

Abstract
The purpose of this work is to define rare variants of cystic fibrosis (CF) that are potential sources of error and can confound molecular genetic testing methods. We performed routine, clinical CF mutation screening using a laboratory-developed test and the oligonucleotide ligation assay reagents from Abbott/Celera. In this report, we describe 11 unique allele drop outs [3849 + 10kb C>T (NM_000492.2:c.3718-2477C>T), V520F (c.1558G>T), 1078delT (c.948delT), A455E (c.1364C>A), R347P (c.1040G>C), 2184delA (c.2052delA), W1282X (c.3846G>A), R117H (c.350G>A), G85E (c.254G>A), 621 + 1G>T (c.489 + 1G>T), and 2789 + 5G>A (c.2657 + 5G>A)] observed with this platform. The allele drop outs account for less than 0.01% of all results reported in our laboratory. Both the recognition and enumeration of such variants along with clinical information in CF testing is valuable in avoiding false-positive and false-negative results.
Cystic fibrosis (CF) is one of the most common autosomal recessive genetic disorders among Caucasians with an estimated incidence of 1 in 3300 live births. Approximately 1000 new cases are reported each year (http://www.cff.org/AboutCF/, accessed September 21, 2008). The CFTR gene, located on chromosome 7q31, is ∼250,000 bp in length and encodes a protein known as the cystic fibrosis transmembrane conductance regulator (CFTR). CFTR is a channel for the transport of chloride ions across the epithelial cell apical membrane that faces the lumen of the duct or airway and a mutation results in a chloride ion channel defect. Mutations in this gene are associated with CF, which is characterized by pulmonary and gastrointestinal complications as well as male infertility attributable to congenital bilateral absence of the vas deferens.1 More than 1500 mutations have been identified in this gene (http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html, accessed September 21, 2008).
The American College of Obstetricians and Gynecologists and the American College of Medical Genetics have published a joint recommendation to offer CF carrier screening for a panel of 23 mutations to all couples for preconception planning.2,3 These recommendations have been well received and the number of women undergoing CF carrier testing has increased considerably.4
Rare variants are potential sources of error that can confound molecular genetic testing. Examples of this include variants that interfere with polymerase chain reaction (PCR) primer binding, hybridization probe binding, and restriction enzyme recognition. Recognizing and enumerating such variants in CF testing is valuable in avoiding false-positive and false-negative results. The significant increase in CF carrier testing has already resulted in identification of variants. For example, a previous study revealed anomalous electropherograms in five cases screened by a PCR/oligonucleotide ligation assay (OLA), which demonstrated the phenomenon of allele drop out. Of 1763 tests performed, 5 patient samples had electropherograms with absent fluorescent signals. Further analysis led to the identification of three CFTR mutations and one variant. The presence of these sequence variations deemed the clinical analysis to be inconclusive by PCR/OLA and thus, sequencing analysis was necessary for further resolution.5 Similar findings from another study using the Inno-LiPa reagents were discovered in a 28-year-old patient with no family history of CF. A homozygous mutation of 3120G>A was detected by sequence analysis of the region initially identified with a lack of hybridization at the 3120 + 1G>A mutation site.6
Our clinical laboratory performs the CF carrier screen using commercially available reagents that detect the 23 American College of Obstetricians and Gynecologists/American College of Medical Genetics recommended and 8 additional mutations by PCR/OLA. Here we report 11 unique types of allele drop out after using our routine clinical assay. We also performed a cross platform comparison for two of these samples [3849 + 10kb C>T (NM_000492.2:c.3718-2477C>T) and 2184delA (c.2052delA) allele drop out] using the Tag-It CF40 + 4 and InPlex platforms to determine whether the variants interfered with the CF analysis using other kits and reagents.
Materials and Methods
Study Population
The study consisted of more than 1,000,000 samples submitted for routine carrier screening as well as diagnostic testing for CF and congenital bilateral absence of the vas deferens. The ethnicity provided was either self-reported or unavailable.
