

Abstract
A CTG repeat amplification is responsible for the dominantly inherited neuromuscular disorder, myotonic dystrophy type 1 (DM1), which is characterized by progressive muscle wasting and weakness. The expanded (CTG)n tract not only alters the myogenic differentiation of the DM1 muscle precursor cells but also reduces their proliferative capacity. In this report, we show that these muscle precursor cells containing large CTG expansion sequences have not exhausted their proliferative capacity, but have entered into premature senescence. We demonstrate that an abnormal accumulation of p16 is responsible for this defect because the abolition of p16 activity overcomes early growth arrest and restores an extended proliferative capacity. Our results suggest that the accelerated telomere shortening measured in DM1 cells does not contribute to the aberrant induction of p16. We propose that a cellular stress related to the amplified CTG repeat promotes premature senescence mediated by a p16-dependent pathway in DM1 muscle precursor cells. This mechanism is responsible for the reduced proliferative capacity of the DM1 muscle precursor cells and could participate in both the impaired regeneration and atrophy observed in the DM1 muscles containing large CTG expansions.
Myotonic dystrophy type 1 (DM1) is the most prevalent form of adult neuromuscular disorder, characterized by myotonia, muscle wasting, and weakness as well as other multisystemic defects.1 DM1 is an autosomal dominant disease caused by the expansion of a CTG repeat in the 3′ untranslated region (3′UTR) of the DMPK gene.2,3,4,5 Unaffected individuals have fewer than 38 CTG repeats whereas DM1 patients have from 100 to several thousand repeats in the most severe cases. In general, the size of the expansion correlates with the age of onset and the severity of the disease.6,7 An increasing amount of evidence supports a RNA gain-of-function mechanism in DM1 disease. The mutant transcripts containing the CUG expansion fold into RNA hairpins that are not exported to the cytoplasm but accumulate in the nuclei as discrete foci. These mutant RNAs interfere with the activities of proteins involved in RNA processing such as MBNL, CUG-BP1, hnRNP H, leading to specific misregulated splicing events.8 As an example, the misregulation of alternative splicing of the chloride channel ClC-1 mRNAs found in DM1 patients has been directly linked to myotonia, a characteristic feature of the disease.9
The CTG mutation is very unstable and its amplification occurs both over generations and in somatic tissues. Intergenerational instability of the expanded microsatellite provides the molecular basis for the anticipation phenomenon described in DM1 disease: the size of the CTG expansion progressively increases in successive generations of DM1 families, and correlates with the severity of the disease.1 After several generations, the patients often develop the severe DM1 congenital form associated with a large CTG expansion (>1000) and characterized by delayed muscle maturation and atrophy. Somatic instability is also measured throughout the life of a DM1 patient with a gradual increase in the average repeat size. Moreover, variable repeat sizes are detected in different tissues of the same patient with the largest size being found in the skeletal muscle.10,11 In a previous study, we have shown that the size of the CTG expansion increases progressively at each cellular division during the proliferative lifespan of DM1 muscle precursor cells, also called satellite cells in vivo or myoblasts in culture, indicating a replication-dependent somatic instability of the expanded microsatellite.12 In addition to myogenic differentiation defects, we showed that the proliferative capacity of the satellite cells isolated from congenital DM1 patients with large CTG repeats was significantly reduced as compared with age-matched controls.
