

Abstract
Patients develop cystic fibrosis because of a variety of homozygous recessive mutations, including single nucleotide polymorphisms, insertions, and deletions, in the cystic fibrosis transmembrane regulator (CFTR) gene, or because of compound heterozygosity for two mutations in the CFTR gene. A false determination of homozygosity for a particular CFTR mutation could negatively affect both carrier screens for a patient's family as well as researchers' ability to study the physiological implications of a particular mutation. We argued previously that homozygosity for rare or novel mutations in the CFTR gene could result from a mutation on one allele and the presence of a large deletion encompassing the same sequence region on the second allele. We present here a patient with classic cystic fibrosis who has a novel microdeletion in exon 7 on one allele and a large deletion encompassing exon 7 on the second allele. These data highlight the need to prevent misdiagnosis of homozygous mutations, which can lead to misinterpretation of mutation penetrance and its effects on protein function.
The majority of cystic fibrosis transmembrane conductance regulator (CFTR) mutations are accounted for by single nucleotide polymorphisms and small base pair insertions and deletions.1 When less than two CFTR mutations are discovered by mutation and sequence analysis, however, rearrangements in the CFTR gene are analyzed. This analysis is becoming a routine part of the molecular evaluation of patients with classic cystic fibrosis (CF).2,3,4,5 To date, ∼40 separate large deletions and duplications have been described in the CFTR gene (http://www.genet.sickkids.on.ca/cftr/app, accessed Nov 27, 2008). Some of these have been detected repeatedly in certain populations sharing identical breakpoints, suggesting these are founder rearrangements.4,6 The frequency of individual rearrangement types among the 40 known rearrangements is yet to be determined, with the exception of the CFTRdele2,3 (21 kb), which accounts for ∼4% of CF chromosomes in people of Slavic origin and 0.2% of CF chromosomes in the United States population.7
When standard molecular analysis reveals apparent homozygosity for a rare CFTR mutation, it is essential to determine whether this is true homozygosity. Homozygosity for ΔF508 mutations is common in CF patients of certain ethnic backgrounds, present in ∼68% of CF chromosomes in Caucasians and in ∼36% of CF chromosomes of Ashkenazi Jews.1,8 True homozygosity for CFTR mutations can also result from consanguinity, a common practice in certain parts of the world. On the other hand, apparent homozygosity can be a result of allele dropout9 because of the presence of polymorphisms that influence primer binding. We previously suggested that apparent homozygous mutations in the CFTR gene, especially rare or novel ones, can also be caused by the presence of a large deletion on one chromosome encompassing the location of the mutation.4,10
Distinguishing true homozygosity from apparent homozygosity has important implications not just for genetic counseling of the patient and family members, but also for prenatal diagnosis and preimplantation genetic diagnosis. Not testing for large deletions in an individual with apparent homozygosity for a mutated CFTR allele could result in false-negative carrier screens in any at-risk family members who test negative for the mutation.
Patients with homozygous CFTR mutations are studied to understand physiological implications of CFTR mutations11 or to examine the role of modifier genes.12,13,14 Assessing true homozygosity for mutations will reduce complications that might affect interpretation of such studies. The stepwise approach for studying homozygous CFTR mutation genotypes described by Stanke and colleagues,11 in which frequent mutations were screened for first, followed by more extensive analysis using DNA sequencing and detection of rearrangements, is an example for comprehensive analysis for CFTR mutations in CF patients. On the other hand, patients with missense mutations that are thought to be disease-causing can harbor undetected CFTR deletions that are more likely to cause disease.4 We describe here a patient with an apparent novel homozygous mutation in the CFTR exon 7 that was resolved to be in compound heterozygosity with a large deletion.
Patient
The proband is a 19-year-old Caucasian female with clinical symptoms of classic CF and sweat chlorides of 90 and 87 mmol/L (normal range, <40 mmol/L). She was initially tested in 1999 with a CFTR mutation panel, and no mutations were found. Unfortunately, we do not have information on the CFTR mutation panel because the test was performed at a different laboratory, and the ordering physician did not provide additional information. The ordering physician recently submitted her blood sample to our laboratory for a comprehensive CFTR analysis that includes extensive sequencing analysis of the CFTR gene and detection of CFTR exon deletions/duplications.