DNA Extraction
Genomic DNA was isolated from 50 μL of whole blood samples using the Qiagen (Valencia, CA) M96 BioRobot.
Abbott/Celera OLA
The CF Abbott/Celera reagents (Alameda, CA) detect 31 clinically relevant mutations and 4 variants. Briefly, genomic DNA is amplified by multiplex PCR and subsequently the mutant and normal target sequences are detected by multiplex oligonucleotide ligation amplification (OLA). The OLA products are then detected on an ABI 3100 genetic analyzer and interpreted using the ABI CF Genotyper software (Applied Biosystems, Foster City, CA).7
CFTR InPlex™ Assay
The CFTR InPlex (Third Wave Technologies, Madison, WI) reagents detect 40 clinically relevant mutations and 6 polymorphisms. Briefly, genomic DNA is amplified and the PCR products are hybridized to Invader reaction mix (Invader Buffer on a CFTR InPlex Card). Results were read on a plate reader and genotypes were determined by the Invader Data Analysis Worksheet software.
Tag-It CFTR 40 + 6 Assay
The Tag-It CFTR 40 + 6 mutation detection kit (Luminex Molecular Diagnostics, Austin, TX) simultaneously screens for 40 mutations and 6 variants (polymorphisms). Genomic DNA was amplified and alleles were determined using allele-specific primer extension and hybridization to a universal microsphere array. Genotypes were detected on a Luminex 100 xMAP System and called using the TDAS software.
CFTR Sequencing
DNA sequence analysis was performed as described by Strom and colleagues.8
Results
More than 1,000,000 patient samples were screened using the CF OLA assay. The patient samples consisted of routine carrier screening as well as diagnostic testing for CF and congenital bilateral absence of the vas deferens. Among the patient samples tested, 37 samples revealed atypical results that required additional testing. The electropherograms for each of these cases revealed the absence of one of the OLA products expected in the reaction (Figure 1). We observed allele drop out for the following mutations: 3849 + 10kb C>T (NM_000492.2:c.3718-2477C>T), V520F (c.1558G>T), 1078delT (c.948delT), A455E (c.1364C>A), R347P (c.1040G>C), 2184delA (c.2052delA), W1282X (c.3846G>A), R117H (c.350G>A), G85E (c.254G>A), 621 + 1G>T (c.489 + 1G>T), and 2789 + 5G>A (c.2657 + 5G>A). The sequencing results and frequency of the variants are shown in Table 1.
Figure 1.
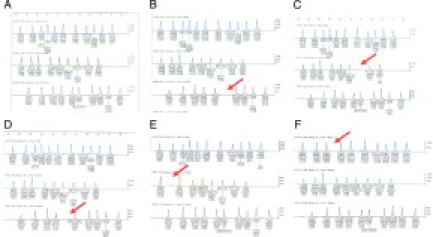
A: A normal electropherogram pattern for no mutation detected. B: The electropherogram in which the 3849 + 10kb C>T peak is missing. C: The electropherogram in which the 621 + 1G>T peak is missing. D: The electropherogram in which the 1078delT peak is missing. E: The electropherogram in which the R117H peak is missing (a slight background peak is present). F: The electropherogram in which the V520F peak is missing. Red arrows indicate missing peaks.
Table 1.
