMA only) of cell or tissue lysates were separated on SDS-PAGE. Mouse anti-collagen I and anti-α-SMA (Sigma, St. Louis, MO), rabbit anti-Notch1 and NIC (Calbiochem), rabbit anti-Jagged1 (Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit anti-Hes1 (Chemicon International, Temecula, CA) antibodies were used as primary antibodies. The immunostained bands were visualized using a chemiluminescent substrate (Cell Signaling Technology, Beverly, MA) followed by exposure to ECL Hyperfilm (Amersham Biosciences, Buckinghamshire, UK). α-SMA immunofluorescence staining was performed on four-well chamber slides with 3 × 104 cells using mouse anti α-SMA −Cy5 (Sigma) as previously described.19 The slides were examined under fluorescence microscopy.
Hydroxyproline Assay
Lung collagen deposition was estimated by measuring the hydroxyproline content of whole lung homogenates as previously described.46 Briefly, the lungs were excised, homogenized in 0.5 mol/L acetic acid, and hydrolyzed in 6 N HCl overnight at 110°C. Hydroxyproline was assessed by colorimetric assay and the results were expressed as μg of hydroxyproline per lung.
Statistical Analysis
All data were expressed as mean ± SE. Differences between means of various treatment groups were assessed for statistical significance by analysis of variance followed by post hoc analysis using Scheffé’s test. A P value <0.05 was considered to indicate statistical significance.
Results
FIZZ1 Activation of Notch1 Signaling in Lung Fibroblasts
FIZZ1 is known to promote myofibroblast differentiation in lung fibroblasts,22 but the intracellular signaling pathway mediating this effect is unknown. To initially assess if FIZZ1 activates Notch1 signaling, isolated murine lung fibroblasts were treated with the indicated doses of recombinant murine FIZZ1 and analyzed for activation of components of Notch signaling. The results showed an increase in protein expression of Notch1, NIC, Jagged 1, and Hes1 after 1 hour of treatment with 25 ng/ml of FIZZ1 (Figure 1A). This increase remained evident at 24 hours of treatment (data not shown). Thus components of this signaling pathway showed evidence of activation, from the generation of NIC to its best-characterized direct transcriptional target, Hes1. Moreover Notch1 signaling is activated through cell-cell interaction via binding its ligands expressed on neighboring cells,5 and in this experiment, the Notch1 ligand Jagged1 was also up-regulated as early as 20 minutes after treatment with FIZZ1 (data not shown). This increase in Jagged1 expression was confirmed by real-time qPCR detection of Jagged1 mRNA expression. The results showed that FIZZ1 treatment caused significant up-regulation as early as 15 minutes, which was sustained up to 30 minutes, and then declined to control levels at 1 hour (Figure 1B). In contrast, Delta4, another Notch ligand, was not induced by FIZZ1 treatment (data not shown). These results taken together indicated that FIZZ1 was able to activate Notch1 signaling, as well as their downstream target Hes1, likely via activation of its ligand, Jagged1.
Figure 1.

Effect of FIZZ1 treatment on lung fibroblast-expressed Notch1 signaling components. Normal murine lung fibroblasts were treated with the indicated doses of rmFIZZ1 for 1 hour, or as indicated. A: Cell lysates (20 μg protein each) were analyzed for Notch1, NIC1, Jagged1, and Hes1 proteins by Western blotting. Representative blots from three independent experiments are shown. The GAPDH protein served as a loading control. B: Total cell RNA was isolated at the indicated time points and analyzed for Jagged1 mRNA by real-time qPCR. The amount of Jagged1 mRNA was calculated as 2−ΔΔCT equivalent to fold change over the untreated control sample (calibrator) using GAPDH as the reference. The effect of FIZZ1 treatment was statistically significant at all time points shown, except for the 1-hour time point. Mean ± SE of triplicate samples are shown.
FIZZ1 Up-Regulation of α-SMA Expression Requires Jagged1
The induction of Jagged1 by FIZZ1 treatment (Figure 1) suggests its importance in mediating the downstream effect on α-SMA expression. To evaluate such a possibility, the effect of induced overexpression of Jagged1 on lung fibroblast α-SMA expression was examined. The results showed that transfection with the Jagged1 expression plasmid increased α-SMA expression in lung fibroblasts 48 hours after transfection compared with the control empty vector (Figure 2A). This Jagged1-induced response was further increased by FIZZ1 treatment, suggesting that indeed FIZZ1 could induce α-SMA expression via up-regulation of Jagged1. To further confirm this possibility, the effect of Jagged1 knockdown using specific siRNA on the FIZZ1 response was evaluated. The results showed that specific knockdown of Jagged1 expression resulted in the suppression of α-SMA expression that was not reversible by treatment with FIZZ1 (Figure 2B). This suppression of α-SMA by Jagged1 siRNA was accompanied by inhibition of NIC expression (Figure 2B). These findings confirmed the importance of Jagged1 in mediating FIZZ1 activation of α-SMA expression. Although activation of Jagged1 is expected to act upstream of Notch activation, transfection of NIC was noted to result also in up-regulation of Jagged1 expression (Figure 2C). In contrast the γ-secretase inhibitor, which prevents release of NIC,11 was able to inhibit FIZZ1-activated Jagged1 (Figure 2D). Because FIZZ1 treatment resulted in generation of NIC (Figure 1A), this finding would suggest that a positive feedback effect of NIC on Jagged1 induction may contribute to the propagation of Notch1 signaling.
Figure 2.
