

Abstract
Apolipoprotein AI-derived (AApoAI) amyloidosis may present either as a non-hereditary form with wild-type protein deposits in atherosclerotic plaques or as a hereditary form due to germline mutations in the APOA1 gene. Currently, more than 50 apoAI variants are known, and 13 are associated with amyloidosis. We describe six patients with AApoAI amyloidosis due to APOA1 germline mutations that affect the larynx, small intestine, large intestine, heart, liver, kidney, uterus, ovary, or pelvic lymph nodes. In each patient, the amyloid showed a characteristic apple green birefringence when viewed under polarized light after Congo red staining and was immunoreactive with antibodies against apoAI. Sequence analyses revealed one known (p.Leu75Pro) and three novel APOA1 mutations that included gene variations leading to two different frameshifts (p.Asn74fs and p.Ala154fs) and one amino acid exchange (p.Leu170Pro). These three novel mutations extend our knowledge about both the location of the mutations and the organ distribution in hereditary AApoAI amyloidosis. Thirteen of the now sixteen amyloidogenic mutations are localized in two hot-spot regions that span residues 50 to 93 and 170 to 178. The organ distribution and clinical presentation of AApoAI amyloidosis seems to depend on the position of the mutation. Patients with alterations in codons 1 to 75 mostly develop hepatic and renal amyloidosis, while carriers of mutations in residues 173 to 178 mainly suffer from cardiac, laryngeal, and cutaneous amyloidosis.
The amyloidoses are a large group of heterogeneous diseases characterized by insoluble protein and peptide aggregates oriented in a β-pleated sheet structure, forming amyloid fibrils of 10 to 12 nm diameter. More than 26 different proteins have been identified to form amyloid. Depending on the histoanatomical distribution and amount, amyloid may cause progressive and life-threatening organ dysfunction.1 Amyloid may be acquired or hereditary in origin, and it can deposit locally or present as a systemic disease. Due to the diversity of the precursor proteins with no sequence homology between them, it has been impossible to find any common primary structural or functional motif that predicts the amyloidogenicity of a peptide or protein. In this respect, hereditary amyloidoses are particularly interesting. They are caused by germline mutations, which increase the propensity of the affected protein to form aggregates under particular circumstances. Variants of transthyretin, apolipoprotein AI (apoAI), apolipoprotein AII, fibrinogen Aα-chain, gelsolin, and lysozyme are some of the proteins known to cause hereditary amyloidosis. The most frequent form of hereditary amyloidosis is the transthyretin-derived ATTR amyloidosis, which clinically presents with polyneuropathy and/or cardiomyopathy.2
Apolipoprotein AI-derived (AApoAI) amyloidosis can be present as a non-hereditary form with wild-type protein deposits in atherosclerotic plaques,3 or as a hereditary form with the variant protein depositing more systemically. The clinical manifestations of hereditary AApoAI amyloidosis frequently involve liver, kidney, larynx, skin, and myocardium. In rarer cases, amyloid is also found in the testes and adrenal glands.4 ApoAI is a plasma protein of 28 kDa synthesized by the liver and the small intestine. It is the main protein of high-density lipoprotein particles and important for the formation and metabolism of high-density lipoprotein cholesterol esters.5 Mature apoAI consist of 243 amino acids encoded by exons 3 and 4 of the APOA1 gene.6 More than 50 apoAI variants have been described,4 and about half of them are associated with a decreased plasma level of high-density lipoprotein-apoAI. These apoAI variants either affect lecithin:cholesterol acyltransferase activity or promote the formation of amyloid.5 To date, 13 mutations are known to be associated with hereditary AApoAI amyloidosis (Table 1).7,8,9,10,11,12,13,14,15,16,17,18,19,20,21 The majority of the germline mutations are nucleotide substitutions, but two variants are caused by deletions13,17 and another one is due to a deletion/insertion mutation.12
Table 1.
