

Abstract
Tissue repair requires that fibroblasts migrate into the wound to produce and remodel extracellular matrix, a process that requires adhesion. Failure to suppress the tissue repair program results in fibrotic disorders that are characterized by excessive adhesive signaling. The role of specific components of adhesive signaling in fibrogenic responses is unclear, but may involve small GTPases such as Rac1. To address the functions of Rac1 in fibroblasts, we generated mice containing a fibroblast-specific deletion of Rac1. These mice show delayed cutaneous wound closure, including reduced collagen production and myofibroblast formation. In cultured Rac1-deficient fibroblasts, adhesion, spreading, and migration were significantly inhibited. Rac1-deficient fibroblasts possessed impaired myofibroblast formation and function as visualized by reduced α-smooth muscle actin expression as well as matrix contraction. Both in vivo and in vitro, Rac1- deficient fibroblasts showed a reduced generation of reactive oxygen species; in vitro, hydrogen peroxide alleviated the phenotype of Rac1-deficient fibroblasts. Thus, Rac1 is an essential signaling integrator that is required for normal wound healing and dermal homeostasis.
Tissue repair requires the reconstitution of the epithelial barrier and the underlying connective tissue.1 During normal skin healing, fibroblasts migrate into the wound area where they synthesize and remodel a collagen- and cellular fibronectin-rich extracellular matrix (ECM).2 This migration involves the extension of lamellopodia and filopodia, accompanied by the assembly and disassembly of focal adhesions at the leading edge of the cell. Focal adhesion formation and turnover is intimately related to their ability, thorough integrins, to mediate adhesive contact with the extracellular matrix and the actin cytoskeleton.3 These focal adhesions typically contain proteins such as β- and γ-cytoplasmic actins, α-smooth muscle actin (α-SMA), αv integrin, vinculin, paxillin, α-actinin, talin, and focal adhesion kinase.4,5 These specialized fibroblasts, termed myofibroblasts, generate the adhesive and tensile forces required for wound closure. Normally myofibroblasts disappear from the healed wound; persistence of the myofibroblast results in fibrosis.4,6 There is no therapy for fibrotic diseases, which can affect the skin and internal organs often resulting in death. Controlling the excessive adhesive and tensile forces mediated by myofibroblasts resident within scars is therefore essential for developing rational anti-fibrotic strategies.
It is unclear what molecules mediating adhesion and tension contribute to fibrogenic responses in vivo. A number of growth factors and cytokines contribute to wound closure in vivo7; however, the signaling mechanisms by which these factors regulate this function remain poorly defined. Focal adhesion proteins serve as a point of convergence for signals resulting from stimulation of growth factor receptors.3,8 For example, paxillin provides a platform for protein tyrosine kinases such as focal adhesion kinase and SRC, which are activated as a result of adhesion or growth factor stimulation.9 Fibroblasts lacking focal adhesion kinase or paxillin spread poorly and cannot migrate, and are deficient in their pro-fibrotic response to transforming growth factor (TGF)β.10,11,12 These processes are controlled by members of the Rho family of small GTPases.13 For example, the small GTPase Rac1 is expressed ubiquitously and is recruited to focal adhesions by paxillin.14 The contribution of Rho GTPases including rac to adhesive signaling has been investigated in mammalian cells predominantly using dominant negative mutants where threonine 17 is mutated to asparagine (T17N). 15 These experiments indicate that Rac1 is indispensable for fibroblast chemotaxis toward platelet-derived growth factor,16 invasion into collagen matrices17 and wound closure in cultured rat embryonic fibroblasts.18 Recently, we showed that Rac1 was involved with the ability of endothelin-1 to induce myofibroblast formation in cultured fibroblasts.19 Although the use of dominant negative mutants has revealed numerous processes where Rac1 is involved, this approach has significant limitations. Most importantly, such constructs may impair GTPase loading of not only Rac isoforms but also Cdc42 and Rho family members.20,21 These considerations emphasize the necessity of evaluating the role of Rac1 in vivo using genetic model systems. Moreover, although there are three different rac proteins, it remains unclear which rac isoforms are specifically responsible for particular in vivo functions.
Investigation of the contribution of Rac1 in vivo requires the generation of knockout mouse models. However, as mouse embryos deficient for Rac1 die at embryonic day 6.5 (E6.5) from incomplete gastrulation,22 the creation of conditional Rac1 mice is essential for studying the effect of Rac1 in more differentiated cell types. Recently, mice containing the Rac1 allele flanked by loxP sites have been generated which can be used to delete Rac1 in specific cell lineages in vivo using mice expressing cre recombinase under the control of a tissue-specific promoter.23 Previously, we have shown that mice containing a fibroblast-specific deletion of Rac1 show resistance to the bleomycin-induced model of skin fibrosis.24 In this report, we use mice with a fibroblast-specific deletion of Rac1 to analyze the contribution of Rac1 in the dermal punch model of cutaneous wound healing and to dermal homeostasis in vivo. Moreover, we probe the mechanism underlying Rac1 action in fibroblasts. Our data provide new insights into the role and mechanism of Rac1 action in fibroblasts.
Materials and Methods
Generation of Rac1 Conditional Knockout Mice
Rac1 conditional knockout mice were generated as described previously.24 Briefly, mice that carry a tamoxifen-inducible Cre-recombinase under the control of a fibroblast-specific regulatory sequence from the proα2(I) collagen gene25 were crossed with mice that carry homozygous conditional Rac1 allele23 to generate Cre/Rac1 heterozygote mice. The second cross obtained Cre/Rac1 homozygote mice. Animals used for experiments were genotyped by PCR to detect Rac1 and Cre as described previously. All animal protocols were approved by the regulatory authority of the experimental animal committee of University of Western Ontario. To delete Rac1, a stock solution of tamoxifen (4-hydroxitamoxifen, Sigma, St. Louis, MO) in ethanol (100 mg/ml) was diluted in corn oil to 10 mg/ml. Adult mice (age 3 weeks) were given intraperitoneal injections of the tamoxifen suspension (0.1 ml of 10 mg/ml) over 10 days. Deletion of Rac1 was tested by PCR genotyping and Western blot with an anti-Rac1 antibody.