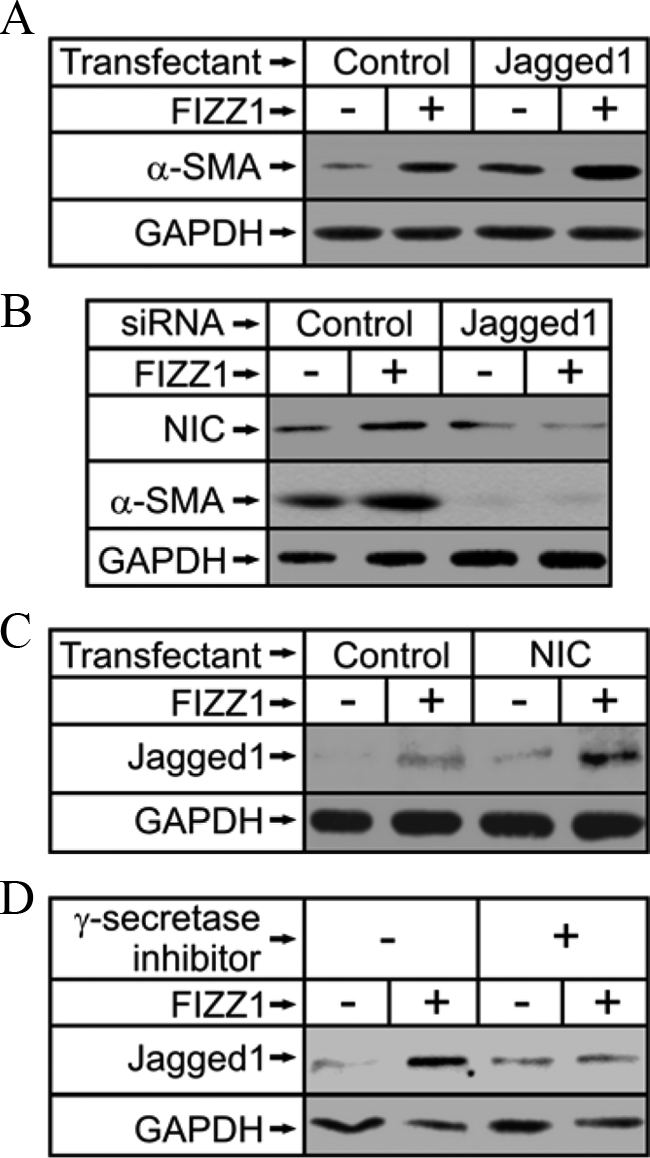
Role of Jagged1 in FIZZ1 induction of α-SMA expression. A: Fibroblasts were transfected as indicated with either Jagged1 expression plasmid or its empty vector control, pcDNA3.1, and then treated with buffer only or FIZZ1. The cells were then harvested for analysis of α-SMA protein expression by Western blotting. B: Alternatively, cells were transfected with Jagged1 or control siRNA as indicated, and then treated with FIZZ1 or buffer only. The cell extracts were then similarly analyzed for α-SMA and NIC1 protein. C: NIC1 (NIC)- or empty vector control (Control)-transfected fibroblasts were treated with rmFIZZ1 or buffer and then analyzed for Jagged1 protein expression by Western blotting. D: Cells were similarly treated as in C, except instead of transfection with the NIC1 expression plasmid, 5 μmol/L of γ-secretase inhibitor X was used to suppress the generation of NIC. Representative blots from at least three independent experiments are shown.
Notch1 Mediation of FIZZ1 Induced α-SMA Expression in Lung Fibroblasts
Given this ability of FIZZ1 to activate Notch1 signaling, its importance in mediating FIZZ1 induction of lung myofibroblast differentiation22 was then evaluated. First, the ability of induced overexpression of NIC to affect α-SMA expression was evaluated. Transfection with a NIC expression plasmid into lung fibroblasts caused an increase in α-SMA protein expression, in the absence or presence of added FIZZ1 treatment (Figure 3A). Overexpression of NIC alone was sufficient to cause an increase in α-SMA expression. The control or empty vector did not show any significant effect on α-SMA expression. This was also observed by immunofluorescence staining showing more α-SMA-expressing cells and brighter stained signals in cells with overexpressed NIC than that in the cells with control vector (Figure 3B). To further confirm the importance of Notch1 signaling in the observed induction of α-SMA, the effect of γ-secretase inhibition on α-SMA expression was evaluated. Treatment with 5 μmol/L of γ-secretase inhibitor X caused a marked reduction in fibroblast α-SMA expression, which could not be reversed by treatment with FIZZ1 (Figure 3C). As expected NIC expression was also markedly reduced by treatment with this inhibitor (data not shown). To further demonstrate that this inhibitor effect was specifically because of reduction in NIC, the effect of a specific siRNA to Notch1 on α-SMA expression was examined. The results showed that specific knockdown of Notch1 by siRNA transfection caused a similar marked reduction in α-SMA expression, which again was not affected by additional treatment with FIZZ1 (Figure 3D). Treatment with the control RNA had no significant effect on α-SMA expression, which responded to FIZZ1 treatment. Transfection with the specific Notch1 siRNA reduced NIC expression by 70% (data not shown).
Figure 3.

Role of NIC1 in regulation of α-SMA gene expression. A: Fibroblasts were transfected as indicated with NIC1 expression plasmid (NIC) or its empty vector control pcDNA3.0 (Control) and then treated with buffer only or rmFIZZ1. Cell extracts were then harvested for determination of α-SMA protein levels by Western blotting. B: The cells were treated as in A and plated on four-well chamber slides. The cells were fixed and stained after 48 hours with anti-α-SMA-Cy5 (red fluorescence). Arrows indicate myofibroblasts with bright staining for α-SMA arranged in fiber bundles. C: Fibroblasts were similarly treated as in A, except instead of transfection with NIC1 expression plasmid, treatment with γ-secretase inhibitor X was used to suppress the generation of NIC from Notch. The effect on α-SMA protein expression was then analyzed by Western blotting. D: As an alternative approach, Notch1 (NIC) or control siRNA was transfected into fibroblasts and then analyzed for α-SMA protein expression by Western blotting. Representative blots from at least three independent experiments are shown in A, C, and D. E: Cells were transfected with the α-SMA promoter construct with luciferase reporter gene, and then co-transfected with either the NIC1 expression plasmid (NIC) or control empty vector (Control). This was followed by treatment with either buffer only or FIZZ1 as indicated in the abscissa. After harvest, the cells were then analyzed for luciferase activity, and the data were expressed as relative light units. F: Fibroblasts were treated similarly as in E, but co-transfected with Notch1 siRNA or their control siRNA instead of NIC plasmid DNA and its control empty vector. G: Fibroblasts were transfected as in A, and total RNA was harvested 8 hours after FIZZ1 addition. α-SMA mRNA expression was analyzed by real time qPCR, and data were shown as fold change (2−ΔΔCT) over control cells transfected with empty vector. Means ± SE of triplicates are shown.