ApoA1 Mutations Associated with AApoAI Amyloidosis
| Variant (mature protein) | Clinical manifestations/found amyloid sites | Reference |
|---|---|---|
| p.Gly26Arg | Renal failure, gastrointestinal amyloid, peripheral neuropathy | 7, 8 |
| p.Trp50Arg | Renal failure | 9 |
| p.Leu60Arg | Renal failure | 10 |
| p.Leu64Pro | Renal failure | 11 |
| p.Leu60_Phe71delinsValThr | Liver failure | 12 |
| p.Glu70_Trp72del | Renal failure | 13 |
| p.Asn74fs | Gastrointestinal amyloid, renal failure, amyloid detected in uterus, ovaries, pelvic lymph nodes | Current report |
| p.Leu75Pro | Renal failure, hepatic amyloid, gastrointestinal amyloid | 14, 15 Current report |
| p.Leu90Pro | Cardiomyopathy, cutaneous amyloid | 16 |
| p.Lys107del | Aortic intimal amyloid | 17 |
| p.Ala154fs | Renal amyloid | Current report |
| p.Leu170Pro | Amyloid in larynx | Current report |
| p.Arg173Pro | Cardiomyopathy, cutaneous and laryngeal amyloid | 18 |
| p.Leu174Ser | Cardiomyopathy | 19 |
| p.Ala175Pro | Laryngeal amyloid | 20 |
| p.Leu178His | Cardiomyopathy, cutaneous and laryngeal amyloid | 21 |
In this report, we describe six German patients with hereditary AApoAI amyloidosis, presenting with one known and three novel mutations in the APOA1 coding sequence.
Case Reports
Six German patients (four women and two men) with hereditary AApoAI amyloidosis were studied. Patients were retrieved from the Amyloid Registries of the University Hospitals in Berlin and Heidelberg between June 2001 and August 2008. Following the identification of cases with AApoAI amyloidosis, all patients gave written informed consent for genomic analyses. The study was performed in accordance with the guidelines set out by the German government and the local Ethics Committee of the University of Berlin.
Patient No. 1
A normotensive woman was admitted with nephrotic syndrome at the age of 58 years, and a kidney biopsy was taken. No further clinical information was available from this patient.
Patient No. 2
A hysteroadnexectomy was performed in a 48-year-old woman because of a serous borderline tumor of the right ovary. She was on hemodialysis twice a week since June 2000 due to terminal renal failure. She also suffered from secondary hyperparathyroidism, anemia, and hypertension. Tissue samples were obtained from the hysteroadnexectomy specimens.
Patient No. 3
A rectal biopsy of this 67-year-old woman was submitted for the immunohistochemical classification of amyloid. Patients No. 2 and No. 3 are related. No further clinical information was available.
Patient No. 4
A 54-year-old woman had a short history of discomfort in the upper abdomen after an episode of diarrhea and nausea nine months earlier. Biopsies retained from a duodenoscopy and colonoscopy revealed amyloid. She also had a history of type I diabetes for 20 years and of renal failure without nephrotic syndrome. No symptoms of dyspnea, polyneuropathy, or other organ involvement were present. Analysis of a kidney biopsy excluded diabetic nephropathy and revealed minimal renal amyloidosis. Vasculopathy was determined as the main cause of renal failure. In the course of the clinical work-up for liver transplantation, a cardiac biopsy was obtained, and cardiac amyloidosis was found. The daughter, sister, mother, and grandmother of the patient all suffer from type I diabetes, while the father died of leukemia.
Patient No. 5
A 52-year-old man was suffering from swallowing complaints for 6 years. A biopsy from the vocal cord revealed amyloid deposits in the larynx. The patient showed no symptoms of systemic amyloidosis. All laboratory values were normal. Ultrasound of the abdomen as well as electrocardiogram and echocardiography were also normal. There was no family history of amyloidosis.
Patient No. 6
A 41-year-old man presented with elevated liver enzymes. The main symptom was itching. A liver biopsy revealed amyloidosis. The patient's grandmother had died of an undiagnosed liver disease at the age of 65 years.