Wound Surgery
Experiments were performed on littermate mice homozygous for the loxP-Rac1 allele and heterozygous for type I collagen-cre that were treated with either tamoxifen (“knockout Rac1,” K/K) or corn oil (“conditional Rac1,” C/C) alone. Wounding experiments were performed after 2 weeks from the last injection of tamoxifen. Mice were anesthetized by intraperitoneal injection of 90 μg ketamine plus 10 μg xylazine/g, and their back skin was shaved, depilated with Nair and cleaned with alcohol. Using a sterile 4-mm biopsy punch, four bilateral full-thickness skin wounds were created on the dorso-rostral back skin without injuring the underlying muscle. Wounds were separated by a minimum of 6 mm of uninjured skin. Wounds were photographed at 0, 2, 4, 7, and 10 days after wounding using a Sony D-9 digital camera. The wound area was analyzed using Northern Eclipse (Empix) software and wound closure was expressed as percentage of initial wound size. Mice were sacrificed by CO2 euthanasia after 3, 7, and 10 days. Wound samples were collected for histology, immunohistochemistry, and hydroxyproline assay.
The H2O2 rescue experiment was performed according to the method previously described.26 Briefly, four full-thickness skin wounds were created on the dorso-rostral back skin on Rac1 K/K mice. Wounds from two sides were topically treated with either low dose of H2O2 (1.25 μmoles/wound; or 25 μl of 0.15% H2O2) or saline (25 μl PBS) once daily. Wound closure was monitored and analyzed as described above.
Immunohistochemistry and Assessment
Sections were cut and processed as described above. Immunolabeling of α-SMA and proliferating cell nuclear antigen was performed using the DakoCytomation LSAB+ System-HRP kit (Carpinteria, CA). Immunohistochemical procedures were performed according to the manufacturer’s recommendations. Briefly, endogenous peroxide was blocked using 0.5% H2O2 in methanol for 5 minutes. Non-specific IgG binding was blocked by incubating sections with bovine serum albumin (0.1%) in PBS for 1 hour and then incubated with primary antibody for α-SMA (1:1000) in a humidified chamber and left overnight at 4°C. Next, sections were incubated with biotinylated link for 30 minutes followed by incubation with streptavidin for 30 minutes. The chromogen diaminobenzidine tetrahydrochloride, was then added until sufficient color development and sections counterstained with Harris’s hematoxylin. The percentage positive cell staining was calculated by numbers of cells per mm2 was detected using image analysis software (Northern Eclipse, Empix). When indicted, tissue was stained with Harris’s H&E, and formation of new epithelia was measured. To assess the effects of Rac-1 deletion on wound collagen synthesis, van Gieson’s collagen stain was used. van Gieson’s stain results in a deep red color for mature collagen fibers and a pink color for immature collagen fibers. Muscle and fibrin appear yellow with black nuclei.
Hydroxyproline Assay
The hydroxyproline assay was performed as a marker of collagen synthesis in wound tissues using the method previously described.27 Wound tissues were homogenized in saline, hydrolyzed with 2N NaOH for 30 minutes at 120°C, followed by the determination of hydroxyproline by modification of the Neumann and Logan’s reaction using Chloramine T and Ehrlich’s reagent using a hydroxyproline standard curve and measuring at 550 nm. Values were expressed as μg of hydroxyproline per mg of protein.
Cell Culture, Immunofluorescence, and Western Analysis
Dermal fibroblasts were isolated from explants (4- to 6-week-old animals) as previously described.6 Cells were subjected to indirect immunofluorescence analysis as previously described6,12 using anti-α-SMA, rhodamine phalloidin (Sigma), and anti-vinculin (Sigma) antibodies, followed by an appropriate secondary antibody (Jackson Immunoresearch), and photography (Zeiss Axiphot B-100) using a digital camera (Empix). Fluorescence intensity of α-SMA fibers was quantified by line scan measurement using Northern Eclipse (Empix) software. Alternatively, cells were lysed in 2% SDS, proteins quantified (Pierce) and subjected to Western blot analysis as previously described.6,12 When indicated, cells were treated in 0.5% fetal bovine serum in the presence or absence of 200 μmol/L H2O2 for 24 hours.
Real-Time PCR
Real-time PCR was performed essentially as previously described.28 A total of 10 control and knockout mice were analyzed independently for each data point. Averages ± SD represent averages from these 10 independent animals. Cells were cultured until 80% confluence, and serum starved for 24 hours. Total RNA was isolated (Qiagen). The integrity of the RNA was verified by gel electrophoresis or Agilent bioanalyzer. Total RNA (25 ng) was reverse transcribed and amplified using TaqMan Assays on Demand (Applied Biosystems) in a 15-μl reaction volume containing two unlabeled primers and 6-carboxyfluoroscein labeled TaqMan MGB probe. Samples were combined with One-step Mastermix (Eurogentech). Amplified sequences were detected using the ABI Prism 7900 HT sequence detector (Perkin-Elmer-Cetus, Vaudreuil, QC) according to the manufacturer’s instructions. Triplicate samples were run, and expression values were standardized to values obtained with control 18 S RNA primers using the delta ct method. Statistical analysis was performed by the Student’s paired t-test. Response to TGFβ stimulation.
For α-SMA expression assay by real-time PCR in response to TGFβ, cells were serum-starved for 24 hours and treated with 4 ng/ml TGFβ for 6 hours. Total RNA was isolated using Trizol (Invitrogen). Real-time PCR for α-SMA was performed as described above. Triplicate samples were run, and transcripts and expression values were standardized to values obtained with control 18 S RNA.