Observed Frequency of Allele Drop Out Using the Abbott/Celera CF Reagents
| Allele dropped out (common name)* | Observed frequency | Sequencing data (observed frequency)* | Ethnicity (observed frequency) | Overall frequency in population screened |
|---|---|---|---|---|
| c.3718-2477C>T (3849 + 10kb C>T) | 19 | c.3718-2530A>G homozygote (13) | African-American (14), Hispanic (2), Haitian (1), Liberian (1), not given (1) | 1.9 × 10−5 |
| c.1558G>T (V520F) | 6 | p.V520I homozygote (2) | Asian (2), Caucasian (1), not given (3) | 0.6 × 10−5 |
| c.948delT (1078delT)† | 3 | p.L320V homozygote (1), p.Y301C/p.L320V(1) | Caucasian (2), not given (1) | 0.3 × 10−5 |
| c.1364C>A (A455E) | 2 | Not performed | African-American (1), Asian (1) | 0.2 × 10−5 |
| c.1040G>C (R347P) | 1 | p.A349V/p.A300G (1) | Caucasian (1) | 0.1 × 10−5 |
| c.2052delA (2184delA) | 1 | p.S686Y homozygote | Ashkenazi Jewish | 0.1 × 10−5 |
| c.3846G>A (W1282X) | 1 | p.A1285V homozygote | Asian Indian | 0.1 × 10−5 |
| c.350G>A (R117H) | 1 | c.492G>A homozygote | African (Sierra Leone) | 0.1 × 10−5 |
| c.254G>A (G85E) | 1 | Not performed | Not given | 0.1 × 10−5 |
| c.489 + 1G>T (621 + 1G>T) | 1 | Not performed | Not given | 0.1 × 10−5 |
| c.2657 + 5G>A (2789 + 5G>A) | 1 | Not performed | Not given | 0.1 × 10−5 |
All cDNA sequence change nomenclature is based on the GenBank cDNA reference sequence NM_000492.3.
c.948delT (1078delT) was removed from the ACOG/ACMG panel in 2004, but is still present in the Abbott/Celera reagents.
Two of these samples (3849 + 10kb C>T and 2184delA allele drop out) were retested using the Tag-It CF40 + 4 and InPlex platforms. Both of these samples were normal by these methods, indicating the OLA probe binding site as the most likely explanation for the loss of the expected OLA products. Additional allele drop out samples were not tested by the Tag-It and InPlex platforms because they were detected after the testing laboratory was acquired and discontinued testing.
Discussion
CF mutation analysis is perhaps the most frequently ordered molecular genetics test. It is used for population carrier screening, carrier status in families with CF, diagnosis of CF and CFTR-related conditions, prenatal diagnosis of high-risk pregnancies, and newborn screening. Most assay platforms are susceptible to interfering variants. A large reference laboratory, such as ours, has the advantage of observing rare anomalies at a significant frequency. Our data show that, although the commonly used PCR/OLA method is robust, in some rare cases, it fails to produce determinate results on mutational analysis resulting from inconclusive electropherograms. Allele drop out accounts for less than 0.01% of all results reported in our laboratory. Approximately 20% of the laboratories that perform CF testing use this method based on the College of American Pathologists survey data, MGL-A 2007.
We suspect the cause of the allele drop outs to be attributable to variants under one of the oligonucleotides in the OLA reaction region rather than variants in PCR primer binding sites. A variant in the PCR primer binding site is expected to result in the loss of multiple loci that are targeted by the PCR fragment rather than just one locus. We have not to date detected a variant in a PCR primer binding site that would be reflected as the loss of multiple OLA products. Sequencing data from 21 of our 37 reference cases support this (Table 1). However, these instances were apparently homozygous for the allele drop out except for two compound heterozygotes (Table 1). Furthermore, because the variants are outside where the oligonucleotides of the Invader platform and the Luminex platform hybridize, they did not interfere with the interpretation of the results using either of these methods. The possibility of a deletion (hemizygosity) in the probe-binding region on one of the two alleles cannot be ruled out.
We were unable to perform studies of parental samples or other family members to determine the cause of apparent homozygosity in most of the samples. Routine CF carrier screening is ordered primarily by obstetricians and gynecologists. In the cases in which we saw allele drop out, the primary practice of these physicians was to screen the partner instead of testing parents for variants.
Almost all of our cases (36 of 37, 97%) were detected in individuals who were referred for population-based carrier screening and who were not symptomatic of CF. The majority of the cases were women (33 of 37, 89%). The frequency of most of these variants is evidently very low. Although a carrier frequency for each can be calculated, the significance of such a figure is unclear.
