

Abstract
Although recent reports suggest that the endoplasmic reticulum (ER) stress response is induced in association with the development of inflammatory bowel disease, its role in the pathogenesis of inflammatory bowel disease remains unclear. The CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) is a transcription factor that is involved in the ER stress response, especially ER stress-induced apoptosis. In this study, we found that experimental colitis was ameliorated in CHOP-null mice, suggesting that CHOP exacerbates the development of colitis. The mRNA expression of Mac-1 (CD11b, a positive regulator of macrophage infiltration), Ero-1α, and Caspase-11 (a positive regulator of interleukin-1β production) in the intestine was induced with the development of colitis, and this induction was suppressed in CHOP-null mice. ERO-1α is involved in the production of reactive oxygen species (ROS); an increase in ROS production, which is associated with the development of colitis in the intestine, was suppressed in CHOP-null mice. A greater number of apoptotic cells in the intestinal mucosa of wild-type mice were observed to accompany the development of colitis compared with CHOP-null mice, suggesting that up-regulation of CHOP expression exacerbates the development of colitis. Furthermore, this CHOP activity appears to involve various stimulatory mechanisms, such as macrophage infiltration via the induction of Mac-1, ROS production via the induction of ERO-1α, interleukin-1β production via the induction of Caspase-11, and intestinal mucosal cell apoptosis.
Inflammatory bowel disease (IBD), Crohn’s disease, and ulcerative colitis, have become substantial health problems with an actual prevalence of 200 to 500 per 100,000 people in western countries, which almost doubles every 10 years.1 Although the etiology of IBD is not clear at present, recent studies suggest that IBD is a disorder involving activation of leukocytes (macrophages, lymphocytes, and neutrophils) and their infiltration into the inflamed intestine, and intestinal mucosal damage induced by reactive oxygen species (ROS).2 To understand the molecular mechanism underlying the pathogenesis of IBD and to establish a clinical protocol for its treatment, it is important to identify proteins that are involved in the pathogenesis of IBD. For this purpose, various experimental animal models of colitis, in particular the dextran sulfate sodium (DSS)- and trinitrobenzene sulfonic acid (TNBS)-induced colitis models, are useful.3
Pro-inflammatory cytokines and cell adhesion molecules (CAMs) play an important role in the activation and infiltration of leukocytes that are associated with IBD. Increases in the intestinal levels of pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β, as well as various CAMs, such as intercellular adhesion molecule-1 (ICAM-1) and Mac-1, have been reported in both IBD patients and animal models of IBD.4,5,6,7,8,9,10,11 TNF-α-deficient mice or ICAM-1-deficient mice show a phenotype resistant to experimental colitis.8,12 A chimeric monoclonal antibody against TNF-α, infliximab, antibody against Mac-1, and alicaforsen (ISIS 2302), an oligodeoxynucleotide that inhibits the expression of ICAM-1, are reported to be effective in the treatment of IBD patients and experimental colitis.8,10,13,14,15
Accumulation of unfolded and misfolded proteins in the endoplasmic reticulum (ER) induces the ER stress response. At the final step of mammalian ER stress response, the apoptotic response is initiated to eliminate cells. C/EBP homologous transcription factor (CHOP) is a transcription factor involved in the ER stress response, especially ER stress-induced apoptosis through various mechanisms such as down-regulation of Bcl-2 and up-regulation of Bim.16,17,18 A close relationship between inflammation and the ER stress response, especially the induction of CHOP, has been suggested. For example, TNF-α was reported to induce the ER stress response and expression of CHOP.19 CREBH was recently identified as a factor connecting the ER stress response and the acute inflammatory response.20 Therefore, it is reasonable to hypothesize that the ER stress response, and CHOP in particular, is involved in the pathogenesis of IBD. In fact, some recent reports support this idea; up-regulation of CHOP and GRP78 was observed in the inflamed intestine in both IBD.21,22 However, the exact role (positive or negative) of the ER stress response (or CHOP) in the pathogenesis of IBD has remained unknown. The analysis of knockout mice is useful in addressing this type of question. For example, we recently suggested, through analysis of DSS-induced colitis in heat shock factor 1 (HSF1, a transcription factor involved in the heat shock response)-null mice, that HSF1 plays a protective role, inhibiting the development of IBD.23 In the present study, we compared the development of DSS- and TNBS-induced colitis between CHOP-null mice and wild-type (WT) mice and obtained genetic evidence that CHOP plays a positive role in the pathogenesis of experimental colitis. Furthermore, results in this study suggest that CHOP achieves this effect through various mechanisms such as stimulation of intestinal ROS production, sensitization of intestinal mucosal cells to ROS-induced apoptosis, stimulation of macrophage infiltration into the inflamed intestine, and stimulation of the intestinal production of IL-1β. Based on these findings, we propose that inhibitors for CHOP may be therapeutically beneficial for the treatment of IBD.
Materials and Methods
Chemicals, Cells, and Animals
Paraformaldehyde, 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), menadione, fetal bovine serum, o-dianisidine, 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF), and TNBS were obtained from Sigma (St. Louis, MO). Thiobarbituric acid (TBA), butylated hydroxytoluene (BHT), n-butanol, and pyridine were from Nacalai Tesque (Kyoto, Japan). DSS (M.W. 5000, 15 to 20% sulfur content) was from Wako Pure Chemicals (Tokyo, Japan). Proteose peptone was from Becton Dickinson (San Jose, CA). Lipopolysaccharide (LPS) was from List Biological Laboratories, Inc (Campbell, CA). Antioxidant Assay Kit was from Cayman (Ann Arbor, MI). An enzyme-linked immunosorbent assay kit for the detection of IL-1β was from Pierce Chemical (Rockford, IL). Optimal cutting temperature (O.C.T.) compound was from Sakura Finetek Japan (Tokyo, Japan). Mayer’s hematoxylin, 1% eosin alcohol solution, and Malinol were from Muto Pure Chemicals (Tokyo, Japan). Terminal deoxynucleotidyl transferase (TdTase) was obtained from Toyobo (Osaka, Japan). The Envision kit was from DAKO (Carpinteria, CA). Biotin 14-ATP and Alexa Fluor 488 conjugated with streptavidin were purchased from Invitrogen (Carlsbad, CA). Vectashield was from Vector Laboratories (Burlingame, CA). HilyMax and 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) were from Dojindo Laboratories (Kumamoto, Japan). The RNeasy kit and HiPerFect were obtained from Qiagen (Valencia, CA), the PrimeScript first strand cDNA synthesis kit was purchased from Takara Bio (Ohtsu, Japan), and iQ SYBR Green Supermix was from Bio-Rad (Hercules, CA). Antibodies against CHOP, actin, and GRP78 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and that against CD68 was from Dack Co. (Carpinteria, CA). α-(4-pyridyl-1-oxide)-N-tert-butylnitrone (POBN) was from Alexis (San Diego, CA). HCT-15 and RAW264 cells were obtained from the Cell Resource Center for Biochemical Research at Tohoku University (Sendai, Japan) and RIKEN BioResource Center (Tsukuba, Japan), respectively. CHOP-null mice that had been backcrossed with WT mice (C57BL/6) for more than 10 times and the WT mice (5 to 7 weeks old, male) were prepared and there was no apparent phenotypes in CHOP-null mice as described previously.24 The experiments and procedures described here were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health, and were approved by the Animal Care Committee of Kumamoto University.