Materials and Methods
DNA was extracted from whole blood using a standard Qiagen (Valencia, CA) protocol and was analyzed by DNA sequencing of the promoter and all coding exons of the CFTR gene, as described previously,15,16 and by semiquantitative fluorescent polymerase chain reaction (SQF PCR) for detection of exon deletions/duplications.4 For the latter method, briefly, fragments representing the promoter and all CFTR coding exons and internal controls (amplified from factor II, factor V, and hexosaminidase genes4), were amplified in a single multiplex PCR reaction using fluorescently labeled primers. Fragments were separated by size and analyzed using the ABI 3100 (Applied Biosystems, Foster City, CA) as previously described.4 DNA sequencing results were analyzed using SeqScape software; SQF PCR results were analyzed using GeneMapper software (both from Applied Biosystems). Dosage equivalents were calculated using the area under the peak of each fragment and normalized to each internal control fragment. The normalized fragment from each patient was then divided by the corresponding normalized fragment from a normal control. The equation to calculate dosage equivalents is
where IC1 is internal control 1 fragment, NC is normal control, P is patient, and AUP is area under peak. The average results from normalization using three internal control fragments for each exon is recorded. All samples were analyzed in duplicates and the patient sample was analyzed twice.
Results and Discussion
Examining the electropherograms from a normal (Figure 1A) and the patient's sample (Figure 1B) indicated the presence of an apparent homozygous deletion of exon 7, as no exon 7 fragment was present; all other fragments of the CFTR exons were amplified. Furthermore, the patient's DNA seemed to harbor a deletion extending from exon 4 to exon 10. The complete absence of the exon 7 fragment was surprising, and we suspected either i) an insertion or a deletion within exon 7 affecting its size, ii) a deletion removing exon 7, or iii) a mutation/polymorphism within the primer binding sites for exon 7. Further examination of the electropherograms also revealed the presence of an increased dosage of exon 19 (dosage equivalent = 1.73). Interestingly, the peak for exon 19 was broader than normal, but closer examination of the electropherogram suggested the presence of two very closely migrating fragments within exon 19 bin. We reasoned that one exon 7 allele harbored a deletion of ∼20 bp that shifted the amplified exon 7 fragment to within the range of exon 19. Therefore, exon 19 is normal but appears duplicated because of the presence of the exon 7-related fragment.
Figure 1.
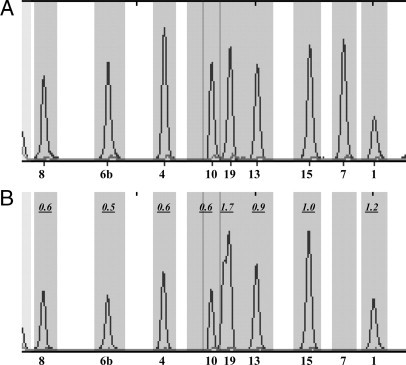
Detection of compound heterozygosity for CF 40-kb del 4-10/1220del20. A: Partial view of the SQF PCR results of a normal electropherogram. Numbers on the bottom indicate the exon. B: Partial view of the SQF PCR results of the patient's electropherogram. Numbers inside the panel show dosage equivalent of exons relative to the normal sample in A. Exons 4, 6b, 8, and 10 show a deletion, whereas exon 19 shows an apparent duplication. Exon 7 is absent, whereas exons 1, 13, and 15 show normal dosage.
Based on the above information, it appeared that the patient harbored two mutations, a deletion of exons 4-10 on one allele, which has been reported previously as CF 40-kb del 4-10,17 and a deletion of ∼20 bp in exon 7 on the second allele. We confirmed the presence the CF 40-kb del 4-10 deletion in the patient's DNA using primers that amplify the junction fragment (Figure 2A) as described by Ferec and colleagues.6 Furthermore, as shown in Figure 2A, amplification of exon 7 using primers described previously15 showed a faster migrating exon 7-related fragment from the patient's DNA, compared with normal control. No normal migrating exon 7 fragment was amplified from the patient's DNA, confirming the apparent homozygosity for the exon 7 fragment.