To further confirm that NIC could regulate α-SMA gene expression, the effect of NIC overexpression on α-SMA promoter activity was examined. Fibroblasts transfected with the α-SMA promoter construct and co-transfected with the NIC expression plasmid showed significantly higher promoter activity than cells co-transfected with the empty vector control (Figure 3E). As expected FIZZ1 stimulated α-SMA promoter activity, which was not significantly affected by NIC overexpression. Thus NIC overexpression alone was sufficient to stimulate promoter activity comparable with that induced by FIZZ1 treatment alone. This NIC-induced stimulation appeared to be maximal because the combined NIC overexpression plus FIZZ1 treatment did not significantly increase activity. The siRNA to Notch1 and Jagged1 strongly diminished α-SMA protein expression (Figures 2B and 3D). To check if they have similar effects on the α-SMA promoter activity, fibroblasts were co-transfected with the α-SMA promoter construct and the Notch1 or Jagged1 siRNA and their controls. The results showed that both siRNAs to Notch1 (Figure 3F) and Jagged1 (data not shown) caused significant inhibition on α-SMA promoter activity alone (P < 0.05), and essentially completely abolished the FIZZ1-induced promoter activity (Figure 3F). Analysis of α-SMA mRNA regulation by NIC also confirmed that NIC transfection was able to significantly up-regulate α-SMA gene expression, whereas FIZZ1 caused a further >50% increase in α-SMA mRNA levels (P < 0.05) (Figure 3G). FIZZ1 alone caused a more than twofold increase in α-SMA mRNA. These results indicated that Notch1 signaling was required in FIZZ1 activation of α-SMA expression.
FX Deficiency Impairs FIZZ1 Up-Regulation of α-SMA Expression
To further clarify that Notch1 signaling is also important for FIZZ1 induction of myofibroblast differentiation in vivo with contribution to pulmonary fibrosis, the effects of the FX mutation with its well-studied deficiency in Notch signaling,47 were investigated. First, to confirm that lung fibroblasts from FX KO mice were deficient in their response to FIZZ1-induced myofibroblast differentiation, in vitro analysis was undertaken using cells isolated from these KO mice. As previously noted, FX KO mice exhibit deficient Notch signaling in the absence of exogenous fucose supplementation, which is reversible by inclusion of dietary fucose.42 The isolated fibroblasts, with (ie, wild-type Notch phenotype) or without (ie, Notch-deficient) fucose supplementation in the medium, were treated with FIZZ1 at the indicated doses, and analyzed for α-SMA by Western blotting. The results showed that the dose-dependent induction of α-SMA by FIZZ1 in Notch-sufficient cells (with fucose supplementation) was essentially abolished in Notch-deficient (without fucose supplementation) (Figure 4A). Thus the dependence on Notch signaling for FIZZ1-induced myofibroblast differentiation was also suggested in this model of Notch deficiency. To confirm that the observed result in this model was specifically because of Notch signaling, the ability of exogenously induced NIC expression to reverse the effect of Notch deficiency in this model was evaluated. Transfection of the NIC expression plasmid into cells grown without fucose supplementation (ie, Notch-deficient) caused a marked increase in α-SMA protein expression relative to that in cells transfected with the control empty vector (Figure 4B). FIZZ1 treatment caused a further increase in α-SMA expression only in the cells transfected with the NIC expression plasmid. Thus the deficit in response to FIZZ1 in the Notch-deficient cells was attributable to specific loss of Notch signaling in these cells.
Figure 4.

Effect of Notch deficiency on FIZZ1 induction of α-SMA in isolated lung fibroblasts. Murine lung fibroblasts were isolated from FX KO mice and were cultured in the presence (Notch-sufficient, indicated with +) or absence (Notch-deficient, indicated with −) of fucose in the media. A: The cells were then treated with the indicated doses of rmFIZZ1, and after harvest, the cell extracts were analyzed for α-SMA protein by Western blotting. B: The Notch-deficient cells (without fucose supplementation) were transfected with the NIC1 expression plasmid (NIC) or empty vector (Control) and then treated with either buffer or rmFIZZ1 as indicated. The cell extracts were then analyzed for α-SMA protein by Western blotting. Representative blots from three independent experiments are shown.
Effects of Notch Deficiency on BLM-Induced Pulmonary Fibrosis
Because FIZZ1 is highly induced and promotes myofibroblast differentiation in BLM-induced pulmonary fibrosis,22 the availability of the FX KO mice afforded the opportunity to study the importance of Notch signaling in vivo in the context of its importance in FIZZ1-induced myofibroblast differentiation. To confirm Notch1 signaling deficiency in FX−/− mice without fucose supplementation, the Notch1 downstream target Hes1 was examined in the lungs of FX−/− mice with and without fucose supplementation (Notch-sufficient and Notch-deficient, respectively). The results showed that Hes1 was basically abolished in saline-injected Notch-deficient mice and not induced by BLM at day 21 after BLM treatment, whereas it was significantly increased in BLM-treated Notch-sufficient mice (Figure 5A). We then evaluated the effect of Notch deficiency on pulmonary fibrosis, BLM was used to induce lung fibrosis in FX KO mice with or without fucose supplementation. Evaluation of gene expression by real-time qPCR 7 days after BLM administration revealed that FIZZ1 mRNA was highly induced in BLM-treated Notch-sufficient mice compared with saline control animals as expected (Figure 5B). Although the absolute lung FIZZ1 mRNA levels were decreased in Notch-deficient versus -sufficient mice, the induction by BLM appeared intact and not significantly affected by Notch deficiency (P > 0.05). However, analysis of lung mRNAs for a number of pro-inflammatory and pro-fibrotic factors, including MCP-1, TNF-α, IL-4, and TGF-β revealed that their induction by BLM was impaired in Notch-deficient mice relative to that in sufficient mice (P < 0.05 between BLM and saline groups for all four factors) (Figure 5C). Because Notch has been implicated in helper T-cell (Th cell) differentiation,9,11 it is especially noteworthy that BLM completely failed to induce the expected increase in expression of the Th2 marker gene, IL-4, in the Notch-deficient mice. Thus Notch deficiency has significant inhibitory effects in the response to BLM treatment, including the Th2-type response.