Development of DSS- or TNBS-Induced Colitis and Measurement of Colon Length and Disease Activity Index (DAI)
DSS-induced colitis was induced in mice by the addition of 3% DSS (w/v, final concentration) to their drinking water as described previously.23 The animals were allowed free access to the DSS-containing water for 7 days. For histopathological observation, measurement of myeloperoxidase (MPO), various mRNAs, ROS, thiobarbituric acid reactant substances (TBARS), as well as apoptosis, we used rectum and distal colon. After 7 days, animals were placed under deep ether anesthesia and sacrificed, the colons were dissected and their length measured from the ileocecal junction to the anal verge. The DAI was determined macroscopically by an observer unaware of the treatment the mice had received, according to previously reported criteria.23,25 Briefly, the DAI was calculated as the sum of the diarrheal stool score (0: normal stool; 1: mildly soft stool; 2: very soft stool; 3: watery stool) and the bloody stool score (0: normal color stool; 1: brown color stool; 2: reddish color stool; 3: bloody stool). TNBS-induced colitis was produced by intrarectal administration of TNBS once as described previously.26
MPO Activity
MPO activity in the colonic tissues was measured as previously described.23,27 After DSS treatment, colons were dissected, rinsed with cold saline, and cut into small pieces. Samples were homogenized and protein concentrations of the supernatants were determined using the Bradford method.28 MPO activity was determined in 10 mmol/L phosphate buffer with 0.5 mmol/L o-dianidisine, 0.00005% (w/v) hydrogen peroxide, and 20 μg of protein. MPO activity was obtained from the slope of the reaction curve and its specific activity was expressed as the number of hydrogen peroxide molecules converted per minute per mg of protein.
Lipid Peroxidation Measured by TBARS
The amounts of TBARS in colonic tissues were measured as previously described.29,30,31 After DSS treatment, colons were dissected, cut into small pieces and weighed. Samples were homogenized and centrifuged. Supernatants were mixed with 20 μl of 8.1% sodium dodecyl sulfate solution, 150 μl of 20% acetic acid solution, and 5 μl of 0.8% BHT solution, then with 150 μl of 0.8% TBA solution, and finally boiled for 1 hour. Samples were mixed with 500 μl of n-butanol/pyridine (15:1) and centrifuged. The absorbance of the supernatant was measured at 532 nm and the amount of TBARS expressed as the number of TBARS molecules per gram of tissue.
Immunoblotting Analysis
Total protein was extracted from the colonic tissues as described previously.23,32 The protein concentration of the samples was determined by the Bradford method.28 Samples were applied to 8% (GRP78 and actin) or 12% (CHOP) polyacrylamide sodium dodecyl sulfate gels and subjected to electrophoresis, after which the proteins were immunoblotted with appropriate antibodies.
Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis
Real-time RT-PCR was done as described33 with some modifications. Total RNA was extracted from intestinal tissues using an RNeasy kit according to the manufacturer’s protocol. Samples (2.5 μg of RNA) were reverse-transcribed using a first-strand cDNA synthesis kit according to the manufacturer’s instructions. Synthesized cDNA was used in real-time RT-PCR (Chromo 4 instrument, Bio-Rad) experiments using iQ SYBR Green Supermix and analyzed with Opticon Monitor Software (Bio-Rad, Hercules, CA). Specificity was confirmed by electrophoretic analysis of the reaction products and by inclusion of template- or reverse transcriptase-free controls. To normalize the amount of total RNA present in each reaction, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA was used as an internal standard.
Primers were designed using the Primer3 website (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). The primers used were (name: forward primer, reverse primer): for mouse; Tnf-α: 5′-CGTCAGCCGATTTGCTATCT-3′, 5′-CGGACTCCGCAAAGTCTAAG-3′; Il-1β: 5′-GATCCCAAGCAATACCCAAA-3′, 5′-GGGGAACTCTGCAGACTCAA-3′; Il-6: 5′-CTGGAGTCACAGAAGGAGTGG-3′, 5′-GGTTTGCCGAGTAGATCTCAA-3′; Vcam-1 (vascular cell adhesion molecule): 5′-CTCCTGCACTTGTGGAAATG-3′, 5′-TGTACGAGCCATCCACAGAC-3′; Icam-1: 5′-TCGTGATGGCAGCCTCTTAT-3′, 5′-GGGCTTGTCCCTTGAGTTTT-3′; Madcam-1 (mucosal addressin cell adhesion molecule): 5′-GCAGGCTGGGAGCTACTCT-3′, 5′-TCCCTCTTGTGGTAGGTTGc-3′; CD49d: 5′-CAGAGCCACACCCAAAAGTT-3′, 5′-TGAAATGTCGTTTGGGTCTTt-3′; CD11b: 5′-TGTGAGCAGCACTGAGATCC-3′, 5′-ATGGCTCCACTTTGGTCTCT-3′; L-selectin: 5′-ATTCCTGTAGCCGTCATGGT-3′, 5′-CATCCTTTCTTGAGATTTCTTGC-3′; Il-10: 5′-GGCCCTTTGCTATGGTGTCC-3′, 5′-AAGCGGCTGGGGGATGAC-3′; Caspase-11: 5′-TGGAAGCTGATGCTGTCAAG-3′, 5′-GAGCCTCCTGTTTTGTCTCG-3′; endoplasmic reticulum oxidoreductin (Ero)-1α: 5′-TTAAGTCTGCGAGCTACAAGTATTC-3′, 5′-AGTAAGTCCACATACTCAGCATCG-3′; Bcl-2: 5′-CCTGTGGATGACTGAGTACC-3′, 5′-GAGACAGCCAGGAGAAAT-3′; Chop: 5′-ACAGAGGTCACACGCACATC-3′, 5′-GGGCACTGACCACTCTGTTT-3′; Grp78: 5′-GCTTCCGATAATCAGCCAAC-3′, 5′-GCAGGAGGAATTCCAGTCA-3′; C/ebp-β: 5′-GCAAGAGCCGCGACAAG-3′, 5′-GGCTCGGGCAGCTGCTT-3′. For human; CHOP: 5′-TGCCTTTCTCTTCGGACACT-3′, 5′-TGTGACCTCTGCTGGTTCTG-3′.
Histological and Immunohistochemical Analysis
Colonic tissue samples were fixed in 4% buffered paraformaldehyde, embedded in O.C.T. compound, and cryosectioned. For histological examination [hematoxylin and eosin (H&E) staining], sections were stained first with Mayer’s hematoxylin and then with 1% eosin alcohol solution. Samples were mounted with Malinol and inspected with the aid of an Olympus (Tokyo, Japan) BX51 microscope. For histological evaluation of the tissue damage (damage score) and areas of lesions (extent of lesion), sections were evaluated microscopically by an observer unaware of the treatment the animals had received, and quantified as described.34,35 Colonic damage (damage score) was categorized into six groups (0: normal mucosa; 1: infiltration of inflammatory cells; 2: shortening of the crypt by less than 50%; 3: shortening of the crypt by more than 50%; 4: crypt loss; 5: destruction of epithelial cells). The extent of lesions in the total colon was categorized into six grades (0: 0%; 1: 1 to 20%; 2: 21 to 40%; 3: 41 to 60%; 4: 61 to 80%; 5: 81 to 100%).
For immunohistochemical analysis, sections were treated in a microwave oven with 0.01 mol/L citric acid buffer for antigen activation and incubated with 0.3% hydrogen peroxide in methanol for removal of endogenous peroxidase. Sections were blocked with 2.5% goat serum for 10 minutes, incubated for 12 hours with each antibody in the presence of 3% bovine serum albumin, and then incubated for 1 hour with peroxidase-labeled polymer conjugated to goat anti-rabbit immunoglobulins (Envision kit). Then, 3,3′-diaminobenzidine was applied to the sections and the sections were finally incubated with Mayer’s hematoxylin. Samples were mounted with malinol and inspected using a fluorescence microscope (BX51; Olympus, Tokyo, Japan).