Materials and Methods
Histology and Immunohistochemistry
The biopsies were fixed in formalin and embedded in paraffin. Serial sections were stained with H&E. Amyloid was analyzed by viewing Congo red-stained sections under polarized light. Immunohistochemistry was performed with commercially available monoclonal antibodies directed against AA amyloid (1:600) and polyclonal antibodies against amyloid P component (1:5000), fibrinogen (1:2000), lysozyme (1:3000), transthyretin (1:4000), λ-light chain (1:160,000), and κ-light chain (1:160,000) (all DAKO, Hamburg, Germany), as well as with non-commercially available polyclonal antibodies directed against apolipoprotein AI (1:1000),22 λ-light chain-derived amyloid proteins (AL1, 1:3000),23 and anti-λ-light chain peptide antibodies (AL3,24 1:250 and AL7, 1:500, peptide used for antibody production as described by others25). Immunostaining was done with the BenchMark XT immunostainer using the ultraView Universal Alkaline Phosphatase Red Detection Kit (both Ventana Medical Systems, Inc., Tucson, Arizona) or the NOVADetect DAB-Substrate Kit (Dianova, Hamburg, Germany). Before the incubation with primary antibodies, sections were pretreated with Cell Conditioning 1 according to the manufacturer's instructions (CC1; Ventana) or with sodium citrate (four times, 5 minutes, 600W, microwave oven). The specificity of the immunostaining was verified using specimens containing known classes of amyloid (AA amyloid, transthyretin, λ-light chain), or by using positive controls recommended by the manufacturers (remaining antibodies). Omission of the primary antibodies served as a negative control.
DNA Sequence Analysis
Genomic DNA was isolated from peripheral blood leukocytes using the QIAamp DNA blood mini kit (Qiagen, Hilden, Germany). In one case, total genomic DNA was extracted from formalin-fixed paraffin-embedded tissue with the QIAamp DNA mini kit (Qiagen) as described by the manufacturer. Amplification of APOA1 exons 1–4 was performed by PCR with the following specific primer pairs (Biotez, Berlin, Germany): exon 1–2 (489 bp) forward 5′-AAGTTCCACATTGCCAGGAC-3′ and reverse 5′-AGAGGCAGCAGGTTTCTCAC-3′; exon 3 (376 bp) forward 5′-AGAGGCAGCAGGTTTCTCAC-3′ and reverse 5′-AATATCTGATGAGCTGGGCC-3′; exon 4a (493 bp) forward 5′-AAGAGAAGCTGAGCCCACT-3′ and reverse 5′-CCCTACAGCGACGAGCTG-3′; and exon 4b (400 bp) forward 5′-CTGGAAATGCTAGGCCAC-3′ and reverse 5′-CAGCTTCTTTCTTTTGGGAGAA-3′. A 30-μl reaction mixture contained 200 ng DNA, 1 μmol/L of the exon-specific primers, 1x PCR buffer, 1.5 U Taq polymerase, 0.2 μmol/L dNTPs, and Q-solution in concentrations recommended by the manufacturer (Qiagen). The following thermocycling conditions were used: initial denaturation at 94°C for 3 minutes, 40 cycles of 94°C for 1 minute, 60°C (61°C for the exon 4a primer pair) for 1.5 minutes, 72°C for 1 minute, and a final elongation step at 72°C for 7 minutes. A negative control with water instead of DNA was included in each PCR run, and blood isolated from an individual with wild-type APOA1 served as a positive control. The quality and size of the generated PCR products were analyzed by agarose gel electrophoresis. Fragments were purified with the ExoSAP-IT kit for PCR product clean-up (USB Corp., Cleveland, OH). The PCR products were sequenced in both directions with the ABI PRISM BigDye Terminator v3.1 Ready Reaction Cycle Sequencing kit (Applied Biosystems, Foster City, CA) using the same set of primers as described above. Sequences were analyzed on an ABI PRISM 3130 Genetic Analyzer.