Figure 5.

Effects of Notch deficiency on lung cytokine and FIZZ1 gene expression in BLM-induced pulmonary fibrosis. FX KO mice were maintained with (Notch-sufficient) or without (Notch-deficient) dietary fucose supplementation, and then treated with saline (SAL) or BLM to induce lung injury and fibrosis. A: Lung Hes1 protein expression in Notch-sufficient and -deficient mice 21 days after BLM treatment. Twenty μg of lung tissue lysates were used for Western analysis. B: At day 7 after BLM or saline treatment, lung tissue total RNA was isolated, and analyzed for mRNA levels of FIZZ1, or the cytokines TGF-β, TNF-α, MCP-1, and IL-4 (C). The results were expressed as 2−ΔΔCT equivalent to fold change, using GAPDH as the reference and the Notch-sufficient saline control as the calibrator. C: The values were expressed as a percentage of their respective saline controls. All mean values from the Notch-deficient mice (without fucose supplementation) were significantly lower than the corresponding ones from sufficient mice. Data are shown as means ± SE with n = 3 in each group of mice.
The extracellular matrix, collagen I, and α-SMA are key parameters for pulmonary fibrosis and myofibroblast differentiation.18,20 Examination of the lung tissue mRNA levels for these markers at day 7 after BLM treatment revealed the expected increases in Notch-sufficient, fucose-fed mice (Figure 6A). These BLM-induced increases were significantly reduced in Notch-deficient mice without dietary fucose supplementation (P < 0.05 for all three parameters). This reduction in fibrotic response to BLM treatment in the Notch-deficient mice was also noted in the lung hydroxyproline content measured at day 21 after model induction (P < 0.05) (Figure 6A). Also, consistent with the mRNA data, Western blotting analysis showed lung collagen I protein content was significantly reduced in Notch-deficient mice in response to BLM treatment when compared with that in Notch-sufficient mice (P < 0.05) (Figure 6B). Moreover, histological examination showed that whereas the lungs from BLM-treated Notch-sufficient mice exhibited typical severe fibrotic lesions, characterized by increased interstitial density, loss of normal alveolar architecture, the lungs from BLM-treated Notch-deficient mice were shown to have reduced fibrosis with scattered lesions that are much smaller in size with lower density than those in Notch-sufficient animals (Figure 7). There were no morphological abnormalities observed in saline-treated control lungs in both Notch-sufficient and -deficient mice. Thus these findings indicated that Notch deficiency was associated with a significant reduction in myofibroblast genesis and pulmonary fibrosis in this model.
Figure 6.

Effects of Notch deficiency on BLM-induced pulmonary fibrosis. As described in the legend to Figure 5, FX KO mice with or without fucose supplementation were treated with saline or BLM by endotracheal injection. A: The lungs were then harvested for analysis of lung hydroxyproline content or mRNA levels for procollagen I and α-SMA. Results were expressed as a percentage of their respective saline-treated controls. All three parameters were significantly lower in the Notch-deficient (without fucose supplementation) versus the sufficient group. Mean ± SE are shown with n = 5 animals per group. B: Lung type I collagen protein was analyzed by Western blotting. A representative blot from three of five animals in each treated group is shown.
Figure 7.

Histopathological evaluation of fibrosis in FX KO mice. Saline (C and D)- and BLM (A and B)-treated FX KO mice without (A and C) or with (B and D) fucose supplementation were examined at day 21 after treatment. The lungs were prepared for H&E staining and representative sections were shown. Original magnifications: ×100; ×400 (insets).
Discussion
Pulmonary fibrosis usually results in end-stage lung disease, respiratory failure, and eventually death. BLM-induced pulmonary fibrosis is a well-characterized animal model in which a progressive fibrotic process develops after initial lung inflammation. The common feature characteristic of progressive pulmonary fibrosis is abnormal deposition of extracellular matrix that effaces the normal lung tissue architecture. A key cellular source of this matrix is the mesenchymal cell population that occupies fibroblastic foci during the active period of fibrosis.15,48 This population is heterogeneous with respect to a number of key phenotypes. One of these phenotypes is the myofibroblast, which appears to play important roles in the pathogenesis of pulmonary fibrosis. Its dual role, as a key source of extracellular matrix and as an inflammatory cell, makes it the key cell in two processes that represent the hallmark of fibrosis. In addition to TGF-β, a novel inducer of myofibroblast differentiation, FIZZ1 was recently demonstrated to induce myofibroblast differentiation by directly stimulating α-SMA expression.22 However, its downstream signaling leading to up-regulation of α-SMA expression is essentially unknown. In the present study, the Notch1 signaling pathway was found to be involved in FIZZ1 induction of myofibroblast differentiation.