Overexpression and Suppression of Expression of Targeting Genes
The CHOP- and ERO-1α-specific siRNAs were purchased from Qiagen. A plasmid expressing CHOP or C/EBP-β was as described.36,37 HCT-15 and RAW264 cells were transfected with these siRNAs or plasmids using HiPerFect or HilyMax (transfection reagents) according to the manufacturer’s instructions. Nonsilencing siRNA (5′-UUCUCCGAACGUGUCACGUDTdT-3′ and 5′-ACGUGACACGUUCGGAGAADTdT-3′) was used as a negative control.
Preparation of Mouse Peritoneal Macrophages
Mouse peritoneal macrophages were prepared as described previously.23,38 Mice were given 0.5 ml of 10% proteose peptone by intraperitoneal injection and peritoneal cells were harvested 3 days later. The cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum. After incubation for 4 hours, nonadherent cells were removed and the adherent cells were cultured for use in the experiments. Virtually all of the adherent cells were macrophages, as previously described.38 Caspase-1 activity was measured as described.39 The amounts of IL-1β secreted into the medium were measured by enzyme-linked immunosorbent assay according to the manufacturer’s protocol.
TdT-Mediated dUTP-Biotin End-Labeling (TUNEL) Assay
Colonic tissue samples were fixed in 4% buffered paraformaldehyde, embedded in O.C.T. compound, and cryosectioned. Sections were incubated first with proteinase K (20 μg/ml) for 15 minutes at 37°C, then with TdTase and biotin 14-ATP for 1 hour at 37°C and finally with Alexa Fluor 488 conjugated with streptavidin and DAPI (5 μg/ml) for 2 hours. Samples were mounted with Vectashield and inspected with the aid of a fluorescence microscope (Olympus BX51).
Flow Cytometric Analysis of ROS Production and Measurement of Intracellular Antioxidant Activity
Flow cytometry was performed on a FACSCalibur cell sorter (Becton Dickinson), as described.40 Briefly cells were incubated with 20 mmol/L of H2DCF in the dark at 37°C for 30 minutes. The shift in green fluorescence is associated with ROS production and was determined from histogram data using CellQuest software (Becton Dickinson). A total of 20,000 cells were collected for each histogram. To assess the antioxidant capacity in HCT-15 cells, antioxidant assay was performed using the antioxidant assay kit from Cayman Chemical following the manufacturer’s protocol.
Determination of ROS Production in Vivo by Electron Spin Resonance (ESR) Analysis
In vivo ESR analysis was performed as described,41,42 with some modifications. After DSS administration for 5 days, animals were placed under deep anesthesia with chloral hydrate (250 mg/kg) and injected with POBN intraperitoneally (4 mmol/kg). After 1 hour, mice were sacrificed, the colons were dissected and lipid phase from samples were extracted as described.41,42 After evaporating the sample, ESR spectra were immediately recorded at room temperature using a quartz flat cell (160 μl) in a JEOL JES-TE200 spectrometer (JEOL, Tokyo, Japan). Operating conditions of ESR; 9.43 GHz, field 335.2 ± 5 mT, 40 m microwave power, 100 kHz modulation frequency, 0.25 field modulation width, 0.3 ms time count and sweep time 2 minutes.
Statistical Analysis
All values are expressed as the mean ± SEM. Two-way analysis of variance followed by the Tukey test or the Student’s t-test for unpaired results was used to evaluate differences between more than three groups or between two groups, respectively. Differences were considered to be significant for values of P < 0.05.
Results
A Phenotype of CHOP-Null Mice Resistant to Experimental Colitis
The severity of DSS-induced colitis can be monitored by various indices, such as body weight, DAI, length of colon, MPO activity, the amount of TBARS, and histological indices. We compared the development of colitis induced by 3% DSS administration in CHOP-null mice and WT mice by measuring body weight and DAI daily. Administration of 3% DSS caused a decrease in body weight and an increase in the DAI of the WT mice (Figure 1, A and B). DSS-induced colitis in this phenotype was significantly ameliorated in CHOP-null mice (Figure 1, A and B). DSS-induced colon shortening, used as a morphometric measure for the degree of inflammation, was less severe in CHOP-null mice than in the WT mice (Figure 1C). Colonic MPO activity, an indicator of infiltration of leukocytes, was lower in DSS-administered CHOP-null mice than in the WT mice (Figure 1D). Colonic TBARS, an index of lipid peroxidation associated with inflammation, was also lower in DSS-administered CHOP-null mice than in the WT mice (Figure 1E). Absence of the Chop gene did not affect the background levels of these indices (colon length, colonic MPO activity, and colonic TBARS) (Figure 1, C–E). Figure 1F shows results of histological analyses of colonic tissues prepared from DSS-administered and control CHOP-null mice and WT mice. Crypt loss and infiltration of leukocytes were more apparent in sections from DSS-administered WT mice than those from CHOP-null mice. Histological score analysis (damage score and extent of lesion) revealed that the histological differences between DSS-administered CHOP-null mice and the WT mice were statistically significant (Figure 1, G and H). Results in Figure 1 show that CHOP-null mice are more resistant to DSS-induced colitis than the WT mice.
Figure 1.

Development of DSS-induced colitis in CHOP-null mice and WT mice. CHOP-null mice (Chop−/−) and WT mice were treated with or without 3% DSS for 7 days. Body weight (A) and DAI (B) were measured daily. After 7 days, length of colon (C), colonic MPO activity (D), and colonic TBARS (E) were determined as described in the Materials and Methods. G and H: After 7 days, sections of colonic tissues were prepared and subjected to histological examination (H&E staining) and the damage score and extent of lesion for seven independent sections were determined. F: One of the sections is shown. Values are mean ± SEM (n = 5 to 13). **P < 0.01; *P < 0.05; n.s., not significant.
We examined the effect of DSS administration on expression of CHOP and GRP78 in the intestine at both mRNA and protein levels. Analysis by real-time RT-PCR revealed that the mRNA expression of Chop in colonic tissues was induced by the DSS administration in the WT mice and that the Chop mRNA was not expressed in CHOP-null mice (Figure 2A). The mRNA expression of Grp78 was also induced by DSS administration (Figure 2A), suggesting that the ER stress response is induced simultaneously with development of DSS-induced colitis. Results in Figure 2A also suggest that CHOP positively regulates the mRNA expression of Grp78 in the intestine.
Figure 2.

Expression of CHOP and GRP78 in colonic tissues of CHOP-null mice and WT mice. DSS was administered to CHOP-null mice (Chop−/−) and WT mice, as described in the legend of Figure 1. Colonic tissues were removed and total RNA was extracted. Samples were subjected to real-time RT-PCR, using a specific primer set for Chop or Grp78. Values were normalized to Gapdh, expressed relative to the control sample (ie, WT mice without DSS treatment), and given as the mean ± SEM (n = 4). A: ** or ##P < 0.01; *P < 0.05; n.s., not significant. Sections of colonic tissues were prepared and subjected to immunohistochemical analysis with an antibody against CHOP (B) or GRP78 (C). B: The right panel in each column is a ×4 magnified image of the boxed area defined in the left panel. D: Total protein was extracted from colonic tissues and analyzed by immunoblotting with an antibody against CHOP, GRP78, or actin.