Results
Histology and Immunohistochemistry
In each patient, the amyloid deposits showed a characteristic apple green birefringence when viewed under polarized light after Congo red staining (Figure 1). Amyloid was present as glomerular (kidney), vascular, or interstitial deposits. In patient No. 1, focal glomerular amyloid deposits were found in a kidney biopsy. Patient No. 2 had massive vascular and interstitial deposits in the uterus, ovaries, soft tissue, and pelvic lymph nodes. Patients No. 3 and No. 4 were found to have massive vascular and interstitial amyloid deposits in the biopsies of the large and small intestine, respectively. Patient No. 4 also had interstitial amyloid deposits in an endomyocardial biopsy, while patient No. 5 had subepithelial deposits in a laryngeal biopsy. In patient No. 6, amyloid was found in a liver biopsy and was restricted to the portal tracts (Figure 1). Table 2 summarizes the biopsy sites where amyloid was found in our patients.
Figure 1.
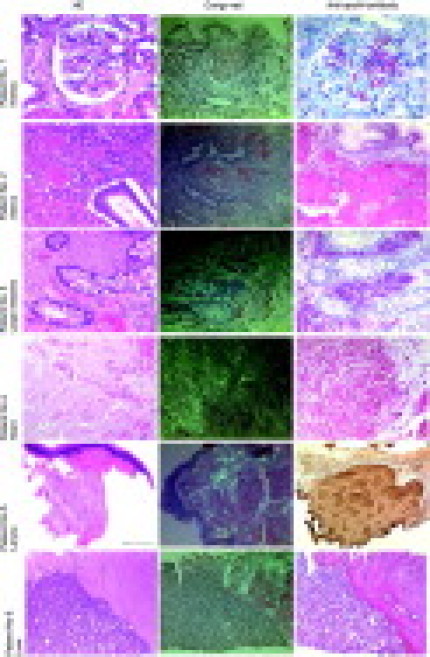
Biopsies from the six patients with AApoAI amyloidosis.Histological examination of the biopsy and resection specimens revealed homogeneous eosinophilic deposits in H&E-stained sections (left panel). Same areas produced a typical apple green birefringence in polarized light after Congo red (CR) staining (center). Staining with an anti-apoAI antibody (right panel) showed a strong and even immunoreactivity of these amyloid deposits in every patient specimen. Sections from the patients 1–4 and 6 were stained with the alkaline phosphatase system (red color), whereas the section from patient No. 5 was stained with a DAB substrate kit (brown color). Scale bar=100 μm.
Table 2.
AApoAI Patients, Mutations, and Tissue Sites
| Patient no. | Sex | Age (y) | Mutation (cDNA) | Mutation (protein) | Analyzed tissue sites | Histology |
|---|---|---|---|---|---|---|
| 1 | Female | 58 | c.460_461dupGC | p.Ala154fsX47 | Kidney | Glomerular amyloid deposits; no renal vascular deposits |
| 2 | Female | 48 | c.293_294insA | p.Asn74Lysfs | Uterus, ovaries, pelvic lymph nodes | Massive vascular and interstitial amyloid deposits |
| 3 | Female | 67 | c.293_294insA | p.Asn74Lysfs | Large intestine | Interstitial amyloid deposits |
| 4 | Female | 54 | c.296T>C | p.Leu75Pro | Small intestine, Heart | Interstitial amyloid deposits |
| 5 | Male | 52 | c.581T>C | p.Leu170Pro | Larynx | Subepithelial and interstitial amyloidosis |
| 6 | Male | 41 | c.296T>C | p.Leu75Pro | Liver | Massive amyloid deposits in portal tracts |
The amyloid deposits in every patient showed an intense and even staining with antibodies directed against amyloid P component and apoAI (Figure 1). No significant immunostaining was found with antibodies directed against AA amyloid, fibrinogen, lysozyme, β2-microglobulin, λ- and κ-light chain, or transthyretin.