Figure 2.
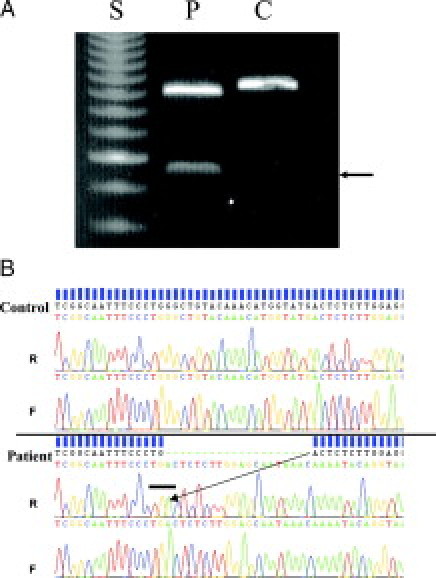
Confirmation of compound heterozygosity for CF 40-kb del 4-10/1220del20. A: Agarose gel electrophoresis showing detection of CF 40-kb del 4-10 in the patient (lane P, arrow) but not in the normal control (lane C) using primers described elsewhere.5 The upper fragments shown are for exon 7 amplicons; notice the slightly faster migration of the apparent homozygous exon 7 (one amplicon) from the patient compared with the control. Lane S, 50-bp size standard. B: DNA sequencing of exon 7 traces (forward, F, and reverse, R) from a normal patient (control, top) and patient (bottom). The 20-bp apparent homozygous deletion in the patient's electropherogram is marked by green dashes. The arrow shows the junction of the 20-bp deletion. The bar in the bottom panel points toward the TGA stop codon at the deletion junction.
Comprehensive DNA sequencing of the patient's CFTR coding exons confirmed our analysis and showed a deletion of 20 bp, leading to the generation of a TGA stop codon immediately at the point of the deletion site (Figure 2B). This would result in a CFTR protein lacking the nucleotide-binding domain 1 and beyond. We designate the mutation c1220del20. Therefore, because the CF 40-kb del 4-10 deletion encompassed exon 7, the novel c1220del20 microdeletion in exon 7 on the other allele appeared homozygous.
This observation that apparent homozygosity for a rare mutation in the CFTR gene was caused by the presence of a large deletion confirms our initial suspicions that such patients can be compound heterozygotes for a rearrangement rather than homozygous for a rare mutation. Interpretation of mutation penetrance, segregation within family and effects on protein function, cannot be made with absolute confidence until the true nature of the patient's CFTR mutation is determined, and this requires both extensive sequencing and deletion/duplication analysis in these cases.
Family studies can also be extremely helpful in these instances for proper genetic and parental counseling, but in this case family members were not available for analysis. Current testing with the American College of Medical Genetics-recommended mutation panel18 would obviously miss both mutations described here, but now that both mutations are identified, relatives of the proband can be tested for the mutations using single exon sequencing for the c1220del20, and deletion-duplication analysis or junction fragment amplification for the CF 40-kb del 4-10.
The detection of deletions and duplications in the CFTR gene is becoming more routine as newer methodologies and software algorithms are developed to replace the laborious Southern blot analysis.4,5,19 Other methods have already been developed for other genes such as exon arrays,20 capillary electrophoresis,21,22 SNP arrays,23 and oligo-microarrays.24,25,26 Facilitating easier and faster methods for detection of CFTR rearrangements will make identification of large rearrangement more feasible. The actual frequency of CFTR rearrangements is not yet fully known, and it will vary depending on the population screened and geographical regions analyzed, but is generally thought to account of less than 2% of CF chromosomes.3,27 However, this value might increase if more cases of apparent homozygosity, like the one presented here, are resolved to be because of a large deletion, or apparent pathogenic missense mutations are shown to be present in cis with a large deletion/duplication. Utilization of rapid high throughput methods will facilitate identification of such rearrangements and determination of their actual frequency in patients and in the general population.