Immunohistochemical and immunoblotting analyses demonstrated that DSS administration increases the level of CHOP in colonic tissues in the WT mice but not in CHOP-null mice (Figure 2, B and D). CHOP expression was localized in nuclei, being consistent that CHOP is a transcription factor. We also found that DSS administration increases the level of GRP78 in colonic tissues in WT mice and that this increase was not so clearcut in CHOP-null mice (Figure 2, C and D).
We also examined the effect of a deficiency in CHOP on the development of TNBS-induced colitis. As shown in Figure 3A, a TNBS (3 mg/kg)-dependent decrease in body weight was less apparent in CHOP-null mice than in WT mice. Administration of a higher dose of TNBS (8 mg/kg) caused the death of some mice, with the survival rate of CHOP-null mice being much higher than that of WT mice (Figure 3B). Histological analysis of colonic tissues revealed that TNBS-dependent intestinal mucosal damage was more severe in the WT mice than in CHOP-null mice (Figure 3, C–E). We also found that this TNBS (3 mg/kg) administration up-regulated the mRNA expression of Chop and Grp78 in the intestine (data not shown).
Figure 3.

Development of TNBS-induced colitis in CHOP-null mice and WT mice. CHOP-null mice (Chop−/−) and WT mice were intrarectally administered with (TNBS) or without (EtOH) TNBS of 3 mg/mouse (A, C–E) or 8 mg/mouse (B) once and cultivated for the indicated days. Body weight (A) and mouse survival rate (B) were measured daily. After 1 day, sections of colonic tissues were prepared and subjected to histological examination (H&E staining) as described in the legend of Figure 1, C–E. Values are mean ± SEM (n = 6 to 12). **P < 0.01; n.s., not significant.
Analysis of the mRNA Expression of Various Genes in DSS-Administered CHOP-Null Mice and WT Mice
To obtain a better understanding of the molecular mechanism governing CHOP-dependent exacerbation of DSS-induced colitis, we compared the mRNA expression of various genes in the intestine of both DSS-administered CHOP-null mice and WT mice. Tested genes included those for pro-inflammatory cytokines (Il-1β, Tnf-α, and Il-6), CAMs expressed on vascular endothelial cells (Madcam-1, Vcam-1, and Icam-1), CAMs expressed on leukocytes (CD11b, CD49d, and L-selectin), an anti-inflammatory cytokine (Il-10), and CHOP-regulated genes (Caspase-11, Ero-1α, and Bcl-2). Mac-1 and very late gene (VLA)-4 are CAMs expressed on leukocytes and Mac-1 is a heterodimer of CD11b and CD18 and VLA-4 is a heterodimer of CD49d and CD29. The mRNA expression of all of the pro-inflammatory cytokine genes tested was up-regulated by DSS administration, whereas the mRNA expression of Tnf-α but not that of Il-1β and Il-6 was significantly lower in DSS-administered CHOP-null mice than in WT mice (Figure 4A). On the other hand, the mRNA expression of Il-10 was higher in DSS-administered CHOP-null mice than in WT mice (Figure 4D).
Figure 4.

The mRNA expression of various genes in colonic tissues. DSS was administered to CHOP-null mice (Chop−/−) and WT mice, as described in the legend of Figure 1. Relative mRNA expression of each gene in colonic tissues was monitored and expressed as described in the legend of Figure 2A. A: IL-1β, TNF-α, IL-6. B: Madcam-1, Vcam-1, Icam-1. C: CD11b, CD49d, L-selectin. D: IL-10. E: Capsase-11. F: Ero-1α, Bcl-2. Values are mean ± SEM (n = 4 to 5). ** or ##P < 0.01; * or #P < 0.05; n.s., not significant.
The mRNA expression of all CAM genes tested was also up-regulated by DSS administration and the mRNA expression of Vcam-1 and CD11b but not other CAM genes was significantly lower in DSS-administered CHOP- null mice than in WT mice (Figure 4, B and C). The mRNA expression of Caspase-11 and Ero-1α but not Bcl-2 was up-regulated by DSS administration in the WT mice, and the mRNA expression of Caspase-11 and Ero-1α was significantly lower in DSS-administered CHOP-null mice than in WT mice (Figure 4, E and F).
The results in Figure 4 suggest that CHOP regulates the mRNA expression of Tnf-α, Il-10, CD11b, Caspase-11, and Ero-1α under inflammatory conditions. To test this idea in vitro, we compared the mRNA expression of these factors in the presence of LPS in peritoneal macrophages prepared from CHOP-null mice and WT mice. As shown in Figure 5A, LPS treatment stimulated the expression of Chop and Grp78 mRNAs in macrophages in a CHOP-dependent manner. The mRNA expression of CD11b, Caspase-11, and Ero-1α was up-regulated by the LPS treatment, and the expression of these genes was significantly lower in LPS-treated CHOP-null macrophages than in the WT macrophages (Figure 5, C, E, and F). The mRNA expression of Il-10 in the presence of LPS was higher in CHOP-null macrophages than in WT macrophages. However, no significant difference was found in the mRNA expression of Tnf-α (Figure 5, B and D). These results suggest that CHOP is involved in the expression of CD11b, Caspase-11, Il-10, and Ero-1α under inflammatory conditions, and that these may play an important role in CHOP-dependent exacerbation of DSS-induced colitis.
Figure 5.

The mRNA expression of various genes in peritoneal macrophages. Peritoneal macrophages were prepared from CHOP-null mice (Chop−/−) and WT mice and incubated with 10 ng/ml of LPS for 12 hours (3 hours for the Tnf-α). Relative mRNA expression of each gene in colonic tissues was monitored and expressed as described in the legend of Figure 2A. A:Chop, Grp78. B: Tnf-α. C:CD11b, D: IL-10. E:Caspase-11. F:Ero-1α. Values are mean ± SEM (n = 3). ** or ##P < 0.01; * or #P < 0.05; n.s., not significant.
Involvement of IL-1β and Infiltration of Macrophages in the CHOP-Dependent Exacerbation of DSS-Induced Colitis
CHOP-induced expression of Caspase-11 and the resulting activation of Caspase-1 and stimulation of production of IL-1β are responsible for CHOP-dependent exacerbation of LPS-induced lung inflammation.39 To test whether a similar mechanism is involved in the CHOP-dependent exacerbation of DSS-induced colitis, we measured the levels of IL-1β and Caspase-1 activity in colonic tissues in DSS-administered mice. As shown in Figure 6, A and B, DSS administration increased the level of IL-1β and Caspase-1 activity in colonic tissues in WT mice. These alterations were significantly suppressed in CHOP-null mice. Similar results were observed in LPS-treated macrophages; the levels of IL-1β and the Caspase-1 activity in LPS-stimulated peritoneal macrophages prepared from CHOP-null mice were lower than in those from the WT mice (Figure 6, C and D).
Figure 6.

Involvement of Caspase-11-Caspase-1-IL-1β pathway in CHOP-dependent exacerbation of DSS-induced colitis. DSS was administered to CHOP-null mice (Chop−/−) and wild-type mice (WT), as described in the legend of Figure 1, A and B. C and D: Peritoneal macrophages were prepared from CHOP-null mice (Chop−/−) and WT mice and incubated with 10 ng/ml of LPS for 24 hours. A and C: The amount of IL-1β was determined by enzyme-linked immunosorbent assay. B and D: The Caspase-1 activity was measured as described in Materials and Methods. Values are mean ± SEM (n = 3 to 4). **P < 0.01; *P < 0.05; n.s., not significant.