The variant (c.3718-2530A>G) causing the allele drop out for 3849 + 10kb C>T was the most common in our data set. This variant has not been previously reported in the literature. Based on Hardy-Weinberg distribution, the calculated carrier frequency of it in the tested population is ∼1 in 115. Interestingly, c.3718-2530A>G was observed in 14 individuals who identified themselves as being African-American, 2 as Hispanic, 1 as Liberian (African), and 1 as Haitian. It therefore appears to be primarily present in individuals of African descent.
Because the c.3718-2530A>G variant is novel, its clinical significance has not been previously described. Of note, two individuals with the 3849 + 10kb C>T (c.3718-2477C>T) allele drop out were males who were a part of a couple being evaluated for infertility. It is possible that the variant contributes to male infertility by affecting splicing similar to the mechanism of 5T in intron 8 of CFTR.9 Although most of the individuals who were homozygous for this variant reported no clinical symptoms, potentially this variant may cause mild symptoms of CF that have not been clinically recognized.
One 3849 + 10kb C>T (c.3718-2477C>T) allele drop out was in a sample from a premature, newborn male twin who had some symptoms consistent with CF (fatty stool, malabsorption, and diarrhea). Sweat chloride testing was not performed. The child was homozygous for the c.3718-2530A>G variant. No deletion/duplication testing was performed on this child's sample. The child's mother was a carrier for the c.3718-2530A>G variant and the father declined testing.
Sequencing performed on the other allele drop out cases identified variants that have been reported in the literature as variants or deleterious mutations. The few cases in which known deleterious mutations were found by follow-up sequencing after an ambiguous screening result demonstrate the importance of the ability of laboratories using the PCR/OLA method to recognize the presence of allele drop out. It is extremely important that these cases get the recommended follow-up testing to avoid falsely reporting an indeterminate result as positive or negative. In our data set (Table 1), several physicians declined the recommended follow-up sequencing of CFTR.
Although the scope for false-positive homozygotes attributable to allele drop out in a heterozygote for one of these variants exists, they have not been reported thus far. This possibility has to be borne in mind in newborn screening, and presymptomatic and prenatal testing. During preparation of this article, we saw such a result from a sample. The asymptomatic individual's sample was sent for routine carrier screening and was homozygous for p.F508del, one of the American College of Obstetricians and Gynecologists/American College of Medical Genetics mutations using the PCR/OLA platform (Figure 2). Reflex testing for presence of the known interfering variants I506V (c.1516A>G) and I507V (c.1519A>G) (data not shown) was performed and not detected. The F508C polymorphism is included in the reagents and therefore is not part of the reflex testing. Because the patient did not have clinical symptoms of CF, repeat testing using the Tag-It assay revealed a heterozygous result. Sequencing the sample identified a variant and a mutation (p.F508del and p.E479D).
Figure 2.
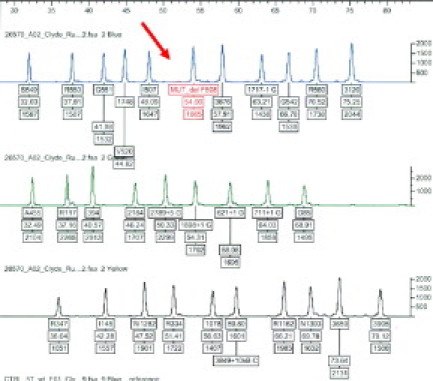
Electropherogram in which the normal peak for F508del is missing (red arrow). The pattern is consistent with an individual who is homozygous for F508del.
When encountering such instances of allele drop out, we recommend confirmation by DNA sequencing of the CFTR gene or by another method. DNA sequencing is indicated if such an event is detected in an individual with a suspected diagnosis of CF. Additionally, it is important to recommend screening on the other partner when allele drop out is detected during evaluation of a pregnant couple.