In conclusion, comprehensive mutation analysis using DNA sequencing and exon deletions/duplications is important to resolve apparent homozygosity for novel and rare mutations. Because some of the mutations tested with the American College of Medical Genetics panel can be considered rare,28 apparent homozygosity for these mutations would benefit from re-examination for the presence of large exon deletions. This case also demonstrates the utility of re-examining older unresolved CFTR patient cases, in this case 9 years after initial examination.
Acknowledgements
We thank the reviewers for their very helpful comments on our manuscript.
Footnotes
Conflict of Interest Statement: the authors declare they are employees of Quest Diagnostics and some of them hold stock in the company.
References
- 1.Cutting GR. In: Cystic Fibrosis. Principles and Practice of Medical Genetics. Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Churchill Livingstone; London: 2002. pp. 2685–2717. [Google Scholar]
- 2.Audrézet MP, Chen JM, Raguenes O, Chuzhanova N, Giteau K, Le Marechal C, Quere I, Cooper DN, Ferec C. Genomic rearrangements in the CFTR gene: extensive allelic heterogeneity and diverse mutational mechanisms. Hum Mutat. 2004;23:343–357. doi: 10.1002/humu.20009. [DOI] [PubMed] [Google Scholar]
- 3.Niel F, Martin J, Dastot-Le Moal F, Costes B, Boissier B, Delattre V, Goossens M, Girodon E. Rapid detection of CFTR gene rearrangements impacts on genetic counselling in cystic fibrosis. J Med Genet. 2004;41:e118. doi: 10.1136/jmg.2004.022400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hantash FM, Redman JB, Starn K, Anderson B, Buller A, McGinniss MJ, Quan F, Peng M, Sun W, Strom CM. Novel and recurrent rearrangements in the CFTR gene: clinical and laboratory implications for cystic fibrosis screening. Hum Genet. 2006;119:126–136. doi: 10.1007/s00439-005-0082-0. [DOI] [PubMed] [Google Scholar]
- 5.Tomaiuolo R, Sangiuolo F, Bombieri C, Bonizzato A, Cardillo G, Raia V, D'Apice MR, Bettin MD, Pignatti PF, Castaldo G, Novelli G. Epidemiology and a novel procedure for large scale analysis of CFTR rearrangements in classic and atypical CF patients: a multicentric Italian study. J Cyst Fibrosis. 2008;7:347–351. doi: 10.1016/j.jcf.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Férec C, Casals T, Chuzhanova N, Macek M, Jr, Bienvenu T, Holubova A, King C, McDevitt T, Castellani C, Farrell PM, Sheridan M, Pantaleo SJ, Loumi O, Messaoud T, Cuppens H, Torricelli F, Cutting GR, Williamson R, Ramos MJ, Pignatti PF, Raguenes O, Cooper DN, Audrezet MP, Chen JM. Gross genomic rearrangements involving deletions in the CFTR gene: characterization of six new events from a large cohort of hitherto unidentified cystic fibrosis chromosomes and meta-analysis of the underlying mechanisms. Eur J Hum Genet. 2006;14:567–576. doi: 10.1038/sj.ejhg.5201590. [DOI] [PubMed] [Google Scholar]
- 7.Dörk T, Macek M, Jr, Mekus F, Tummler B, Tzountzouris J, Casals T, Krebsova A, Koudova M, Sakmaryova I, Macek M, Sr, Vavrova V, Zemkova D, Ginter E, Petrova NV, Ivaschenko T, Baranov V, Witt M, Pogorzelski A, Bal J, Zekanowsky C, Wagner K, Stuhrmann M, Bauer I, Seydewitz HH, Neumann T, Jakubiczka S. Characterization of a novel 21-kb deletion, CFTRdele2,3(21 kb), in the CFTR gene: a cystic fibrosis mutation of Slavic origin common in Central and East Europe. Hum Genet. 2000;106:259–268. doi: 10.1007/s004390000246. [DOI] [PubMed] [Google Scholar]