Adhesion Assay
Fibroblasts were isolated and cultured as described above. Adhesion assays were performed essentially as previously described.6 Wells of 96 well plates overnight, 4°C, with 10 μg/ml fibronectin (Sigma) or type I collagen (First Link) in 0.5% bovine serum albumin (BSA), 1× PBS. Wells were blocked for 1 hour in 10% BSA in PBS, room temperature. Fibroblasts were harvested with 2 mmol/L EDTA in PBS (20 minutes, room temperature), washed twice with Dulbecco’s Modified Eagle’s Medium serum-free medium containing 1% BSA (Sigma, St. Louis, MO), resuspended in the same medium at 2.5 × 105 cells/ml and 100 μl of suspension was incubated in each well for 1 hour. Non-adherent cells were subsequently removed by washing with PBS. To detect cell adhesion, an acid phosphatase assay was used, adherent cells were quantified by incubation with 100 μl substrate solution (0.1 M/L sodium acetate, pH 5.5; 10 mmol/L –p-nitophenylphosphate and 0.1% Triton X-100) for 2 hours at 37°C. The reaction was stopped by the addition of 15 μl 1N NaOH/well and A450 was measured. Comparison of adhesive abilities was performed by using Student’s unpaired t-test. A P value <0.05 was considered as statistically significant.
Collagen Gel Contraction
Experiments were performed essentially as described previously.19 Briefly, 24-well tissue culture plates were pre-coated with BSA. Cells were used at passage 3. Trypsinized fibroblasts were suspended in MCDB medium (Sigma) and mixed with collagen solution (one part of 0.2 M/L HEPES, pH 8.0; four parts collagen [Vitrogen-100, 3 mg/ml] and five parts of MCDB X 2) yielding a final concentration of 80,000 cells per ml and 1.2 mg/ml collagen. Collagen/cell suspension (1 ml) was added to each well. After polymerization, gels were detached from wells by adding 1 ml of MCDB medium. Contraction of the gel was quantified by measuring changes in diameter using image analysis software (Empix) over a 24-hour period.
Migration and Proliferation Assays
For in vitro wounding (migration) experiments, cultured fibroblasts obtained from Rac1 conditional (C/C) and knockout (K/K) mice were grown in 12-well plates. Medium was removed and cells were once rinsed with serum-free medium + 0.1% BSA and were cultured for 24 hours in serum-free medium + 0.1% BSA. The monolayer was artificially injured by scratching across the plate with a blue pipette tip (approximately 1.3-mm width). The wells were washed two times to remove detached cells or cell debris. The cells were then cultured in serum-free medium. Mitomycin C (10 μg/ml, Sigma) was always included in the media to prevent cell proliferation. After 24 and 48 hours, images of the scratched areas under each condition were photographed. For the proliferation assay, cells were seeded in 96 well plates at 2000 cells/well. Cell number was determined after 24, 48, and 72 hours incubation using MTT kit (Roche).
Measurement of Reactive Oxygen Species Production
The in vivo and in vitro reactive oxygen species (ROS) production were measured using a fluorescent dye, 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Sigma). DCFH-DA is a nonpolar compound that is readily diffusible into cells, where it is hydrolyzed to the nonfluorescent polar derivative DCFH and thereby trapped within cells.29 In the presence of an oxidant, DCFH is converted into the highly fluorescent 2′,7′-dichlorofluorescein. For in vitro assay, freshly frozen, enzymatically intact, 10-μm thick sections from mice of 7 days after wounding were incubated with DCFH (10 μmol/L) in PBS for 30 minutes at 37°C protected from light. Sections were then fixed with 4% paraformaldehyde and photographed under Zeiss B-100 fluorescence microscope using a digital camera. The percentage of ROS-positive cells were calculated by counting ROS-positive cell and total cell numbers in the same field. For in vitro assays, cells were loaded with 10 μmol/L DCFH-DA. After 30 minutes incubation in the dark, cells were fixed and photographed under Zeiss B-100 fluorescence microscope using a digital camera (Empix). The ROS fluorescent intensity in individual cells was quantified by Northern Eclipse (Empix) software.
Measurement of Intracellular ROS by Flow Cytometry
For flow cytometric analysis, cells were loaded with 10 μmol/L DCFH-DA. After 60 minutes incubation in the dark, cells were detached by trypsinization and analyzed immediately by flow cytometry (Becton Dickinson, Franklin Lakes, NJ) equipped with an Argon lamp (FL-1; emmision, 480 nm, band pass filter, 530 nm) and data were analyzed using Cell Quest software (BD Biosciences, San Diego, CA).
Results
Deletion of Rac1 Results in Delayed Cutaneous Wound Repair
To generate mice deleted for Rac1 in fibroblasts, we crossed mice harboring a Rac1 allele bordered by loxP sites23 with mice containing a cre recombinase gene located downstream of a collagen I promoter/enhancer, which confers fibroblast-specific gene expression.25 The expression of cre is dependent on the presence of tamoxifen.25 Littermate mice homozygous for the loxP-rac 1 that were heterozygous for collagen I-cre were generated and were treated with or without tamoxifen, as described in the Materials and Methods and in our previous publication.24 As a result, mice deleted for Rac1 (K/K) and control mice (C/C) that were otherwise genetically identical were generated. Deletion of the Rac1 gene was verified using polymerase chain reaction analysis of tail DNA (Figure 1A), and by subjecting dermal fibroblasts cultured from explants derived from animals to Western blot analysis using an anti-Rac1 antibody (Figure 1B).
Figure 1.

Conditional deletion of Rac1 in fibroblasts results in impaired cutaneous wound healing. Mice deleted for Rac1 (K/K) and control mice (C/C) that were otherwise genetically identical were generated as described in the Materials and Methods and previously.24 A: Mice were genotyped by PCR. Bands corresponding to cre, deleted (null) and undeleted (conditional) alleles are shown. B: Fibroblasts from Rac1 (K/K) and control mice (C/C) were cultured. Whole cell protein extracts were made from these cells and subjected to Western blot analysis with anti-Rac1 and anti-β-actin antibodies. C: Mice were wounded and wound closure was measured as described in Materials and Methods. For all assays, six mice per group were analyzed. Data represent averages and SD from all these mice (*P < 0.05, Student’s t-test). D: Sections from mice 3 days post-wounding were subjected to H&E staining as described in Materials and Methods. Note that Rac1-deficient wounds had defects in wound closure as revealed by reduced granulation tissue in the wound as well as modest, yet significant, defects in re-epithelialization (*P < 0.05, Student’s t-test), likely occurring secondarily to defects in granulation tissue formation.