To arrive at this conclusion, the effect of FIZZ1 on Notch 1 signaling activation was first examined. Our data showed that FIZZ1 was able to activate Notch 1 signaling by inducing expression of Jagged 1, NIC, and Hes 1, a direct NIC downstream target. The effects of Notch 1 activation on induction of α-SMA were then determined. The findings showed that overexpression of NIC (activated Notch 1) or Jagged 1 caused significantly increased expression of α-SMA in lung fibroblasts, which was enhanced by FIZZ1 stimulation. In contrast FIZZ1 induction of α-SMA expression was significantly diminished when NIC or Jagged 1 expression was inhibited. Given that α-SMA is a key marker for myofibroblast differentiation, our data indicated that Notch 1 signaling is important for FIZZ1 induction of myofibroblast differentiation (Figure 8). The ligand Jagged 1 is also a transmembrane protein,49 therefore, cell-cell contact is necessary for the activation of Notch 1 signaling.50 Our results showed that Jagged1 could be activated by FIZZ1, and FIZZ1 induction of α-SMA was clearly decreased by inhibition of Jagged1, suggesting Jagged 1 may mediate FIZZ1 activation of Notch 1. Activation of this pathway in turn induces α-SMA expression in fibroblasts. Lung fibroblasts are heterogeneous,48 so it is possible that ligand-expressing cells and Notch 1 receptor-expressing cells may represent different subpopulations. It might explain how Notch 1 is activated by FIZZ1-induced Jagged 1 on neighboring cells through cell-cell contact interaction. Additionally a positive feedback loop because of NIC induction of Jagged 1 ligand might also act to amplify Notch1 signaling in a field of cells on continuous and prolonged exposure to ligand via cell-to-cell relay, which is having an effect possibly beyond the initial ligand-expressing cells. Similar mechanisms have been observed in NIH 3T3 cells.51
Figure 8.
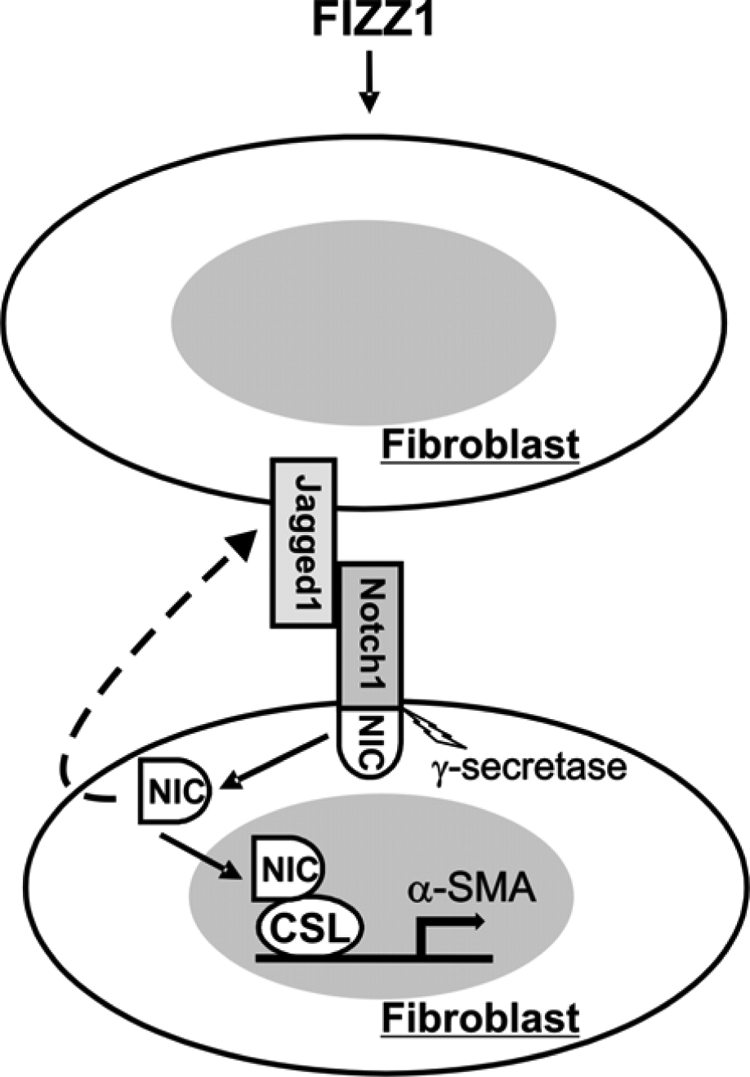
Schematic illustration of the proposed mechanism by which Notch1/Jagged1 mediates FIZZ1 induction of α-SMA gene expression. FIZZ1 is primarily secreted by alveolar epithelial cells in the lung, and activates Jagged1 expression through unknown mechanisms. The activated Jagged1 extracellular domain on fibroblasts interacts with Notch1 receptor on neighboring cells and causes the γ-secretase-mediated cleavage of NIC1, the intracellular domain of Notch1. The released NIC then translocates into the nucleus where it binds to the transcription factor CSL on the α-SMA promoter and converts CSL from a repressor to an activator with subsequent activation of α-SMA gene transcription. There is evidence to suggest that NIC could also activate Jagged1 expression with a potential positive feedback loop mechanism.
To seek out the possible mechanism for the noted induction of α-SMA after overexpression of NIC in fibroblast, the effect of NIC overexpression on α-SMA promoter was examined in lung fibroblasts. The findings showed that NIC overexpression alone caused significant up-regulation of α-SMA promoter activity to a level that was comparable with that in cells treated with FIZZ1. Although the induction by NIC overexpression was further enhanced by FIZZ1 stimulation, this was not statistically significant. This finding is consistent with a previous study showing that in vascular smooth muscle cells, α-SMA is shown to be a direct target of Notch via activation of its major effecter CSL.38 It appears that CSL could bind directly to a conserved cis-element in the α-SMA promoter, and this consensus sequence is required for Notch-mediated α-SMA induction. All these data confirmed the direct NIC induction of α-SMA gene transcription. However, other possible mechanism(s) involved in FIZZ1 induction of α-SMA cannot be ruled out based on present data. Taken together these findings suggest that Notch signaling in response to FIZZ1 may play an alternative or additional role to TGF-β in induction of myofibroblast differentiation in lung fibrosis.