DNA Sequence Analysis
Sequence analysis revealed a mutation in the coding sequence of the APOA1 gene in all six patients. Patient No. 1 had a GC duplication (c.460_461dupGC), leading to a frameshift (p.Ala154fsX48) resulting in a truncated protein of 200 amino acids instead of 243 amino acids. Two of the patients (2 and 3) carried an adenine insertion at nucleotide position 294 (c.293_294insA), which also leads to a frameshift (p.Asn74fsX106). Theoretically, this mutation would result in a mutant protein of 178 amino acids, where the first 73 residues are identical to the wild-type apoAI, and the remaining 105 amino acids are completely different. Patient No. 5 was heterozygous for a c.581T>C transition, resulting in a p.Leu170Pro substitution. These three mutations have heretofore not been described. One known mutation (c.296T>C or p.Leu75Pro) was found in patients 4 and 6.
Discussion
Hereditary amyloidosis was first described by Ostertag in 1932 and again in the early 1950's.26,27 Even if it is considered to be a rare disease, it has to be separated from other forms of systemic amyloidosis, such as the more common immunoglobulin-derived AL amyloidosis, as these diseases require a different patient management. Lachmann et al reported that about 10% of the patients diagnosed with AL amyloidosis were misdiagnosed and had to be reclassified as hereditary amyloidosis, eg, transthyretin-derived ATTR amyloidosis or fibrinogen Aα-chain-derived AFib amyloidosis.20 In the patient series of the Amyloid Registry of the Charité University Hospital and of the University Hospital in Heidelberg, approximately 5% and 3% of the patients were found to suffer from hereditary amyloidosis, respectively (unpublished observation). Recent reports describe relatively large patient cohorts and families with hereditary amyloidosis22,28,29,30 emphasizing the importance of genetic analyses in the diagnostic work-up of amyloidosis.
We here present six patients with mutations in the coding sequence of the APOA1 gene leading to AApoAI amyloidosis. Three of the mutations have not been described hitherto and extend the number of APOA1 mutations associated with AApoAI amyloidosis to sixteen (Table 1).7,8,9,10,11,12,13,14,15,16,17,18,19,20,21 Half of these mutations affect residues 50 to 93.3,4 One of the novel frameshift mutations, p.Asn74fs is also located within this region. The bp exchange affecting residue 170, in contrast, is present in a region where four other amyloidogenic mutations have been reported. Three of the sixteen mutations, including one of the novel mutations described here, are affecting residues outside these two hot spot regions (Figure 2).
Figure 2.
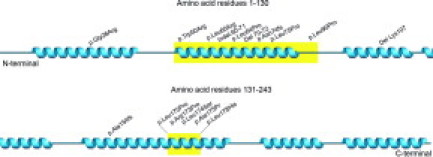
Distribution of ApoAI mutations with regard to the proteins secondary structure. The hot spot regions spanning residues 50 to 93 and 170 to 178 are marked in yellow and the twisted blue areas represent the α-helices. The picture of the secondary structure of ApoAI was obtained from the database Jpred at the European Bioinformatics Institute and modified.
The N-terminal peptide fragments 1 to 100 of apoAI have been found in the amyloid deposits of other AApoAI patients,12,19,31,32 which illustrates the propensity of this region for fibrillogenesis. Amyloid deposits consisting mainly of N-terminal fragments of the precursor protein are also common in other types of amyloid.23,33 The antibody used in this study is directed against an epitope within the first 40 amino acid residues of apoAI.22 Strong immunostaining of the amyloid deposits with our anti-apoAI antibody therefore indicates that the N-terminal region of apoAI is deposited in all our six patients.
The histoanatomical distribution and clinical manifestation of hereditary AApoAI amyloidosis seems to depend on the location of the APOA1 mutation. Patients with gene mutations affecting residues 1 to 75 mainly suffer from hepatic and renal amyloidosis, while mutations in codons 173 to 178 mainly cause AApoAI amyloidosis of the heart, larynx, and skin (Table 1).7,8,9,10,11,12,13,14,15,16,17,18,19,20,21 Correspondingly, the patient carrying p.Leu170Pro suffered from laryngeal amyloidosis. The frameshift mutation with the first affected amino acid at position 74 resulted in massive amounts of amyloid being deposited in the uterus and pelvic soft tissue in one patient and in the gastrointestinal tract in another. No other organ or biopsy site was available for analysis and the extent of amyloid infiltration in these patients cannot be determined. Nevertheless, it is likely that this highly altered apoAI variant can affect several organs and tissue sites.