To investigate whether Rac1-deficient mice displayed a delay in cutaneous wound closure, we subjected 8-week-old mice homozygous for deletions in the Rac1 gene, or their wild-type littermate counterparts, to the dermal punch wound model of wound healing. We monitored wound closure over a 10-day period, and found that Rac1-deficient (K/K) animals displayed a marked reduction in the rate of wound closure relative to control animals (C/C) (Figure 1, C and D). Note that 3-day Rac1-deficient wounds had defects in wound closure as revealed by reduced granulation tissue in the wound as well as modest, yet significant, defects in re-epithelialization, likely occurring secondarily to defects in granulation tissue formation (see Discussion). Please note that we conducted parallel studies with wild-type mice treated with or without tamoxifen. Tamoxifen alone had no impact on wound closure (not shown). Staining of tissues taken from Rac1-deficient animals 7 days post-wounding showed reduced collagen production, as detected by van Gieson stain (Figure 2, van Gieson) and hydroxy-proline levels (Figure 2, hydroxyproline). Moreover, within the dermis Rac1-deficient animals possessed reduced numbers of α-SMA-and proliferating cell nuclear antigen-expressing myofibroblasts (Figure 2).
Figure 2.

Conditional deletion of Rac1 in fibroblasts results in impaired cutaneous wound healing: histological analysis. Mice were wounded and quality of wound closure was measured as described in the Materials and Methods. Mice 7 days post-wounding were measured as described in the Materials and Methods using van Gieson’s stain, and hydroxyproline levels were measured to detect collagen, proliferating cell nuclear antigen to detect proliferation, and α-SMA antibody to detect myofibroblasts. For all assays, six mice per group were analyzed. Maturity of collagen fibers was ascertained as described in Materials and Methods. (*P < 0.05, **P < 0.01, Student’s t-test).
Rac1 Is Essential for Migration, Adhesion, Proliferation, and Spreading
To further investigate the basis for the wound-healing phenotype of the Rac1-deficient animals, we explanted fibroblasts from animals deleted for Rac1 (K/K) and their tamoxifen-untreated counterparts (C/C) and subjected them to the scratch wound assay of in vitro wound repair (Figure 3A). Rac1-deficient cells migrated more slowly (Figure 3A). Rac1-deficient cells also showed a reduction in proliferation (Figure 3B). To further investigate the contribution of Rac1 to the function of fibroblasts, we assessed whether Rac1-deficient cells showed a reduction in their ability to adhere to fibronectin. To perform this analysis, a standard quantitative adhesion assay was performed as described in Materials and Methods, which showed that Rac1-deficient cells showed a reduction in adhesion on fibronectin up to 90 minutes, at which time the adhesion of Rac1-deficient and wild-type cells were identical (Figure 3C). Cell spreading was monitored postadhesion microscopically, using rhodamine phalloidin and anti-vinculin antibodies. We found loss of Rac1 also resulted in delayed spreading on fibronectin, as revealed with anti-vinculin antibody (green) and rhodamine-phalloidin (red) staining to detect actin (Figure 3D).
Figure 3.

Loss of Rac1 results in a reduced ability of fibroblasts to migrate, adhere to ECM, proliferate and spread on ECM. Fibroblasts were isolated by explant culture from mice containing the Rac1 gene (C/C) or not (K/K). Cells were subjected to (A) a scratch wound assay of cell migration, (B) a proliferation assay, and (C) a fibronectin adhesion assay as described in the Materials and Methods. D: Cell spreading was monitored by plating cells on fibronectin and fixing and staining cells with anti-vinculin antibody (to detect focal adhesions) and rhodamine-phalloidin to detect actin. For all assays, fibroblasts from 6 mice per group were analyzed. Data represent averages and SD from all these mice. The number of focal adhesions per cell were counted (30 C/C or K/K cells per mouse line; *P < 0.05, Student’s t-test).
Rac1 Is Essential for Myofibroblast Differentiation and ECM Contraction
The α-SMA-expressing myofibroblast is the cell type responsible for ECM remodeling in wound healing.4,6 To investigate whether Rac1-deficient cells showed defects in myofibroblast differentiation, we used real-time PCR analysis to show that Rac1-deficient cells possessed reduced mRNA expression for α-SMA and type I collagen (Figure 4A). Moreover, we used an anti-α-SMA antibody to show that Rac1-deficient cells expressed reduced amounts of α-SMA protein (Figure 4B). In addition, indirect immunofluorescence analysis with an anti-α-SMA antibody was also used to show that Rac1-deficient cells lacked α-SMA-containing stress fibers (Figure 4C) and a reduction in so-called ‘supermature’ focal adhesions, a key feature of myofibroblasts indicated by intense foci of anti-vinculin staining (Figure 4C). To provide a functional context for our studies, Rac1-deficient cells were less effective at contracting a floating collagen gel matrix (Figure 5). Thus Rac1 was required for myofibroblast formation and action in vitro.
Figure 4.

Loss of Rac1 results in a reduction in myofibroblast formation. Fibroblasts were isolated by explant culture from mice containing the Rac1 gene (C/C) or not (K/K). A: Real-time PCR analysis. mRNA harvested from cells (10 mice total per group, each assay performed in triplicate, average ± SD is shown) and subjected to real-time PCR analysis to detect the mRNAs indicated. For all assays, fibroblasts from 10 mice were analyzed. Data represent averages and SD from all these mice. *P < 0.05, students t-test. B: Cells were subjected to Western blot analysis with an anti-α-SMA antibody. C: Cells were fixed and stained with anti-vinculin antibody to detect focal adhesion and rhodamine phalloidin to detect actin and α-SMA. Rac1-deficient cells showed reduced actin stress fibers indicating reduced myofibroblast formation. Fluorescence intensity of actin fibers was quantified by line scan measurement using Northern Eclipse (Empix) software and showed reduced α-SMA stress fiber formation in Rac1-deficient fibroblasts.
Figure 5.