The importance of Notch signaling in myofibroblast differentiation suggests its potential significance in fibrotic responses in vivo. A recent study revealed reduced hepatic fibrosis in patients with Alagille syndrome (AGS), a genetic disorder of Notch signaling, caused by mutations in the genes encoding Jagged1 or Notch2 receptor itself.52 This was thought to be attributable to an accumulation of intermediate hepatobiliary cells unable to transdifferentiate into biliary cells because of the defective Jagged1/Notch2 signaling, which then proceeds with a form of fibrotic reaction characterized by thin septa and pericellular distribution. However, the effects on myofibroblast differentiation (from hepatic stellate cells) are not studied in these patients. To evaluate out the potential importance of Notch 1 signaling in pulmonary fibrosis, the effects of deficiency in Notch signaling in FX−/− mice without exogenous fucose supplementation was examined.47 Because Notch signaling is required for normal development, the use of KO animals deficient in this critical function is not feasible for studies to demonstrate its in vivo importance in fibrosis. The availability of the FX−/− mouse affords a means to study this because Notch deficiency is only evident if the animals are deprived of fucose in their diet, ie, this is effectively a conditional KO. These animals have an induced null mutation in the GDP-4-keto-6-deoxymannose 3, 5-epimerase-4-reductase (FX) gene, which encodes an enzyme in the de novo pathway for GTP-fucose synthesis. The FX−/− mice exhibit a virtually complete dependence on a fucose-dependent salvage pathway for GDP-fucose synthesis, which enables reversible control of glycan fucosylation by manipulating dietary fucose. Because fucosylation is essential for Notch function, in the absence of exogenous fucose, Notch signaling is deficient in these animals.47 The de novo appearance of myofibroblasts and their persistence are key features for progressive fibrotic diseases.21 Our data showed that after BLM treatment, significantly reduced pulmonary fibrosis in Notch-deficient mice was observed with decreased α-SMA expression and collagen deposition, suggesting a reduction in genesis of myofibroblasts. In view of the evidence of Notch1 activation myofibroblast differentiation, a potential mechanism for the protection from fibrosis afforded by Notch deficiency might be attributable to a reduction in myofibroblast differentiation in the deficient mice. However, in addition to the effects on myofibroblast differentiation, there was also significant reduction in expression of FIZZ1 and a number of profibrogenic factors including IL-4, TGF-β, MCP-1, and TNF-α. Moreover there is evidence that Notch signaling through different ligands induces distinct CD4+ T-cell cytokine production.8 Notch 1 is able to induce Th2 differentiation by direct up-regulation of Gata 3 via either an IL-4-dependent or -independent mechanism.9,10,53 In the present study, Th2 cytokine (IL-4) expression was impaired in Notch-deficient mice, and a Th2-type response is predominant in pulmonary fibrosis. Deficiency of the Th2 cytokines IL-4 and/or IL-13 abrogated FIZZ1 induction, myofibroblast differentiation, and fibrosis in the BLM model.28 Thus the additional contribution of Notch deficiency on impaired Th2 cytokine expression represented an additional mechanism by which fibrosis could be impaired. Finally, a variety of cytokines play important roles in pulmonary fibrosis, and the myofibroblast is a noninflammatory source for some of them, eg, TGF-β and MCP-1.54,55 The decreased myofibroblast differentiation in Notch-deficient mice potentially contributed to the observed reduction in lung profibrogenic cytokine expression. These cytokines may interact with others, such as TNF-α in complex cytokine networks that could ultimately serve in the manner of a positive feedback loop to cause further myofibroblast differentiation and stimulation of extracellular matrix production.54,55,56,57 However, the direct effects of Notch1 on cytokine expression cannot be ruled out.
Although Notch signaling is shown to be important in mediating myofibroblast differentiation in fibroblasts in this and other studies,38 its role in smooth muscle cell differentiation is not always consistent. Thus in human aortic smooth muscle cell line as well as primary human foreskin fibroblasts, α-SMA is shown to be directly up-regulated by Notch1 via its major effector CSL, also referred to as CBF-1.38 However, in 10T1/2 fibroblasts and A10 rat aortic smooth muscle cells, Notch1, acting through another effector, the HRT factor family, is found to repress smooth muscle cell differentiation by inhibiting myocardin-dependent transcription of SMC-restricted genes including α-SMA and SM22α.41 Additionally overexpression of active Notch2 is shown to inhibit TGF-β-induced α-SMA expression, whereas transient knockdown of Notch2 by siRNA in C2C12 myoblasts results in differentiation into myofibroblastic cells without TGF-β treatment.40 In contrast, Notch3 is required for TGF-β-induced myofibroblastic differentiation, and counterregulated by Notch2 in these cells, indicating a countervailing regulatory system dependent on which Notch receptor is being activated. A similar mechanism is suggested by a more recent study showing that whereas Notch signaling via CSL up-regulates α-SMA gene transcription, signaling via HRT1 or HRT2 suppresses transcription.58 Because Notch in a CSL-dependent manner up-regulates expression of HRTs, the authors suggest that this HRT-mediated suppression may represent a negative feedback pathway on actin expression. Thus the ultimate effect on actin expression is dependent on the balance of direct activation via CSL versus indirect suppression via HRT generation. Because our data revealed a Notch-mediated up-regulation in actin expression, the direct effects of CSL on the α-SMA promoter appear to be the dominant effect. Further studies are necessary to assess the relative contributions of these divergent pathways.
Footnotes
Address reprint requests to Sem H. Phan, M.D., Ph.D., Department of Pathology, University of Michigan Medical School, 109 Zina Pitcher Pl., Ann Arbor, MI 48109-2200. E-mail: shphan@umich.edu.
Supported by the National Institutes of Health (grants HL28737, HL31963, HL52285, and HL77297) and the Sandler Foundation (award to S.H.P.).
Current address of M.C.: Department of Pathology, Chonbuk National University Medical School, Jeonju, Republic of Korea.