Interestingly, four of the six patients described here were diagnosed within the last 1.5 years. This was achieved by a combination of extensive and thorough immunohistochemical staining, using a large panel of antibodies directed against eight amyloid precursor proteins followed by molecular biological studies on genomic DNA. Genetic analysis is crucial to detect hereditary amyloidosis in time, since some types can be treated by liver or kidney transplantation. It is also of central prognostic value for family members potentially carrying the same mutation. Correlating the different APOA1 mutations with clinical features may, in addition, also help to improve the early detection of mutation carriers.
In conclusion, the three novel mutations described here extend our knowledge about the underlying mutations and the affected organs in patients with hereditary AApoAI amyloidosis. Thirteen of the sixteen mutations described in the APOA1 gene are located in one of the two hot spot regions spanning residues 50 to 93 and 170 to 178.
Footnotes
Supported by grants of the European Union (EU FP6 EURAMY).
M.E. and S.S. contributed equally to the manuscript.
References
- 1.Westermark P. Aspects on human amyloid forms and their fibril polypeptides. FEBS J. 2005;272:5942–5949. doi: 10.1111/j.1742-4658.2005.05024.x. [DOI] [PubMed] [Google Scholar]
- 2.Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36:411–423. doi: 10.1002/mus.20821. [DOI] [PubMed] [Google Scholar]
- 3.Mucchiano GI, Jonasson L, Häggqvist B, Einarsson E, Westermark P. Apolipoprotein A-I-derived amyloid in atherosclerosis. Its association with plasma levels of apolipoprotein A-I and cholesterol. Am J Clin Pathol. 2001;115:298–303. doi: 10.1309/PJE6-X9E5-LX6K-NELY. [DOI] [PubMed] [Google Scholar]
- 4.Obici L, Franceschini G, Calabresi L, Giorgetti S, Stoppini M, Merlini G, Bellotti V. Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid. 2006;13:191–205. doi: 10.1080/13506120600960288. [DOI] [PubMed] [Google Scholar]
- 5.Sorci-Thomas MG, Thomas MJ. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121–128. doi: 10.1016/s1050-1738(01)00163-3. [DOI] [PubMed] [Google Scholar]
- 6.Karathanasis SK, Zannis VI, Breslow JL. Isolation and characterization of the human apolipoprotein A-I gene. Proc Natl Acad Sci USA. 1983;80:6147–6151. doi: 10.1073/pnas.80.20.6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Allen MW, Frohlich JA, Davis JR. Inherited predisposition to generalized amyloidosis. Clinical and pathological study of a family with neuropathy, nephropathy, and peptic ulcer. Neurology. 1969;19:10–25. doi: 10.1212/wnl.19.1.10. [DOI] [PubMed] [Google Scholar]
- 8.Nichols WC, Gregg RE, Brewer HB, Jr, Benson MD. A mutation in apolipoprotein A-I in the Iowa type of familial amyloidotic polyneuropathy. Genomics. 1990;8:318–323. doi: 10.1016/0888-7543(90)90288-6. [DOI] [PubMed] [Google Scholar]
- 9.Booth DR, Tan SY, Booth SE, Hsuan JJ, Totty NF, Nguyen O, Hutton T, Vigushin DM, Tennent GA, Hutchinson WL. A new apolipoprotein Al variant, Trp50Arg, causes hereditary amyloidosis. QJM. 1995;88:695–702. [PubMed] [Google Scholar]