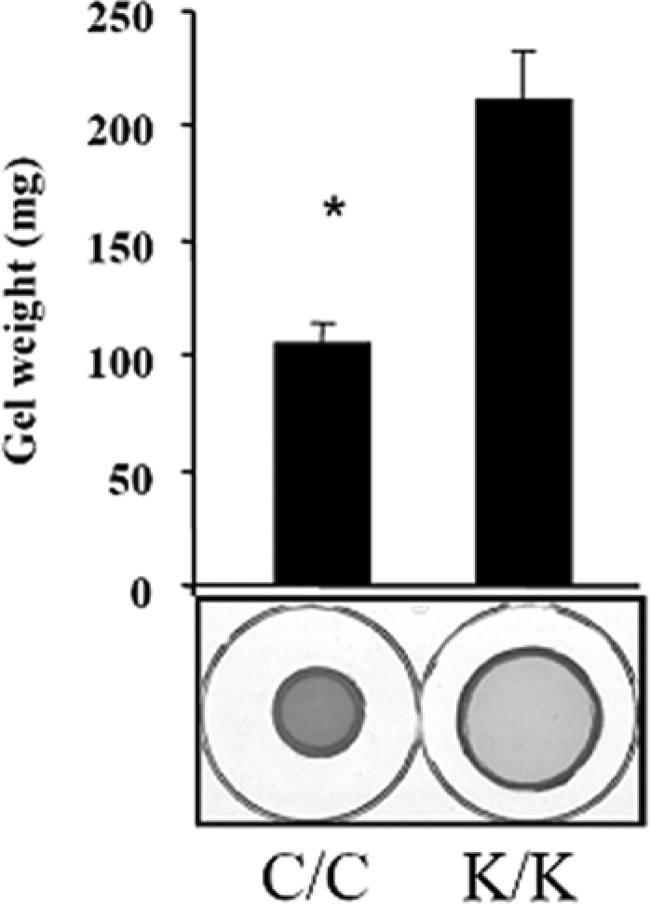
Loss of Rac1 results in a reduction in ECM contraction. Loss of Rac1 results in a reduced ability of fibroblasts to contract a collagen gel matrix: Floating gel analysis. The effect of loss of Rac1 expression on ECM contraction generated by fibroblasts embedded in a floating collagen gel matrix was assessed over a 24 hours period. Contraction was assessed photographically and by measuring diameter of contracted gels (Fibroblasts from three separate animals were used, and experiments were performed in triplicate; Average ± SD is indicated). Note that wild-type (C/C) fibroblasts were able to contract a collagen gel matrix (Student’s t-test, *P < 0.05), relative to Rac1-deficient cells (K/K).
Loss or Rac1 Results in Reduced ROS Production
Fibroblast activation is associated with TGFβ.6 However, TGFβ showed a similar fold increase in α-SMA mRNA expression in Rac1-deficient and wild-type fibroblasts (Figure 6A) To begin to assess whether altered responses to TGFβ may play a role in the phenotype of Rac1-deficient fibroblasts, we first used real time PCR analysis to show that Rac1-deficient fibroblasts possessed a modest, yet significant, reduction in TGFβ2 mRNA expression (Figure 6B) suggesting that the primary defect in Rac1-deficient fibroblasts was not a defect in TGFβ signaling.
Figure 6.

Loss of Rac1 does not result in impairment of TGFβ-induced α-SMA mRNA expression. Fibroblasts were isolated by explant culture from mice containing the Rac1 gene (C/C) or not (K/K). The effect of loss of Rac1 expression on (A) TGFβ1-induced α-SMA mRNA (B) TGFβ2 mRNA expression were assessed as described in Materials and Methods. mRNA harvested from cells (10 mice total per group, each assay performed in triplicate, average ± SD is shown) and subjected to real-time PCR analysis to detect the mRNAs indicated. Data represent averages and SD from all these mice (A) Note that both wild-type (C/C) and Rac1-deficient (K/K) fibroblasts could respond to TGFβ1(Student’s t-test, *P < 0.05), yet the fold-increase response to TGFβ1 was identical in wild-type (C/C) and Rac1-deficient (K/K) fibroblasts. B: Note that wild-type (C/C) fibroblasts possessed elevated TGFβ2 mRNA (Student’s t-test, *P < 0.05), relative to Rac1-deficient cells (K/K).
Rac1 has been shown to stimulate oxidative stress.30 To begin to assess whether defective ROS production may contribute to the phenotype of Rac1-deficient fibroblasts, we first showed that the quantity of ROS-positive fibroblasts was reduced both in vivo and in vitro (Figure 7, A and B). Based on these results, we attempted to rescue the in vitro phenotype of Rac1-deficient fibroblasts by exposing cells to H2O2 for 24 hours. We showed that H2O2 restored the ability of Rac1-deficient fibroblasts to express α-SMA and type I collagen mRNAs, display α-SMA stress fibers and contract a collagen gel matrix (Figure 8, A–C). Consistent with this result, we found that application of H2O2 rescued the wound closure defects of Rac1-deficient mice (Figure 9). Collectively, these results indicate that Rac1 is essential for fibrogenic responses in fibroblasts via a ROS-dependent mechanism.
Figure 7.

Loss of Rac1 results in a reduction in ROS production. A: The in vivo (tissue section) and in vitro (fibroblast) ROS production in Rac1-deficient mice (tissue section) and cultured cells isolated from Rac1-deficient mice (fibroblast) was measured using a fluorescent dye, 2′,7′-dichlorofluorescein diacetate (DCFH-DA) as described in Materials and Methods. The number of ROS-positive fibroblasts in tissue sections was quantified as described in Materials and Methods (average of six mice per group and SD is shown). The average fluorescent intensity over fixed areas in cultured fibroblasts was quantified as described in Materials and Methods (average of data obtained from fibroblasts isolated from four mice and SD is shown). Note that wild-type (C/C) fibroblasts showed reduced ROS production (*P < 0.05, Student’s t-test), relative to Rac1-deficient cells (K/K). B: Fluorescence-activated cell sorting analysis of Rac1-deficient and wild-type fibroblasts was performed as described in Materials and Methods.
Figure 8.