Mac-1 is a CAM expressed on macrophages; its binding to ICAM-1 expressed on vascular endothelial cells is important for infiltration of blood-circulating macrophages into inflamed tissues.43 To test whether infiltration of macrophages is involved in the CHOP-dependent exacerbation of DSS-induced colitis, we compared infiltration of macrophages between DSS-administered CHOP-null mice and the WT mice by immunohistochemical analysis with an antibody against CD68, which is expressed on lysosomal membranes of macrophages.44 As shown in Figure 7A (see magnified panels), the antibody targeted mononuclear cells, confirming that this antibody can be used to detect macrophages. Results in Figure 7A show that DSS-induced infiltration of macrophages into colonic tissues was suppressed in CHOP-null mice compared with the WT mice. To confirm a role for CHOP in the expression of CD11b (Mac-1) in vitro, we used RAW264 cells (mouse leukemic monocyte) and plasmids for overexpression of CHOP and C/EBP-β, known to act co-ordinately with CHOP in the transcription of some genes.45 As shown in Figure 7B, CD11b mRNA expression was up-regulated by transfection of cells with the plasmid with Chop, and this up-regulation was further stimulated by simultaneous transfection with a plasmid with C/ebp-β. We confirmed the overexpression of Chop and C/ebp-β, depending on the transfection of each plasmid (Figure 7, C and D). We found the CHOP-binding consensus sequences in the promoter of CD11b (Figure 7E). Overexpression of C/ebp-β did not affect the mRNA expression of Chop. Results in Figure 7 suggest that up-regulation of the expression of CD11b (Mac-1) by CHOP is involved in CHOP-dependent exacerbation of DSS-induced colitis through stimulation of macrophage infiltration into the intestine.
Figure 7.

Involvement of infiltration of macrophages in CHOP-dependent exacerbation of DSS-induced colitis. DSS was administered to CHOP-null mice (Chop−/−) and WT mice, as described in the legend of Figure 1. A: Sections of colonic tissues were prepared and subjected to immunohistochemical analysis with an antibody against CD68. The bottom panel in each column is a ×4 magnified image of the boxed area defined in the middle panel. RAW264 cells were transiently transfected with expression plasmid for CHOP and C/EBP-β and cultured for 12 hours. The relative mRNA expression of each gene was monitored and expressed as described in the legend of Figure 2A. B–D: Values are mean ± SEM (n = 3). **P < 0.01; *P < 0.05; n.s. not significant. E: The structure and sequences of the CD11b promoter are shown.
Involvement of ROS Production and ROS-Induced Apoptosis in Exacerbation of DSS-Induced Colitis
We measured ROS production in the intestine by measuring the lipid-derived free radical spin adduct with ESR spectroscopy and spin trap POBN, which reacts with ROS to form a radical spin adduct. This method, in vivo free radical production ex vivo detection, was shown to be effective for monitoring ROS production in the lung in vivo.41,42 However, this is the first attempt to use this technique on intestinal tissue. As shown in Figure 8A, a radical spin adduct of ESR spectrum similar to that reported in the lung was obtained. The hyperfine coupling constants for the POBN radical adducts were αN = 14.91 ± 0.08 G and 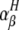 = 2.45 ± 0.04 G, which are similarto data to data previously reported in the lung,41,42 suggesting that this ESR spectrum is derived from lipid-derived free radicals and that this method can be used for the monitoring the ROS production at the intestine in vivo. As shown in Figure 8B, the level of ROS production in the intestine was elevated by DSS administration in the WT mice and lower in DSS-administered CHOP-null mice than in the WT mice. We also found that administration of vitamin E, an antioxidant, decreased the intestinal level of ROS production and the DAI in DSS-treated WT mice (Supplemental Figure 1 at http://ajp.amjpathol.org). Results suggest that the DSS-induced production of ROS in the intestine was suppressed in CHOP-null mice. Production of ROS in peritoneal macrophages prepared from CHOP-null mice and WT mice was compared using fluorescence-activated cell sorting analysis. The cell-permeable fluorescent dye, H2DCF, can be converted to a fluorescent product, DCF, in a ROS-dependent manner. Therefore, the increase in the green fluorescence (x axis in Figure 8, C, D, and G) is indicative of an increase in the level of ROS production.40 Treatment of WT macrophages with LPS stimulated ROS production (Figure 8C), as has been described previously.46 However, this stimulation was not clearly observed in CHOP-null macrophages, indicating that CHOP is responsible for LPS-stimulated ROS production (Figure 8C). We also examined the effect of siRNA for CHOP on LPS-stimulated ROS production in RAW264 cells (Figure 8D). We confirmed that treatment of RAW264 cells with LPS up-regulated the expression of CHOP mRNA and that transfection of cells with siRNA for CHOP significantly suppressed the expression of not only CHOP but also Ero-1α mRNA (Figure 8, E and F). As is the case with peritoneal macrophages, treatment of RAW264 cells with LPS stimulated ROS production and transfection of cells with siRNA for CHOP partially suppressed this LPS-stimulated ROS production (Figure 8D). To examine the role of ERO-1α in this CHOP-dependent production of ROS, we also examined the effect of siRNA for ERO-1α on LPS-stimulated ROS production in RAW264 cells. We confirmed that transfection of cells with siRNA for ERO-1α significantly suppressed the expression of Ero-1α mRNA (Figure 8H). Transfection of cells with siRNA for ERO-1α partially suppressed this LPS-stimulated ROS production (Figure 8G). Results in Figure 8 suggest that CHOP stimulates ROS production in macrophages at least partly through the up-regulation of ERO-1α.
= 2.45 ± 0.04 G, which are similarto data to data previously reported in the lung,41,42 suggesting that this ESR spectrum is derived from lipid-derived free radicals and that this method can be used for the monitoring the ROS production at the intestine in vivo. As shown in Figure 8B, the level of ROS production in the intestine was elevated by DSS administration in the WT mice and lower in DSS-administered CHOP-null mice than in the WT mice. We also found that administration of vitamin E, an antioxidant, decreased the intestinal level of ROS production and the DAI in DSS-treated WT mice (Supplemental Figure 1 at http://ajp.amjpathol.org). Results suggest that the DSS-induced production of ROS in the intestine was suppressed in CHOP-null mice. Production of ROS in peritoneal macrophages prepared from CHOP-null mice and WT mice was compared using fluorescence-activated cell sorting analysis. The cell-permeable fluorescent dye, H2DCF, can be converted to a fluorescent product, DCF, in a ROS-dependent manner. Therefore, the increase in the green fluorescence (x axis in Figure 8, C, D, and G) is indicative of an increase in the level of ROS production.40 Treatment of WT macrophages with LPS stimulated ROS production (Figure 8C), as has been described previously.46 However, this stimulation was not clearly observed in CHOP-null macrophages, indicating that CHOP is responsible for LPS-stimulated ROS production (Figure 8C). We also examined the effect of siRNA for CHOP on LPS-stimulated ROS production in RAW264 cells (Figure 8D). We confirmed that treatment of RAW264 cells with LPS up-regulated the expression of CHOP mRNA and that transfection of cells with siRNA for CHOP significantly suppressed the expression of not only CHOP but also Ero-1α mRNA (Figure 8, E and F). As is the case with peritoneal macrophages, treatment of RAW264 cells with LPS stimulated ROS production and transfection of cells with siRNA for CHOP partially suppressed this LPS-stimulated ROS production (Figure 8D). To examine the role of ERO-1α in this CHOP-dependent production of ROS, we also examined the effect of siRNA for ERO-1α on LPS-stimulated ROS production in RAW264 cells. We confirmed that transfection of cells with siRNA for ERO-1α significantly suppressed the expression of Ero-1α mRNA (Figure 8H). Transfection of cells with siRNA for ERO-1α partially suppressed this LPS-stimulated ROS production (Figure 8G). Results in Figure 8 suggest that CHOP stimulates ROS production in macrophages at least partly through the up-regulation of ERO-1α.