- 10.Soutar AK, Hawkins PN, Vigushin DM, Tennent GA, Booth SE, Hutton T, Nguyen O, Totty NF, Feest TG, Hsuan JJ. Apolipoprotein AI mutation Arg-60 causes autosomal dominant amyloidosis. Proc Natl Acad Sci USA. 1992;89:7389–7393. doi: 10.1073/pnas.89.16.7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy CL, Wang S, Weaver K, Gertz MA, Weiss DT, Solomon A. Renal apolipoprotein A-I amyloidosis associated with a novel mutant Leu64Pro. Am J Kidney Dis. 2004;44:1103–1109. doi: 10.1053/j.ajkd.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 12.Booth DR, Tan SY, Booth SE, Tennent GA, Hutchinson WL, Hsuan JJ, Totty NF, Truong O, Soutar AK, Hawkins PN, Bruguera M, Caballeria J, Sole M, Campistol JM, Pepys MB. Hereditary hepatic and systemic amyloidosis caused by a new deletion/insertion mutation in the apolipoprotein AI gene. J Clin Invest. 1996;97:2714–2721. doi: 10.1172/JCI118725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Persey MR, Booth DR, Booth SE, van Zyl-Smit R, Adams BK, Fattaar AB, Tennent GA, Hawkins PN, Pepys MB. Hereditary nephropathic systemic amyloidosis caused by a novel variant apolipoprotein A-I. Kidney Int. 1998;53:276–281. doi: 10.1046/j.1523-1755.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- 14.Coriu D, Dispenzieri A, Stevens FJ, Murphy CL, Wang S, Weiss DT, Solomon A. Hepatic amyloidosis resulting from deposition of the apolipoprotein A-I variant Leu75Pro. Amyloid. 2003;10:215–223. doi: 10.3109/13506120309041738. [DOI] [PubMed] [Google Scholar]
- 15.Obici L, Palladini G, Giorgetti S, Bellotti V, Gregorini G, Arbustini E, Verga L, Marciano S, Donadei S, Perfetti V, Calabresi L, Bergonzi C, Scolari F, Merlini G. Liver biopsy discloses a new apolipoprotein A-I hereditary amyloidosis in several unrelated Italian families. Gastroenterology. 2004;126:1416–1422. doi: 10.1053/j.gastro.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Hamidi AL, Liepnieks JJ, Hamidi AK, Uemichi T, Moulin G, Desjoyaux E, Loire R, Delpech M, Grateau G, Benson MD. Hereditary amyloid cardiomyopathy caused by a variant apolipoprotein A1. Am J Pathol. 1999;154:221–227. doi: 10.1016/S0002-9440(10)65268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amarzguioui M, Mucchiano G, Häggqvist B, Westermark P, Kavlie A, Sletten K, Prydz H. Extensive intimal apolipoprotein A1-derived amyloid deposits in a patient with an apolipoprotein A1 mutation. Biochem Biophys Res Commun. 1998;242:534–539. doi: 10.1006/bbrc.1997.8005. [DOI] [PubMed] [Google Scholar]
- 18.Hamidi AK, Liepnieks JJ, Nakamura M, Parker F, Benson MD. A novel apolipoprotein A-1 variant. Arg173Pro, associated with cardiac and cutaneous amyloidosis. Biochem Biophys Res Commun. 1999;257:584–588. doi: 10.1006/bbrc.1999.0518. [DOI] [PubMed] [Google Scholar]
- 19.Obici L, Bellotti V, Mangione P, Stoppini M, Arbustini E, Verga L, Zorzoli I, Anesi E, Zanotti G, Campana C, Vigano M, Merlini G. The new apolipoprotein A-I variant Leu(174) → Ser causes hereditary cardiac amyloidosis, and the amyloid fibrils are constituted by the 93-residue N-terminal polypeptide. Am J Pathol. 1999;155:695–702. doi: 10.1016/S0002-9440(10)65167-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, Pepys MB, Hawkins PN. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786–1791. doi: 10.1056/NEJMoa013354. [DOI] [PubMed] [Google Scholar]