References
- Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Deftos ML, Huang E, Ojala EW, Forbush KA, Bevan MJ. Notch1 signaling promotes the maturation of CD4 and CD8 SP thymocytes. Immunity. 2000;13:73–84. doi: 10.1016/s1074-7613(00)00009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Li D, Winoto A, Robey EA. Distinct transcriptional programs in thymocytes responding to T cell receptor, Notch, and positive selection signals. Proc Natl Acad Sci USA. 2004;101:4936–4941. doi: 10.1073/pnas.0401133101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Reizis B, Leder P. Direct induction of T lymphocyte-specific gene expression by the mammalian Notch signaling pathway. Genes Dev. 2002;16:295–300. doi: 10.1101/gad.960702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumortier A, Jeannet R, Kirstetter P, Kleinmann E, Sellars M, dos Santos NR, Thibault C, Barths J, Ghysdael J, Punt JA, Kastner P, Chan S. Notch activation is an early and critical event during T-cell leukemogenesis in Ikaros-deficient mice. Mol Cell Biol. 2006;26:209–220. doi: 10.1128/MCB.26.1.209-220.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu L, Fang TC, Artis D, Shestova O, Pross SE, Maillard I, Pear WS. Notch signaling is an important regulator of type 2 immunity. J Exp Med. 2005;202:1037–1042. doi: 10.1084/jem.20050923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsen D, Antov A, Jankovic D, Sher A, Radtke F, Souabni A, Busslinger M, McCright B, Gridley T, Flavell RA. Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity. 2007;27:89–99. doi: 10.1016/j.immuni.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang TC, Yashiro-Ohtani Y, Del Bianco C, Knoblock DM, Blacklow SC, Pear WS. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity. 2007;27:100–110. doi: 10.1016/j.immuni.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117:515–526. doi: 10.1016/s0092-8674(04)00451-9. [DOI] [PubMed] [Google Scholar]
- Maekawa Y, Tsukumo S, Chiba S, Hirai H, Hayashi Y, Okada H, Kishihara K, Yasutomo K. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity. 2003;19:549–559. doi: 10.1016/s1074-7613(03)00270-x. [DOI] [PubMed] [Google Scholar]
- Jurynczyk M, Jurewicz A, Bielecki B, Raine CS, Selmaj K. Inhibition of Notch signaling enhances tissue repair in an animal model of multiple sclerosis. J Neuroimmunol. 2005;170:3–10. doi: 10.1016/j.jneuroim.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301–1315. doi: 10.1164/ajrccm.157.4.9707039. [DOI] [PubMed] [Google Scholar]
- Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. doi: 10.1056/NEJMra003200. [DOI] [PubMed] [Google Scholar]
- King TE, Jr, Schwarz MI, Brown K, Tooze JA, Colby TV, Waldron JA, Jr, Flint A, Thurlbeck W, Cherniack RM. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001;164:1025–1032. doi: 10.1164/ajrccm.164.6.2001056. [DOI] [PubMed] [Google Scholar]
- Zhang K, Gharaee-Kermani M, McGarry B, Phan SH. In situ hybridization analysis of rat lung alpha 1(I) and alpha 2(I) collagen gene expression in pulmonary fibrosis induced by endotracheal bleomycin injection. Lab Invest. 1994;70:192–202. [PubMed] [Google Scholar]
- Zhang K, Rekhter MD, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am J Pathol. 1994;145:114–125. [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Kuhn C, McDonald JA. The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol. 1991;138:1257–1265. [PMC free article] [PubMed] [Google Scholar]
- Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, Phan SH. FIZZ1 stimulation of myofibroblast differentiation. Am J Pathol. 2004;164:1315–1326. doi: 10.1016/S0002-9440(10)63218-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb IN, Kabakoff RC, Chan B, Baker TW, Gurney A, Henzel W, Nelson C, Lowman HB, Wright BD, Skelton NJ, Frantz GD, Tumas DB, Peale FV, Jr, Shelton DL, Hebert CC. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000;19:4046–4055. doi: 10.1093/emboj/19.15.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubzick C, Choi ES, Kunkel SL, Joshi BH, Puri RK, Hogaboam CM. Impact of interleukin-13 responsiveness on the synthetic and proliferative properties of Th1- and Th2-type pulmonary granuloma fibroblasts. Am J Pathol. 2003;162:1475–1486. doi: 10.1016/S0002-9440(10)64280-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs NW, Hogaboam C, Chensue SW, Blease K, Kunkel SL. Type 1/type 2 cytokine paradigm and the progression of pulmonary fibrosis. Chest. 2001;120:5S–8S. doi: 10.1378/chest.120.1_suppl.s5. [DOI] [PubMed] [Google Scholar]
- Kunkel SL, Lukacs NW, Strieter RM, Chensue SW. The role of chemokines in the immunopathology of pulmonary disease. Forum (Genova) 1999;9:339–355. [PubMed] [Google Scholar]
- Liu T, Jin H, Ullenbruch M, Hu B, Hashimoto N, Moore B, McKenzie A, Lukacs NW, Phan SH. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J Immunol. 2004;173:3425–3431. doi: 10.4049/jimmunol.173.5.3425. [DOI] [PubMed] [Google Scholar]
- Stütz AM, Pickart LA, Trifilieff A, Baumruker T, Prieschl-Strassmayr E, Woisetschlager M. The Th2 cell cytokines IL-4 and IL-13 regulate found in inflammatory zone 1/resistin-like molecule alpha gene expression by a STAT6 and CCAAT/enhancer-binding protein-dependent mechanism. J Immunol. 2003;170:1789–1796. doi: 10.4049/jimmunol.170.4.1789. [DOI] [PubMed] [Google Scholar]
- Teng X, Li D, Champion HC, Johns RA. FIZZ1/RELMalpha, a novel hypoxia-induced mitogenic factor in lung with vasoconstrictive and angiogenic properties. Circ Res. 2003;92:1065–1067. doi: 10.1161/01.RES.0000073999.07698.33. [DOI] [PubMed] [Google Scholar]