- 8.Ogino S, Wilson RB, Gold B, Hawley P, Grody WW. Bayesian analysis for cystic fibrosis risks in prenatal and carrier screening. Genet Med. 2004;6:439–449. doi: 10.1097/01.gim.0000139511.83336.8f. [DOI] [PubMed] [Google Scholar]
- 9.Rechitsky S, Strom C, Verlinsky O, Amet T, Ivakhnenko V, Kukharenko V, Kuliev A, Verlinsky Y. Allele dropout in polar bodies and blastomeres. J Assist Reprod Genet. 1998;15:253–257. doi: 10.1023/A:1022532108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hantash FM, Redman JB, Goos D, Kammesheidt A, McGinniss MJ, Sun W, Strom CM. Characterization of a recurrent novel large duplication in the cystic fibrosis transmembrane conductance regulator gene. J Mol Diagn. 2007;9:556–560. doi: 10.2353/jmoldx.2007.060141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stanke F, Ballmann M, Bronsveld I, Dork T, Gallati S, Laabs U, Derichs N, Ritzka M, Posselt HG, Harms HK, Griese M, Blau H, Mastella G, Bijman J, Veeze H, Tummler B. Diversity of the basic defect of homozygous CFTR mutation genotypes in humans. J Med Genet. 2008;45:47–54. doi: 10.1136/jmg.2007.053561. [DOI] [PubMed] [Google Scholar]
- 12.Dorfman R, Sandford A, Taylor C, Huang B, Frangolias D, Wang Y, Sang R, Pereira L, Sun L, Berthiaume Y, Tsui LC, Pare PD, Durie P, Corey M, Zielenski J. Complex two-gene modulation of lung disease severity in children with cystic fibrosis. J Clin Invest. 2008;118:1040–1049. doi: 10.1172/JCI33754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bremer LA, Blackman SM, Vanscoy LL, McDougal KE, Bowers A, Naughton KM, Cutler DJ, Cutting GR. Interaction between a novel TGFB1 haplotype and CFTR genotype is associated with improved lung function in cystic fibrosis. Hum Mol Genet. 2008;17:2228–2237. doi: 10.1093/hmg/ddn123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F, Zariwala M, Fargo D, Xu A, Dunn JM, Darrah RJ, Dorfman R, Sandford AJ, Corey M, Zielenski J, Durie P, Goddard K, Yankaskas JR, Wright FA, Knowles MR. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443–1453. doi: 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- 15.Strom CM, Huang D, Chen C, Buller A, Peng M, Quan F, Redman J, Sun W. Extensive sequencing of the cystic fibrosis transmembrane regulator gene: assay validation and unexpected benefits of developing a comprehensive test. Genet Med. 2003;5:9–14. doi: 10.1097/00125817-200301000-00002. [DOI] [PubMed] [Google Scholar]
- 16.McGinniss MJ, Chen C, Redman JB, Buller A, Quan F, Peng M, Giusti R, Hantash FM, Huang D, Sun W, Strom CM. Extensive sequencing of the CFTR gene: lessons learned from the first 157 patient samples. Hum Genet. 2005;118:331–338. doi: 10.1007/s00439-005-0065-1. [DOI] [PubMed] [Google Scholar]
- 17.Chevalier-Porst F, Bonardot AM, Chazalette JP, Mathieu M, Bozon D. 40 kilobase deletion (CF 40 kb del 4-10) removes exons 4 to 10 of the cystic fibrosis transmembrane conductance regulator gene. Hum Mutat. 1998;(Suppl 1):S291–S294. doi: 10.1002/humu.1380110191. [DOI] [PubMed] [Google Scholar]