Acknowledgements
We thank all of the people in the laboratory who perform CF testing each day: Suzanne M. Allen, Erica S. Banks, Charles A. Belanger, Amanda L. Field, Roshanna C. Fisher, Justin A. Frye, Nancy M. Fuentes, Jenny R. Hottel, M. Theresa Huffman, Andrea U. Johnston, Man T. Lam, Ancy T. Manalel, Elizabeth B. Medeiros, Joseph Murray, Dorla M. Myvett, Izumi T. Nakamura, Janice M. Priest, Tara E. Simpson, and Mona S. Tint.
Footnotes
Current addresses of K.M.S.: George Group, Arlington, VA; J.A.W.: Berkeley Heart Laboratory, Berkeley, CA; M.J.: Quest Diagnostics, San Juan Capistrano, CA.
References
- 1.Moskowitz Samuel M, Chmiel James F, Sternen Darci L, Cheng Edith, Cutting Garry R. CFTR Related Disorders. GeneReviews at GeneTests: Medical Genetics Information Resource. University of Washington, Seattle. Updated February 19, 2008; accessed September 29, 2008. Available from http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=cf
- 2.ACOG Committee Opinion #325 Update on carrier screening for cystic fibrosis. Obstet Gynecol. 2005;106:1465–1468. doi: 10.1097/00006250-200512000-00055. [DOI] [PubMed] [Google Scholar]
- 3.Watson MS, Cutting GR, Desnick RJ, Driscoll DA, Klinger K, Mennuti MT, Palomaki GE, Popovich BW, Pratt VM, Rohlfs EM, Strom CM, Richards CS, Witt DR, Grody WW. Cystic fibrosis (CF) population carrier screening: 2004 revision of ACMGs mutation panel. Genet Med. 2004;6:387–391. doi: 10.1097/01.GIM.0000139506.11694.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strom CM, Huang D, Buller A, Redman J, Crossley B, Anderson B, Entwistle T, Sun W. Cystic fibrosis screening using the College panel: platform comparison and lessons learned from the first 20,000 samples. Genet Med. 2002;4:289–296. doi: 10.1097/00125817-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Stanziale P, Savino M, De Bonis P, Granatiero M, Zelante L, Bisceglia L. Indirect CFTR mutation identification by PCR/OLA anomalous electropherograms. Genet Test. 2005;9:285–291. doi: 10.1089/gte.2005.9.285. [DOI] [PubMed] [Google Scholar]
- 6.Heaney DL, Flume P, Hamilton L, Lyon E, Wolff DJ. Detection of an apparent homozygous 3120G>A cystic fibrosis mutation on a routine carrier screen. J Mol Diag. 2006;8:137–140. doi: 10.2353/jmoldx.2006.050065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brinson EC, Adriano T, Bloch W, Brown CL, Chang CC, Chen J, Eggerding FA, Grossman PD, Iovannisci DM, Madonik AM, Sherman DG, Tam RW, Winn-Deen ES, Woo SL, Fung S. Introduction to PCR/OLA/SCS, a multiplex DNA test, and its application to cystic fibrosis. Genet Test. 1997;1:61–88. doi: 10.1089/gte.1997.1.61. [DOI] [PubMed] [Google Scholar]
- 8.Strom CM, Huang D, Chen C, Buller A, Peng M, Quan F, Redman J, Sun W. Extensive sequencing of the cystic fibrosis transmembrane regulator gene: assay validation and unexpected benefits of developing a comprehensive test. Genet Med. 2003;5:9–14. doi: 10.1097/00125817-200301000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Chillón M, Casals T, Mercier B, Bassas L, Lissens W, Silber S, Romey MC, Ruiz-Romero J, Verlingue C, Claustres M, Nunes V, Ferec C, Estivill X. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332:1475–1480. doi: 10.1056/NEJM199506013322204. [DOI] [PubMed] [Google Scholar]