H2O2 rescues the phenotype of Rac1-deficient fibroblasts. Fibroblasts were isolated by explant culture from mice containing the Rac1 gene (C/C) or not (K/K). A: Cells incubated in the presence or absence of 200 μmol/L H2O2 for 24 hours were fixed and stained with anti-α-SMA antibody to detect α-SMA. Rac1-deficient cells in the presence of H2O2 possessed α-SMA stress fibers indicating restored myofibroblast formation. Fibroblasts from four mice per group were used. Representative data are shown. B: Real-time PCR analysis. mRNA harvested from cells in the presence or absence of 200 μmol/L H2O2 for 24 hours (Fibroblasts from four mice per group were used, each assay was performed in triplicate, average ± SD is shown) and subjected to real-time PCR analysis to detect the mRNAs indicated. Data represent averages and SD from all these mice. (*P < 0.05, Student’s t-test). C: Addition of 200 μmol/L H2O2 to Rac1-deficient fibroblasts rescues the ability of fibroblasts to contract a collagen gel matrix: Floating gel analysis. The effect of loss of Rac1 expression on ECM contraction generated by fibroblasts embedded in a floating collagen gel matrix was assessed over a 24 hour period in the presence and absence of 200 μmol/L H2O2. Contraction was assessed photographically (N fibroblasts from three separate animals were used, and experiments were performed in triplicate; Average ± SD is indicated). Note that Rac1-deficient cells (K/K) had decreased ability to contract a collagen gel matrix (*P < 0.05) as compared with wild-type (C/C) fibroblasts and that addition of H2O2 to Rac1-deficient cells (K/K) restored this ability (*P < 0.05, Student’s t-test).
Figure 9.

H2O2 rescues the reduction in wound closure observed in Rac1-deficient mice. Mice deleted for Rac1 (K/K) and control mice (C/C) that were otherwise genetically identical were generated as described in methods and previously.24 Mice were wounded and wound closure was measured in the presence or absence of H2O2 as described in the Materials and Methods. For all assays, six mice per group were analyzed. Data represent averages and SD from all these mice (*P < 0.05, Student’s t-test).
Discussion
In this study, we tested the effect of loss of Rac1 in fibroblasts on tissue repair, dermal homeostasis, and fibrotic responses in vivo. We performed these analyses using mice homozygous for a Rac1 gene flanked by loxP sites and heterozygous for a transgene encoding a tamoxifen-dependent cre recombinase driven by a fibroblast-specific type I collagen promoter/enhancer.23,24,25 The tissue specificity of this promoter/enhancer construct has been confirmed in vivo.31 Rac1-deficient mice showed reduced rates of wound closure. Accompanying this, Rac1 deficient mice showed diminished myofibroblast production and collagen deposition. Isolated Rac1-deficient fibroblasts also showed diminished α-SMA expression and stress fiber formation as well as ECM contraction. Rac1-deficient fibroblasts in vitro and in vivo showed reduced ROS production, and H2O2 rescued the in vitro phenotype of Rac1-deficient fibroblasts. These results collectively suggest that expression of Rac1 by fibroblasts is essential for wound healing via a ROS-dependent mechanism.
Although a potential function for Rac1 for fibroblasts to participate in wound healing has been predicted from previous in vitro studies,18,31,32 and from loss-of-function or inhibition experiments in Drosophila,33 the complexity of the wound healing process in mammals cannot truly be represented by these models. Moreover, dominant-negative mutants of Rac1 have also been suggested to inhibit non-Rac dependent cellular functions.34 A specific role for Rac1 in wound healing is, however, suggested by our finding that wound repair is severely disturbed in mice carrying a dermal-specific deletion of Rac1. Our results demonstrate for the first time that wound repair in skin requires Rac1 expression by dermal fibroblasts. These results are consistent with and significantly extend recent findings that loss of Rac1 in keratinocytes results in delayed wound closure35 and resistance to bleomycin-induced scleroderma.24 However, in these latter two reports no mechanism was proposed. For the first time, our results collectively point to Rac1 as an essential integrator required for connective tissue repair and fibrogenesis. Re-epithelialization also appeared to be somewhat impaired in Rac1-deficient mice, although it is unclear as to whether this was due to differences in gene expression between Rac1 wild-type and Rac1 knockout fibroblasts, or whether this was a secondary effect arising due to the defect in connective tissue formation. Investigation of this interesting question is beyond the scope of the current study, which focuses specifically on the effects of loss of Rac1 on restoration of the dermis post-wound healing.
Fibrotic diseases are characterized by the failure to terminate normal tissue repair and the persistence of myofibroblasts within lesions.6,19,36 Myofibroblasts can form through cooperation among several extracellular signaling molecules, including TGFβ, endothelin-1, and connective tissue growth factor 2, all of which act at least in part through adhesive mechanisms.6,19,37,38,39 Within the cell, Rho GTPases, such as Rac1, are involved in the integration of signaling pathways. Rho GTPases have been implicated in the pathology of various human diseases including cancer, and are considered attractive drug targets in future targeted therapy.40 Our results add to recent reports using conditionally deleted Rac1 alleles showing roles for Rac1 in the immune system, epithelial stem cells, and nervous system and vascular development.41,42,43,44 Showing that Rac1 is an essential mediator of myofibroblast activity and fibrogenesis provide not only a useful step in identifying the fundamental process underlying normal tissue repair and fibrogenesis, but also strongly suggests that targeting Rac1 may be a suitable strategy for anti-fibrotic drug intervention.
That function of Rac1 is involved with ROS production has been described previously. Hematopoietic cells like neutrophils and macrophages use Rac1 and especially Rac2 (the hematopoietic-specific Rac isoform) for the activation of gp91phox [NADPH oxidase (Nox) 2) and oxidative burst.45 Rac1 binds the Nox activator Noxa1/p67phox; activated Rac1 is necessary for Nox1 and Nox3 activation.46 Indeed, Nox1 is constitutively activated by Rac1.47 Elevation of ROS by treatment with H2O2 has been shown to induce epithelial mesenchymal transition-related cell scattering and invasiveness, and Rac1-induced ROS were found to be specifically required for induction of the mesenchymal vimentin, as well as other myofibroblast genes.48 It cannot be excluded that ROS modulate many effectors mediating changes in actin cytoskeleton organization and specificity of signaling can depend on local ROS concentrations at different cell compartments. Further experiments are needed to shed light on the detailed mechanisms underlying the regulation of cell architecture and motility by ROS; however, that Rac1 seems to modulate expression of α-SMA and type I collagen seems to suggest that alterations in gene expression are likely to play a key role.