- 18.Watson MS, Cutting GR, Desnick RJ, Driscoll DA, Klinger K, Mennuti M, Palomaki GE, Popovich BW, Pratt VM, Rohlfs EM, Strom CM, Richards CS, Witt DR, Grody WW. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med. 2004;6:387–391. doi: 10.1097/01.GIM.0000139506.11694.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider M, Joncourt F, Sanz J, von Kanel T, Gallati S. Detection of exon deletions within an entire gene (CFTR) by relative quantification on the LightCycler. Clin Chem. 2006;52:2005–2012. doi: 10.1373/clinchem.2005.065136. [DOI] [PubMed] [Google Scholar]
- 20.Dhami P, Coffey AJ, Abbs S, Vermeesch JR, Dumanski JP, Woodward KJ, Andrews RM, Langford C, Vetrie D. Exon array CGH: detection of copy-number changes at the resolution of individual exons in the human genome. Am J Hum Genet. 2005;76:750–762. doi: 10.1086/429588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashton EJ, Yau SC, Deans ZC, Abbs SJ. Simultaneous mutation scanning for gross deletions, duplications and point mutations in the DMD gene. Eur J Hum Genet. 2008;16:53–61. doi: 10.1038/sj.ejhg.5201916. [DOI] [PubMed] [Google Scholar]
- 22.Weber J, Miserere S, Champ J, Looten R, Stoppa-Lyonnet D, Viovy JL, Houdayer C. High-throughput simultaneous detection of point mutations and large-scale rearrangements by CE. Electrophoresis. 2007;28:4282–4288. doi: 10.1002/elps.200700010. [DOI] [PubMed] [Google Scholar]
- 23.Garshasbi M, Motazacker MM, Kahrizi K, Behjati F, Abedini SS, Nieh SE, Firouzabadi SG, Becker C, Ruschendorf F, Nurnberg P, Tzschach A, Vazifehmand R, Erdogan F, Ullmann R, Lenzner S, Kuss AW, Ropers HH, Najmabadi H. SNP array-based homozygosity mapping reveals MCPH1 deletion in family with autosomal recessive mental retardation and mild microcephaly. Hum Genet. 2006;118:708–715. doi: 10.1007/s00439-005-0104-y. [DOI] [PubMed] [Google Scholar]
- 24.Rouleau E, Lefol C, Tozlu S, Andrieu C, Guy C, Copigny F, Nogues C, Bieche I, Lidereau R. High-resolution oligonucleotide array-CGH applied to the detection and characterization of large rearrangements in the hereditary breast cancer gene BRCA1. Clin Genet. 2007;72:199–207. doi: 10.1111/j.1399-0004.2007.00849.x. [DOI] [PubMed] [Google Scholar]
- 25.del Gaudio D, Yang Y, Boggs BA, Schmitt ES, Lee JA, Sahoo T, Pham HT, Wiszniewska J, Chinault AC, Beaudet AL, Eng CM. Molecular diagnosis of Duchenne/Becker muscular dystrophy: enhanced detection of dystrophin gene rearrangements by oligonucleotide array-comparative genomic hybridization. Hum Mutat. 2008;29:1100–1107. doi: 10.1002/humu.20841. [DOI] [PubMed] [Google Scholar]
- 26.Staaf J, Torngren T, Rambech E, Johansson U, Persson C, Sellberg G, Tellhed L, Nilbert M, Borg A. Detection and precise mapping of germline rearrangements in BRCA1, BRCA2, MSH2, and MLH1 using zoom-in array comparative genomic hybridization (aCGH) Hum Mutat. 2008;29:555–564. doi: 10.1002/humu.20678. [DOI] [PubMed] [Google Scholar]
- 27.Paracchini V, Seia M, Coviello D, Porcaro L, Costantino L, Capasso P, Degiorgio D, Padoan R, Corbetta C, Claut L, Costantini D, Colombo C. Molecular and clinical features associated with CFTR gene rearrangements in Italian population: identification of a new duplication and recurrent deletions. Clin Genet. 2008;73:346–352. doi: 10.1111/j.1399-0004.2007.00957.x. [DOI] [PubMed] [Google Scholar]
- 28.Tsongalis GJ, Belloni DR, Grody WW. Cystic fibrosis mutation analysis: how many is enough? Genet Med. 2004;6:456–458. [PubMed] [Google Scholar]