Figure 8.

Involvement of ROS production in CHOP-dependent exacerbation of DSS-induced colitis. DSS was administered to CHOP-null mice (Chop−/−) and wild-type mice (WT), as described in the legend of Figure 1. A: After administration of POBN, the colons were dissected and subjected to radical adduct ESR spectrum analysis. The intensity of the ESR signal of radical adduct (shown by bars) was determined, expressed relative to the control sample (ie, wild-type mice without DSS treatment), and given as the mean ± SEM (n = 3). B: *P < 0.05; n.s., not significant. C: Peritoneal macrophages were prepared from CHOP-null mice (Chop−/−) and WT mice and incubated with 10 ng/ml of LPS for 24 hours. D–H: RAW264 cells were transiently transfected with siRNA for CHOP (D–F) or ERO-1α (G, H) or nonsilencing (ns) siRNA (D–H) and incubated with 50 ng/ml of LPS for 24 hours. The production of ROS was monitored by fluorescence-activated cell sorting analysis with H2DCF, as described in the Materials and Methods (C, D, and G). Relative mRNA expression of the CHOP (E) or Ero-1α (F, H) was monitored and expressed as described in the legend of Figure 2A. Values are mean ± SEM (n = 3). ** or ##P < 0.01.
Next, we compared the level of apoptosis in the colonic mucosa of DSS-administered CHOP-null mice and WT mice by use of the TUNEL assay. More TUNEL-positive cells (apoptosis) were observed in the colonic mucosa of DSS-administered WT mice than in CHOP-null mice (Figure 9A), suggesting that ROS-induced apoptosis associated with DSS-induced colitis is suppressed in CHOP-null mice.
Figure 9.

Involvement of ROS-induced apoptosis in CHOP-dependent exacerbation of DSS-induced colitis. DSS was administered to CHOP-null mice (Chop−/−) and WT mice, as described in the legend of Figure 1. Sections of colonic tissues were prepared and subjected to TUNEL assay and DAPI staining. A: The right panel in each column is a ×3 magnified image of the boxed area defined in the left panel. HCT-15 cells were transfected with siRNA for CHOP (siCHOP) or nonsilencing (ns) siRNA and were incubated with indicated concentrations of menadione for 12 hours. B: Relative mRNA expression of the CHOP was monitored as described in the legend of Figure 2A. C: Cell viability was determined using the MTT method. D: The intracellular antioxidant activity was measured as described in the Materials and Methods. Values shown are mean ± SEM (n = 3). ** or ##P < 0.01; n.s., not significant.
To test the role of CHOP in ROS-induced apoptosis in vitro, we examined the effect of siRNA for CHOP on cell death induced by menadione, a superoxide anion (a representative ROS) releasing drug, in a colonic cancer cell line (HCT-15). We confirmed that transfection of cells with siRNA for CHOP inhibited the mRNA expression of CHOP in both the presence and absence of menadione (Figure 9B). As shown in Figure 9C, treatment of cells with menadione induced cell death in a dose-dependent manner, whereas transfection of cells with siRNA for CHOP significantly suppressed this menadione-induced cell death. We confirmed that cell death such as that evident in Figure 9C was mediated by apoptosis by showing that apoptotic DNA fragmentation and chromatin condensation accompanied the cell death (data not shown). The results in Figure 9 suggest that CHOP makes colonic cells sensitive to ROS-induced apoptosis, maybe through modulation of apoptosis-inducing pathway such as down-regulation of Bcl-2 and up-regulation of Bim.16,17,18 We also examined the effect of CHOP on intracellular antioxidant activity. As shown in Figure 9D, the level of antioxidant activity that was measured by extinction of hydrogen peroxide was not affected by transfection of siRNA for CHOP. Results in Figures 8 and 9 imply that CHOP stimulates ROS-induced apoptosis through both increasing the ROS production and modulating the apoptosis-inducing pathway.
Discussion
CHOP is a transcription factor involved in the ER stress response, which has been recently revealed to play an important role in various diseases, including neurodegenerative diseases, diabetes, gastric ulcer, and lung inflammation.24,39,47,48,49,50 However, its role in IBD has remained unknown. In this study, we obtained direct genetic evidence that CHOP stimulates the development of DSS- and TNBS-induced colitis, animal models of IBD, by showing that CHOP is up-regulated by DSS or TNBS administration and that CHOP-null mice are resistant to development of experimental colitis in these models. It was recently reported that IRE1β knockout mice are sensitive to DSS-induced colitis51 and that up-regulation of GRP78 in IL-10 knockout mice contributes to spontaneous development of colitis through activation of nuclear factor-κB.21 Thus, it seems that some aspects of the ER stress response are positively involved, and other aspects negatively involved, in the development of experimental colitis.
Our in vivo and in vitro analyses suggested that CHOP stimulates the development of DSS-induced colitis via several mechanisms. One of these is the Caspase-11-Casapse-1-IL-1β pathway, which was recently shown to play an important role in LPS-induced lung inflammation.39 It is well-known that IL-1β stimulates the development of IBD and experimental colitis,9 that CHOP positively regulates the transcription of Caspase-11, and that Caspase-11 activates Caspase-1, and that the production of IL-1β from pro-IL-1β is catalyzed by Caspase-1.39 In this study, our in vivo analysis showed that the intestinal level of IL-1β and the activity of Caspase-1 was lower in DSS-administered CHOP-null mice than in WT mice. Because the production of IL-1β is stimulated under inflammatory conditions, it is possible that this decreased level of IL-1β is not a cause but a result of the amelioration of colitis in CHOP-null mice. However, we showed that up-regulation of Caspase-11 mRNA expression and the activity of Caspase-1 in the intestine after DSS administration was suppressed in CHOP-null mice and in macrophages prepared from CHOP-null mice, compared with the respective WT control. These results suggest that the Caspase-11-Casapse-1-IL-1β pathway is involved in the CHOP-dependent exacerbation of DSS-induced colitis.
We also postulated that CHOP positively regulates the expression of CD11b (Mac-1) and that this mechanism is involved in CHOP-dependent exacerbation of DSS-induced colitis thorough stimulation of macrophage infiltration into the intestine. Mac-1 is expressed on the macrophage cell surface and stimulates the infiltration of blood-circulating macrophages into inflamed tissues.43 In this study, we showed that CD11b (Mac-1) mRNA expression and infiltration of macrophages into the intestine were both reduced in DSS-administered CHOP-null mice compared with WT mice. We also showed that up-regulation or down-regulation of expression of Chop stimulates or suppresses, respectively, the expression of CD11b (Mac-1). Furthermore, we found the consensus DNA sequence for CHOP binding in the promoter region of CD11b. Because IL-1β was reported to stimulate infiltration of macrophages,52 lower levels of IL-1β in DSS-administered CHOP-null mice may contribute to their lower levels of macrophage infiltration. However, it is also possible that the CHOP-dependent inductions of CD11b (Mac-1) expression is attributable to the increased recruitment of Mac-1-positive myeloid cells.