In summary, our studies examining the involvement in Rac1 in dermal function may have profound implications for both homeostatic and pathological processes by contributing to our understanding of basic mechanisms regarding wound healing. As a consequence, our results may have future therapeutic implications for the treatment of non-healing or chronic skin wounds, and of fibroproliferative disease.
Footnotes
Address reprint requests to Andrew Leask, Canadian Institute of Health Research Group in Skeletal Development and Remodeling, Division of Oral Biology and Department of Physiology and Pharmacology, Schulich School of Medicine and Dentistry, University of Western Ontario, Dental Sciences Bldg., London, ON, Canada, N6A 5C1. E-mail: Andrew.leask@schulich.uwo.ca.
Supported by grants from the Canadian Foundation for Innovation and the Canadian Institutes of Health Research and the Ontario Thoracic Society. A.L. is a New Investigator of the Arthritis Society (Scleroderma Society of Ontario), the recipient of an Early Researcher Award and a member of the Canadian Scleroderma Research Group New Emerging Team. M.K. is the recipient of postdoctoral fellowships from the Canadian Arthritis Network and the Ontario Ministry of Innovation.
References
- Martin P. Wound healing–aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- Eckes B, Kessler D, Aumailley M, Krieg T. Interactions of fibroblasts with the extracellular matrix: implications for the understanding of fibrosis. Springer Semin Immunopathol. 1999;21:415–429. doi: 10.1007/s002810000034. [DOI] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Dugina V, Fontao L, Chaponnier C, Vasiliev J, Gabbiani G. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J Cell Sci. 2001;114:3285–3296. doi: 10.1242/jcs.114.18.3285. [DOI] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Chen Y, Shiwen X, van Beek J, Kennedy L, McLeod M, Renzoni EA, Bou-Gharios G, Wilcox-Adelman S, Goetinck PF, Eastwood M, Black CM, Abraham DJ, Leask A. Matrix contraction by dermal fibroblasts requires TGFbeta/ALK5, heparan sulfate containing proteoglycans and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol. 2005;167:1699–1711. doi: 10.1016/s0002-9440(10)61252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835–870. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001;114:3583–3590. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- Turner CE. Paxillin and focal adhesion signaling. Nat Cell Biol. 2000;2:E231–E236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- Hagel M, George EL, Kim A, Tamimi R, Opitz SL, Turner CE, Imamoto A, Thomas SM. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol Cell Biol. 2002;22:901–915. doi: 10.1128/MCB.22.3.901-915.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Liu S, Shi-wen X, Kennedy L, Pala D, Carter DE, Black CM, Abraham DJ, Leask A. FAK is required for TGFβ-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix remodeling phenotype. Mol Biol Cell. 2007;18:2169–2178. doi: 10.1091/mbc.E06-12-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Ishibe S, Joly D, Liu ZX, Cantley LG. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol Cell. 2004;6:257–267. doi: 10.1016/j.molcel.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Anand-Apte B, Zetter BR, Viswanathan A, Qiu RG, Chen J, Ruggieri R, Symons M. Platelet-derived growth factor and fibronectin-stimulated migration are differentially regulated by the Rac and extracellular signal-regulated kinase pathways. J Biol Chem. 1997;272:30688–30692. doi: 10.1074/jbc.272.49.30688. [DOI] [PubMed] [Google Scholar]
- Banyard J, Anand-Apte B, Symons M, Zetter BR. Motility and invasion are differentially modulated by Rho family GTPases. Oncogene. 2000;19:580–591. doi: 10.1038/sj.onc.1203338. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-Wen X, Chen Y, Denton CP, Eastwood M, Renzoni EA, Bou-Gharios G, Pearson JD, Dashwood M, du Bois RM, Black CM, Leask A, Abraham DJ. Endothelin-1 promotes mechanoregulation and myofibroblast formation in fibroblasts through the ETA receptor via Akt/PI3 kinase: implications for lung fibrosis. Mol Biol Cell. 2004;15:2707–2719. doi: 10.1091/mbc.E03-12-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- Guo X, Stafford LJ, Bryan B, Xia C, Ma W, Wu X, Liu D, Songyang Z, Liu M. A Rac/Cdc42-specific exchange factor GEFT, induces cell proliferation, transformation, and migration. J Biol Chem. 2003;278:13207–13215. doi: 10.1074/jbc.M208896200. [DOI] [PubMed] [Google Scholar]
- Sugihara K, Nakatsuji N, Nakamura K, Nakao K, Hashimoto R, Otani H, Sakagami H, Kondo H, Nozawa S, Aiba A, Katsuki M. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene. 1998;17:3427–3433. doi: 10.1038/sj.onc.1202595. [DOI] [PubMed] [Google Scholar]
- Sun CX, Downey GP, Zhu F, Koh AL, Thang H, Glogauer M. Rac1 is the small GTPase responsible for regulating the neutrophil chemotaxis compass. Blood. 2004;104:3758–3765. doi: 10.1182/blood-2004-03-0781. [DOI] [PubMed] [Google Scholar]
- Liu S, Kapoor M, Shi-Wen X, Kennedy L, Denton CP, Glogauer M, Abraham DJ, Leask A. Role of Rac1 in a bleomycin-induced scleroderma model using fibroblast-specific Rac1-knockout mice. Arthritis Rheum. 2008;58:2189–2195. doi: 10.1002/art.23595. [DOI] [PubMed] [Google Scholar]