- Tong Q, Zheng L, Li B, Wang D, Huang C, Matuschak GM, Li D. Hypoxia-induced mitogenic factor enhances angiogenesis by promoting proliferation and migration of endothelial cells. Exp Cell Res. 2006;312:3559–3569. doi: 10.1016/j.yexcr.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tong Q, Zheng L, Lin L, Li B, Wang D, Huang C, Li D. VEGF is upregulated by hypoxia-induced mitogenic factor via the PI-3K/Akt-NF-kappaB signaling pathway. Respir Res. 2006;7:37. doi: 10.1186/1465-9921-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji-Kegan K, Su Q, Angelini DJ, Champion HC, Johns RA. Hypoxia-induced mitogenic factor has proangiogenic and proinflammatory effects in the lung via VEGF and VEGF receptor-2. Am J Physiol. 2006;291:L1159–L1168. doi: 10.1152/ajplung.00168.2006. [DOI] [PubMed] [Google Scholar]
- Su Q, Zhou Y, Johns RA. Bruton’s tyrosine kinase (BTK) is a binding partner for hypoxia induced mitogenic factor (HIMF/FIZZ1) and mediates myeloid cell chemotaxis. FASEB J. 2007;21:1376–1382. doi: 10.1096/fj.06-6527com. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- Doi H, Iso T, Sato H, Yamazaki M, Matsui H, Tanaka T, Manabe I, Arai M, Nagai R, Kurabayashi M. Jagged1-selective notch signaling induces smooth muscle differentiation via a RBP-Jkappa-dependent pathway. J Biol Chem. 2006;281:28555–28564. doi: 10.1074/jbc.M602749200. [DOI] [PubMed] [Google Scholar]
- Noseda M, Fu Y, Niessen K, Wong F, Chang L, McLean G, Karsan A. Smooth muscle alpha-actin is a direct target of Notch/CSL. Circ Res. 2006;98:1468–1470. doi: 10.1161/01.RES.0000229683.81357.26. [DOI] [PubMed] [Google Scholar]
- Morrow D, Scheller A, Birney YA, Sweeney C, Guha S, Cummins PM, Murphy R, Walls D, Redmond EM, Cahill PA. Notch-mediated CBF-1/RBP-J{kappa}-dependent regulation of human vascular smooth muscle cell phenotype in vitro. Am J Physiol. 2005;289:C1188–C1196. doi: 10.1152/ajpcell.00198.2005. [DOI] [PubMed] [Google Scholar]
- Ono Y, Sensui H, Okutsu S, Nagatomi R. Notch2 negatively regulates myofibroblastic differentiation of myoblasts. J Cell Physiol. 2007;210:358–369. doi: 10.1002/jcp.20838. [DOI] [PubMed] [Google Scholar]
- Proweller A, Pear WS, Parmacek MS. Notch signaling represses myocardin-induced smooth muscle cell differentiation. J Biol Chem. 2005;280:8994–9004. doi: 10.1074/jbc.M413316200. [DOI] [PubMed] [Google Scholar]
- Smith PL, Myers JT, Rogers CE, Zhou L, Petryniak B, Becker DJ, Homeister JW, Lowe JB. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol. 2002;158:801–815. doi: 10.1083/jcb.200203125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan SH, Varani J, Smith D. Rat lung fibroblast collagen metabolism in bleomycin-induced pulmonary fibrosis. J Clin Invest. 1985;76:241–247. doi: 10.1172/JCI111953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Wu Z, Liu T, Ullenbruch MR, Jin H, Phan SH. Gut-enriched Kruppel-like factor interaction with Smad3 inhibits myofibroblast differentiation. Am J Respir Cell Mol Biol. 2007;36:78–84. doi: 10.1165/rcmb.2006-0043OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Edwards CA, O'Brien WD., Jr Modified assay for determination of hydroxyproline in a tissue hydrolyzate. Clin Chim Acta. 1980;104:161–167. doi: 10.1016/0009-8981(80)90192-8. [DOI] [PubMed] [Google Scholar]
- Zhou L, Li LW, Yan Q, Petryniak B, Man Y, Su C, Shim J, Chervin S, Lowe JB. Notch-dependent control of myelopoiesis is regulated by fucosylation. Blood. 2008;112:308–319. doi: 10.1182/blood-2007-11-115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan SH. The myofibroblast in pulmonary fibrosis. Chest. 2002;122:286S–289S. doi: 10.1378/chest.122.6_suppl.286s. [DOI] [PubMed] [Google Scholar]
- Lissemore JL, Starmer WT. Phylogenetic analysis of vertebrate and invertebrate Delta/Serrate/LAG-2 (DSL) proteins. Mol Phylogenet Evol. 1999;11:308–319. doi: 10.1006/mpev.1998.0588. [DOI] [PubMed] [Google Scholar]
- Fiúza UM, Arias AM. Cell and molecular biology of Notch. J Endocrinol. 2007;194:459–474. doi: 10.1677/JOE-07-0242. [DOI] [PubMed] [Google Scholar]
- Ross DA, Kadesch T. Consequences of Notch-mediated induction of Jagged1. Exp Cell Res. 2004;296:173–182. doi: 10.1016/j.yexcr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Fabris L, Cadamuro M, Guido M, Spirli C, Fiorotto R, Colledan M, Torre G, Alberti D, Sonzogni A, Okolicsanyi L, Strazzabosco M. Analysis of liver repair mechanisms in Alagille syndrome and biliary atresia reveals a role for notch signaling. Am J Pathol. 2007;171:641–653. doi: 10.2353/ajpath.2007.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo M. Notch: filling a hole in T helper 2 cell differentiation. Immunity. 2007;27:3–5. doi: 10.1016/j.immuni.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Zhang K, Phan SH. Cytokines and pulmonary fibrosis. Biol Signals. 1996;5:232–239. doi: 10.1159/000109195. [DOI] [PubMed] [Google Scholar]
- Phan SH. Role of the myofibroblast in pulmonary fibrosis. Kidney Int Suppl. 1996;54:S46–S48. [PubMed] [Google Scholar]
- Zhang K, Gharaee-Kermani M, McGarry B, Remick D, Phan SH. TNF-alpha-mediated lung cytokine networking and eosinophil recruitment in pulmonary fibrosis. J Immunol. 1997;158:954–959. [PubMed] [Google Scholar]
- Gharaee-Kermani M, Phan SH. Molecular mechanisms of and possible treatment strategies for idiopathic pulmonary fibrosis. Curr Pharm Des. 2005;11:3943–3971. doi: 10.2174/138161205774580561. [DOI] [PubMed] [Google Scholar]
- Tang Y, Urs S, Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle {alpha}-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ Res. 2008;102:661–668. doi: 10.1161/CIRCRESAHA.107.165134. [DOI] [PMC free article] [PubMed] [Google Scholar]