We showed that intestinal ROS production is lower in DSS-administered CHOP-null mice than in WT mice and propose that this is one of the mechanisms governing the CHOP-dependent exacerbation of DSS-induced colitis. Although a number of in vitro studies have suggested that CHOP is involved in ROS production,40,53 this is the first evidence showing that CHOP is involved in ROS production in vivo. We revealed this by use of radical spin adduct ESR spectrum analysis. This analysis should be useful for detecting intestinal ROS production in vivo. Furthermore, we suggested that the lower levels of ROS production in CHOP-null mice are attributable to the lower level of expression of ERO-1α; Ero-1α mRNA expression in the intestine in DSS-administered CHOP-null mice and ROS production in LPS-stimulated macrophages prepared from the mice were lower than in the respective WT control samples. Furthermore, we showed that siRNA for ERO-1α suppresses LPS-stimulated ROS production in RAW264 cells.
Analysis using the TUNEL assay revealed that DSS-induced apoptosis in colonic mucosa was inhibited in CHOP-null mice. This correlates with other parameters for DSS-induced colitis. However, it was not clear whether the alteration to apoptosis is caused by or is a result of the inhibition of DSS-induced colitis. Given that transfection with siRNA for CHOP inhibited ROS-induced apoptosis in vitro, this result suggests that CHOP stimulates ROS-induced apoptosis, which seems to contribute to the lower level of apoptosis in the colonic mucosa and to a phenotype resistant to DSS-induced colitis as seen in CHOP-null mice. A number of mechanisms have been proposed for the stimulation of apoptosis by CHOP, such as down-regulation of Bcl-2, translocation of BAX to mitochondria, and activation of Bim.16,40,54 Because we showed that Bcl-2 mRNA expression was not affected by the lack of Chop, other mechanisms seem to be involved. It is also possible that activation of Caspase-11 is involved in the stimulation of ROS-induced apoptosis by CHOP, because Caspase-11 stimulates the activation of capsase-3 and Caspase-7, both of which are directly involved in the induction of apoptosis.55
In addition to the mechanisms described above, other mechanisms may be involved in the CHOP-dependent exacerbation of DSS-induced colitis, such as down-regulation of IL-10 and up-regulation of GRP78. IL-10 was reported to suppress development of IBD and experimental colitis,56 whereas expression of GRP78 stimulates the development of DSS-induced colitis through activation of nuclear factor-κB.21 We showed here that expression of Il-10 or Grp78 mRNA in the intestine of DSS-administered CHOP-null mice was higher or lower, respectively, than in the WT control.
Glucocorticoids, 5-aminosalicylic acid (5-ASA), and immunosuppressive drugs are currently used for the clinical treatment of IBD.2 Although some new types of drugs, such as infliximab, have been developed recently for the treatment of IBD, a number of clinical problems, such as side effects, are yet to be addressed.57 Thus, IBD remains an uncured disease for which the development of new types of drugs is clinically important. As described above, some aspects of the ER stress response are positively and other aspects negatively involved in IBD development. As such, factors located downstream (such as CHOP) rather than upstream (such as ATF6 and IRE1) of the ER stress response may be better drug targets for IBD. Results in this study suggest that inhibitors of CHOP may be therapeutically beneficial for IBD.
In summary, results in this study show that CHOP is positively involved in the development of DSS-induced colitis. Furthermore, the results suggest that this effect involves various mechanisms, such as Mac-1-induced infiltration of macrophages, ERO-1α-induced ROS production, Caspase-11-induced production of IL-1β, and stimulation of mucosal apoptosis.
Acknowledgments
We thank Dr. Akira Shizuo (Osaka University, Osaka, Japan) for generously providing the mice.
Footnotes
Address reprint requests to Tohru Mizushima, Graduate School of Medical and Pharmaceutical Sciences, Kumamoto University, 5-1 Oe-honmachi, Kumamoto 862-0973, Japan. E-mail: mizu@gpo.kumamoto-u.ac.jp.
Supported by the Ministry of Health, Labor, and Welfare of Japan (grants-in-aid for scientific research); the Japan Science and Technology Agency; and the Ministry of Education, Culture, Sports, Science, and Technology of Japan (grants-in-aid for scientific research).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Cuzzocrea S. Emerging biotherapies for inflammatory bowel disease. Expert Opin Emerg Drugs. 2003;8:339–347. doi: 10.1517/14728214.8.2.339. [DOI] [PubMed] [Google Scholar]
- Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- Jurjus AR, Khoury NN, Reimund JM. Animal models of inflammatory bowel disease. J Pharmacol Toxicol Methods. 2004;50:81–92. doi: 10.1016/j.vascn.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Danese S, Semeraro S, Marini M, Roberto I, Armuzzi A, Papa A, Gasbarrini A. Adhesion molecules in inflammatory bowel disease: therapeutic implications for gut inflammation. Dig Liver Dis. 2005;37:811–818. doi: 10.1016/j.dld.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP, Raedler A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin Exp Immunol. 1993;94:174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology. 1995;109:1344–1367. doi: 10.1016/0016-5085(95)90599-5. [DOI] [PubMed] [Google Scholar]
- Koizumi M, King N, Lobb R, Benjamin C, Podolsky DK. Expression of vascular adhesion molecules in inflammatory bowel disease. Gastroenterology. 1992;103:840–847. doi: 10.1016/0016-5085(92)90015-q. [DOI] [PubMed] [Google Scholar]
- Kinoshita K, Hori M, Fujisawa M, Sato K, Ohama T, Momotani E, Ozaki H. Role of TNF-alpha in muscularis inflammation and motility disorder in a TNBS-induced colitis model: clues from TNF-alpha-deficient mice. Neurogastroenterol Motil. 2006;18:578–588. doi: 10.1111/j.1365-2982.2006.00784.x. [DOI] [PubMed] [Google Scholar]
- Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- Palmen MJ, Dijkstra CD, van der Ende MB, Pena AS, van Rees EP. Anti-CD11b/CD18 antibodies reduce inflammation in acute colitis in rats. Clin Exp Immunol. 1995;101:351–356. doi: 10.1111/j.1365-2249.1995.tb08363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon F, Contractor N, Fuss I, Marth T, Lahey E, Iwaki S, la Sala A, Hoffmann V, Strober W, Kelsall BL. Antibodies to complement receptor 3 treat established inflammation in murine models of colitis and a novel model of psoriasiform dermatitis. J Immunol. 2006;177:6974–6982. doi: 10.4049/jimmunol.177.10.6974. [DOI] [PubMed] [Google Scholar]
- Bendjelloul F, Maly P, Mandys V, Jirkovska M, Prokesova L, Tuckova L, Tlaskalova-Hogenova H. Intercellular adhesion molecule-1 (ICAM-1) deficiency protects mice against severe forms of experimentally induced colitis. Clin Exp Immunol. 2000;119:57–63. doi: 10.1046/j.1365-2249.2000.01090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacyshyn BR, Bowen-Yacyshyn MB, Jewell L, Tami JA, Bennett CF, Kisner DL, Shanahan WR., Jr A placebo-controlled trial of ICAM-1 antisense oligonucleotide in the treatment of Crohn’s disease. Gastroenterology. 1998;114:1133–1142. doi: 10.1016/s0016-5085(98)70418-4. [DOI] [PubMed] [Google Scholar]
- Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1036. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- Rutgeerts P, Van Assche G, Vermeire S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology. 2004;126:1593–1610. doi: 10.1053/j.gastro.2004.02.070. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem. 2005;280:33917–33925. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
- Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- Shkoda A, Ruiz PA, Daniel H, Kim SC, Rogler G, Sartor RB, Haller D. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132:190–207. doi: 10.1053/j.gastro.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Burczynski ME, Peterson RL, Twine NC, Zuberek KA, Brodeur BJ, Casciotti L, Maganti V, Reddy PS, Strahs A, Immermann F, Spinelli W, Schwertschlag U, Slager AM, Cotreau MM, Dorner AJ. Molecular classification of Crohn’s disease and ulcerative colitis patients using transcriptional profiles in peripheral blood mononuclear cells. J Mol Diagn. 2006;8:51–61. doi: 10.2353/jmoldx.2006.050079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Namba T, Arai Y, Fujimoto M, Adachi H, Sobue G, Takeuchi K, Nakai A, Mizushima T. Genetic evidence for a protective role for heat shock factor 1 and heat shock protein 70 against colitis. J Biol Chem. 2007;282:23240–23252. doi: 10.1074/jbc.M704081200. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada I, Akira S, Araki E, Mori M. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA. 2001;98:10845–10850. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumi G, Tsubouchi R, Nishio H, Kato S, Mozsik G, Takeuchi K. Dual role of endogenous nitric oxide in development of dextran sodium sulfate-induced colitis in rats. J Physiol Pharmacol. 2004;55:823–836. [PubMed] [Google Scholar]
- Nishihara T, Matsuda M, Araki H, Oshima K, Kihara S, Funahashi T, Shimomura I. Effect of adiponectin on murine colitis induced by dextran sulfate sodium. Gastroenterology. 2006;131:853–861. doi: 10.1053/j.gastro.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Krawisz JE, Sharon P, Stenson WF. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Rumi G, Tsubouchi R, Okayama M, Kato S, Mozsik G, Takeuchi K. Protective effect of lactulose on dextran sulfate sodium-induced colonic inflammation in rats. Dig Dis Sci. 2004;49:1466–1472. doi: 10.1023/b:ddas.0000042248.48819.ad. [DOI] [PubMed] [Google Scholar]
- Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Tsutsumi S, Arai Y, Hoshino T, Suzuki K, Takaki E, Ito T, Takeuchi K, Nakai A, Mizushima T. Genetic evidence for a protective role of heat shock factor 1 against irritant-induced gastric lesions. Mol Pharmacol. 2007;71:985–993. doi: 10.1124/mol.106.033282. [DOI] [PubMed] [Google Scholar]
- Tsutsumi S, Tomisato W, Takano T, Rokutan K, Tsuchiya T, Mizushima T. Gastric irritant-induced apoptosis in guinea pig gastric mucosal cells in primary culture. Biochim Biophys Acta. 2002;1589:168–180. doi: 10.1016/s0167-4889(02)00171-4. [DOI] [PubMed] [Google Scholar]
- Mima S, Tsutsumi S, Ushijima H, Takeda M, Fukuda I, Yokomizo K, Suzuki K, Sano K, Nakanishi T, Tomisato W, Tsuchiya T, Mizushima T. Induction of claudin-4 by nonsteroidal anti-inflammatory drugs and its contribution to their chemopreventive effect. Cancer Res. 2005;65:1868–1876. doi: 10.1158/0008-5472.CAN-04-2770. [DOI] [PubMed] [Google Scholar]
- Ohkawara T, Nishihira J, Takeda H, Hige S, Kato M, Sugiyama T, Iwanaga T, Nakamura H, Mizue Y, Asaka M. Amelioration of dextran sulfate sodium-induced colitis by anti-macrophage migration inhibitory factor antibody in mice. Gastroenterology. 2002;123:256–270. doi: 10.1053/gast.2002.34236. [DOI] [PubMed] [Google Scholar]
- Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- Nishiyori A, Tashiro H, Kimura A, Akagi K, Yamamura K, Mori M, Takiguchi M. Determination of tissue specificity of the enhancer by combinatorial operation of tissue-enriched transcription factors. Both HNF-4 and C/EBP beta are required for liver-specific activity of the ornithine transcarbamylase enhancer. J Biol Chem. 1994;269:1323–1331. [PubMed] [Google Scholar]
- Gotoh T, Oyadomari S, Mori K, Mori M. Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J Biol Chem. 2002;277:12343–12350. doi: 10.1074/jbc.M107988200. [DOI] [PubMed] [Google Scholar]
- Salimuddin , Nagasaki A, Gotoh T, Isobe H, Mori M. Regulation of the genes for arginase isoforms and related enzymes in mouse macrophages by lipopolysaccharide. Am J Physiol. 1999;277:E110–E117. doi: 10.1152/ajpendo.1999.277.1.E110. [DOI] [PubMed] [Google Scholar]
- Endo M, Mori M, Akira S, Gotoh T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J Immunol. 2006;176:6245–6253. doi: 10.4049/jimmunol.176.10.6245. [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Kadiiska MB, Ghio AJ, Corbett J, Fann YC, Holland SM, Thurman RG, Mason RP. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: a model for ARDS. FASEB J. 2002;16:1713–1720. doi: 10.1096/fj.02-0331com. [DOI] [PubMed] [Google Scholar]
- Sato K, Akaike T, Kohno M, Ando M, Maeda H. Hydroxyl radical production by H2O2 plus Cu,Zn-superoxide dismutase reflects the activity of free copper released from the oxidatively damaged enzyme. J Biol Chem. 1992;267:25371–25377. [PubMed] [Google Scholar]
- Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, Hibbs ML, Springer TA. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18). J Cell Biol. 1990;111:3129–3139. doi: 10.1083/jcb.111.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah YM, Morimura K, Gonzalez FJ. Expression of peroxisome proliferator-activated receptor-gamma in macrophage suppresses experimentally induced colitis. Am J Physiol. 2007;292:G657–G666. doi: 10.1152/ajpgi.00381.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HY, Wen MH. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem. 2002;277:22131–22139. doi: 10.1074/jbc.M111883200. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–345. doi: 10.1023/a:1016175429877. [DOI] [PubMed] [Google Scholar]
- Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ, Takenaka H, Tsuchiya T, Mori M, Mizushima T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004;11:1009–1016. doi: 10.1038/sj.cdd.4401436. [DOI] [PubMed] [Google Scholar]
- Milhavet O, Martindale JL, Camandola S, Chan SL, Gary DS, Cheng A, Holbrook NJ, Mattson MP. Involvement of Gadd153 in the pathogenic action of presenilin-1 mutations. J Neurochem. 2002;83:673–681. doi: 10.1046/j.1471-4159.2002.01165.x. [DOI] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–19377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, West AB, Ron D. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. 2001;107:585–593. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bry K, Whitsett JA, Lappalainen U. IL-1beta disrupts postnatal lung morphogenesis in the mouse. Am J Respir Cell Mol Biol. 2007;36:32–42. doi: 10.1165/rcmb.2006-0116OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bek MF, Bayer M, Muller B, Greiber S, Lang D, Schwab A, August C, Springer E, Rohrbach R, Huber TB, Benzing T, Pavenstadt H. Expression and function of C/EBP homology protein (GADD153) in podocytes. Am J Pathol. 2006;168:20–32. doi: 10.2353/ajpath.2006.040774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh T, Terada K, Oyadomari S, Mori M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004;11:390–402. doi: 10.1038/sj.cdd.4401369. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Kuida K, Yuan J. Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ. 2002;9:1115–1125. doi: 10.1038/sj.cdd.4401087. [DOI] [PubMed] [Google Scholar]
- Kühn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–1104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]