- 21.de Sousa MM, Vital C, Ostler D, Fernandes R, Pouget-Abadie J, Carles D, Saraiva MJ. Apolipoprotein AI and transthyretin as components of amyloid fibrils in a kindred with apoAI Leu178His amyloidosis. Am J Pathol. 2000;156:1911–1917. doi: 10.1016/S0002-9440(10)65064-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregorini G, Izzi C, Obici L, Tardanico R, Röcken C, Viola BF, Capistrano M, Donadei S, Biasi L, Scalvini T, Merlini G, Scolari F. Renal apolipoprotein A-I amyloidosis: a rare and usually ignored cause of hereditary tubulointerstitial nephritis. J Am Soc Nephrol. 2005;16:3680–3686. doi: 10.1681/ASN.2005040382. [DOI] [PubMed] [Google Scholar]
- 23.Bohne S, Sletten K, Menard R, Bühling F, Vöckler S, Wrenger E, Roessner A, Röcken C. Cleavage of AL amyloid proteins and AL amyloid deposits by cathepsins B, K, and L. J Pathol. 2004;203:528–537. doi: 10.1002/path.1553. [DOI] [PubMed] [Google Scholar]
- 24.Kuci H, Ebert MP, Röcken C. Anti-lambda-light chain-peptide antibodies are suitable for the immunohistochemical classification of AL amyloid. Histol Histopathol. 2007;22:379–387. doi: 10.14670/HH-22.379. [DOI] [PubMed] [Google Scholar]
- 25.Hoshii Y, Setoguchi M, Iwata T, Ueda J, Cui D, Kawano H, Gondo T, Takahashi M, Ishihara T. Useful polyclonal antibodies against synthetic peptides corresponding to immunoglobulin light chain constant region for immunohistochemical detection of immunoglobulin light chain amyloidosis. Pathol Int. 2001;51:264–270. doi: 10.1046/j.1440-1827.2001.01198.x. [DOI] [PubMed] [Google Scholar]
- 26.Ostertag Familiäre Amyloid-Erkrankung. Z Mensch Vererb Konstitutionsl. 1950;30:105–115. [Google Scholar]
- 27.Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75:408–427. doi: 10.1093/brain/75.3.408. [DOI] [PubMed] [Google Scholar]
- 28.Caballeria J, Bruguera M, Sole M, Campistol JM, Rodes J. Hepatic familial amyloidosis caused by a new mutation in the apolipoprotein AI gene: clinical and pathological features. Am J Gastroenterol. 2001;96:1872–1876. doi: 10.1111/j.1572-0241.2001.03887.x. [DOI] [PubMed] [Google Scholar]
- 29.Eriksson M, Schönland S, Bergner R, Hegenbart U, Lohse P, Schmidt H, Röcken C. Three German fibrinogen Aalpha-chain amyloidosis patients with the p.Glu526Val mutation. Virchows Arch. 2008;453:25–31. doi: 10.1007/s00428-008-0619-4. [DOI] [PubMed] [Google Scholar]
- 30.Eriksson M, Büttner J, Todorov T, Yumlu S, Schönland S, Hegenbart U, Kristen AV, Dengler T, Lohse P, Helmke B, Schmidt H, Röcken C. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with ATTR amyloid. Am J Surg Pathol. 2009;33:58–65. doi: 10.1097/PAS.0b013e3181788566. [DOI] [PubMed] [Google Scholar]
- 31.Solomon A, Murphy CL, Kestler D, Coriu D, Weiss DT, Makovitzky J, Westermark P. Amyloid contained in the knee joint meniscus is formed from apolipoprotein A-I. Arthritis Rheum. 2006;54:3545–3550. doi: 10.1002/art.22201. [DOI] [PubMed] [Google Scholar]
- 32.Nichols WC, Dwulet FE, Liepnieks J, Benson MD. Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem Biophys Res Commun. 1988;156:762–768. doi: 10.1016/s0006-291x(88)80909-4. [DOI] [PubMed] [Google Scholar]
- 33.Röcken C, Menard R, Bühling F, Vöckler S, Raynes J, Stix B, Krüger S, Roessner A, Kähne T. Proteolysis of serum amyloid A and AA amyloid proteins by cysteine proteases: cathepsin B generates AA amyloid proteins and cathepsin L may prevent their formation. Ann Rheum Dis. 2005;64:808–815. doi: 10.1136/ard.2004.030429. [DOI] [PMC free article] [PubMed] [Google Scholar]