- Zheng B, Zhang Z, Black CM, de Crombrugghe B, Denton CP. Ligand-dependent genetic recombination in fibroblasts: a potentially powerful technique for investigating gene function in fibrosis. Am J Pathol. 2002;160:1609–1617. doi: 10.1016/S0002-9440(10)61108-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Khanna S, Nallu K, Hunt TK, Sen CK. Dermal wound healing is subject to redox control. Mol Ther. 2006;13:211–220. doi: 10.1016/j.ymthe.2005.07.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy GK, Enwemeka CS. A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem. 1996;29:225–229. doi: 10.1016/0009-9120(96)00003-6. [DOI] [PubMed] [Google Scholar]
- Kennedy L, Liu S, Shi-wen X, Carter D, Lyons K, Black CM, Abraham DJ, Leask A. CCN2 is essential for fibroblast function. Exp Cell Res. 2007;313:952–964. doi: 10.1016/j.yexcr.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Junn E, Lee KN, Ju HR, Han SH, Im JY, Kang HS, Lee TH, Bae YS, Ha KS, Lee ZW, Rhee SG, Choi I. Requirement of hydrogen peroxide generation in TGF-beta 1 signal transduction in human lung fibroblast cells: involvement of hydrogen peroxide and Ca2+ in TGF-beta 1-induced IL-6 expression. J Immunol. 2000;165:2190–2197. doi: 10.4049/jimmunol.165.4.2190. [DOI] [PubMed] [Google Scholar]
- Khanday FA, Yamamori T, Mattagajasingh I, Zhang Z, Bugayenko A, Naqvi A, Santhanam L, Nabi N, Kasuno K, Day BW, Irani K. Rac1 leads to phosphorylation-dependent increase in stability of the p66shc adaptor protein: role in Rac1-induced oxidative stress. Mol Biol Cell. 2006;17:122–129. doi: 10.1091/mbc.E05-06-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponticos M, Abraham D, Alexakis C, Lu QL, Black C, Partridge T, Bou-Gharios G. Col1a2 enhancer regulates collagen activity during development and in adult tissue repair. Matrix Biol. 2004;22:619–628. doi: 10.1016/j.matbio.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Farooqui R, Fenteany G. Multiple rows of cells behind an epithelial wound edge extend cryptic lamellipodia to collectively drive cell-sheet movement. J Cell Sci. 2005;118:51–63. doi: 10.1242/jcs.01577. [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S, Ng J, Tzu J, Dietzl G, Sun Y, Harms M, Nardine T, Luo L, Dickson BJ. Rac function and regulation during Drosophila development. Nature. 2002;416:438–442. doi: 10.1038/416438a. [DOI] [PubMed] [Google Scholar]
- Vidali L, Chen F, Cicchetti G, Ohta Y, Kwiatkowski DJ. Rac1-null mouse embryonic fibroblasts are motile and respond to platelet-derived growth factor. Mol Biol Cell. 2006;17:2377–2390. doi: 10.1091/mbc.E05-10-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscharntke M, Pofahl R, Chrostek-Grashoff A, Smyth N, Niessen C, Niemann C, Hartwig B, Herzog V, Klein HW, Krieg T, Brakebusch C, Haase I. Impaired epidermal wound healing in vivo upon inhibition or deletion of Rac1. J Cell Sci. 2007;120:1480–1490. doi: 10.1242/jcs.03426. [DOI] [PubMed] [Google Scholar]
- Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- Leask A. Scar wars: is TGFβ the phantom menace in scleroderma? Arthritis Res Ther. 2006;8:213. doi: 10.1186/ar1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-wen X, Rodrigues-Pascual F, Lamas S, Holmes A, Howat S, Pearson JD, Dashwood MR, du Bois RM, Denton CP, Black CM, Abraham DJ, Leask A. Constitutive ALK5-indepenent JNK activation contributes to endothelin-1 over-expression in pulmonary fibrosis. Mol Cell Biol. 2006;26:5518–5527. doi: 10.1128/MCB.00625-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-wen X, Stanton L, Kennedy L, Pala D, Chen Y, Howat SL, Renzoni EA, Carter DE, Bou-Gharios G, Stratton RJ, Pearson JD, Beier F, Lyons KM, Black CM, Abraham DJ, Leask A. CCN2 is necessary for adhesive responses to TGFß1 in embryonic fibroblasts. J Biol Chem. 2006;281:10715–10726. doi: 10.1074/jbc.M511343200. [DOI] [PubMed] [Google Scholar]
- Fritz G, Kaina B. Rho GTPases: promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets. 2006;6:1–14. [PubMed] [Google Scholar]
- Walmsley MJ, Ooi SK, Reynolds LF, Smith SH, Ruf S, Mathiot A, Vanes L, Williams DA, Cancro MP, Tybulewicz VL. Critical roles for Rac1 and Rac2 GTPases in B cell development and signaling. Science. 2003;302:459–462. doi: 10.1126/science.1089709. [DOI] [PubMed] [Google Scholar]
- Castilho RM, Squarize CH, Patel V, Millar SE, Zheng Y, Molinolo A, Gutkind JS. Requirement of Rac1 distinguishes follicular from interfollicular epithelial stem cells. Oncogene. 2007;26:5078–5085. doi: 10.1038/sj.onc.1210322. [DOI] [PubMed] [Google Scholar]
- Benninger Y, Thurnherr T, Pereira JA, Krause S, Wu X, Chrostek-Grashoff A, Herzog D, Nave KA, Franklin RJ, Meijer D, Brakebusch C, Suter U, Relvas JB. Essential and distinct roles for cdc42 and Rac1 in the regulation of Schwann cell biology during peripheral nervous system development. J Cell Biol. 2007;177:1051–1061. doi: 10.1083/jcb.200610108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W, Palmby TR, Gavard J, Amornphimoltham P, Zheng Y, Gutkind JS. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008;22:1829–1838. doi: 10.1096/fj.07-096438. [DOI] [PubMed] [Google Scholar]
- Minakami R, Sumimotoa H. Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (nox) family. Int J Hematol. 2006;84:193–198. doi: 10.1532/IJH97.06133. [DOI] [PubMed] [Google Scholar]
- Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol. 2006;26:2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox1-dependent reactive oxygen generation is regulated by Rac1. J Biol Chem. 2006;281:17718–17726. doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]