The proliferative capacity of human satellite cells, like the majority of human diploid somatic cells, is limited by cellular senescence. One major mechanism implicated in the replicative senescence of human cells is the progressive erosion of their telomeres at each cell division. Once a critically short telomere length is reached, replicative senescence is triggered through a p53-dependent pathway.13 The introduction of the catalytic subunit of the telomerase (hTERT) gene into human fibroblasts is sufficient to block telomere shortening and prevent replicative senescence in these cells, and lead to their immortalization.14 However, expression of hTERT is not sufficient to confer immortality to several types of human cells including satellite cells, indicating the existence of cell-type-specific differences in the regulation of the proliferative capacity.15 The p16-dependent pathway has been shown to provoke proliferative arrest before telomeres reach their critical value, and inhibition of p16 in addition to hTERT activity results in the immortalization of keratinocytes, epithelial cells, and satellite cells.16,17 p16 is up-regulated in response to several telomere-independent stress mechanisms including DNA damage, oncogenic signals, and oxidative stress,18 but has also been proposed to be involved in response to telomere damage.19 Recently several reports have indicated that an increase in p16 expression in the brain, bone marrow, and pancreas progenitor cells during aging contributes to stem cell decline, senescence, and aging.20
In this report, we have analyzed the premature growth arrest of DM1 satellite cells carrying large CTG expansions. Our results indicate that a mechanism of premature senescence limits the proliferative capacity of the DM1 satellite cells. We demonstrate that the p16 pathway is responsible for this premature growth arrest in response to a CTG-related stress. In addition our results indicate that the accelerated telomere shortening measured in DM1 cells is not involved in the early accumulation of p16 but is rather a consequence of the cellular stress induced by the CTG mutation.
Materials and Methods
Materials and Cell Culture
The human satellite cells were isolated from muscle biopsies obtained from the Bank of Tissues for Research (a partner in the European Union network EuroBioBank) in accordance with European recommendations and French legislation. Three DM1 fetuses (28-, 34-, and 37-weeks old) showing clinical symptoms of the congenital form with varus feet, arthrogryposis, muscular hypotrophy, and carrying more than 2000 CTG were included in this study. Three individuals (2 fetuses of 29 and 37 weeks of age, and one 5-day-old infant) showing no sign of neuromuscular disease were used as controls. The muscle biopsies were finely minced and explants were plated onto noncoated Petri dishes in drops of fetal calf serum (Invitrogen, Carlsbad, CA). The cells were cultivated at 37°C in a humid atmosphere containing 5% CO2. Once mononucleated cells had migrated out from the explants, growth medium consisting of Ham’s F10 medium (Invitrogen) supplemented with 20% fetal calf serum and 5 μg/ml gentamicin was added. At the time of cell isolation, all cell populations were considered to be at 1 mean population doubling. Cell populations were trypsinized when they reached 80% of confluence. At each passage, the number of divisions was calculated according to the formula: log (N/n)/log 2 where N is the number of cells counted and n is the number of cells initially plated. The cultures were considered to be in an irreversible growth arrest when they failed to divide during 3 weeks in proliferative conditions. Viability was controlled by trypan blue (Sigma, St. Louis, MO) exclusion. The myogenic purity of the populations was monitored by immunocytochemistry using desmin as a marker because it is only expressed in myogenic cells. All cell populations used in this study had a myogenic purity greater than 75%. For 5-aza-2′deoxycytidine experiment, cells were treated with 3 μmol/L 5-aza-2′deoxycytidine (Sigma) for 7 days as described by Darbro and colleagues.21
Cell Transduction
hTERT and Cdk4 cDNA were cloned into distinct pBABE retroviral vectors containing, respectively, the puromycin selection marker and a neomycin resistance gene. Transduced cell cultures were submitted to selection in the presence of puromycin (0.5 μg/ml) and/or neomycin (0.3 mg/ml) for 10 days.
Immunocytochemistry
For bromodeoxyuridine (BrdU) labeling, cell cultures were grown for 72 hours in the presence of 10 μg/ml BrdU and then fixed for 10 minutes with ethanol. Cells that had incorporated BrdU were detected using the monoclonal antibody Bu20a (DAKO, Glostrup, Denmark), revealed by a specific secondary antibody directly coupled to Alexa Fluor 488 (Molecular Probes, Eugene, OR). DAPI (Calbiochem, San Diego, CA) was used to visualize the nuclei. All images were digitalized using the MetaView image analysis system (Universal Imaging Corp., Downington, PA). To determine the percentage of positive cells, at least 500 cells were counted. The expression of desmin was revealed using the antibody D33 (1/50, DAKO) and specific antibody binding was revealed with peroxidase (Vectastain; Vector Laboratories, Burlingame, CA). SA-ß-galactosidase activity was revealed as described by Dimri and colleagues.22
Telomere Length Analysis
Two μg of genomic DNA were digested with Hinf1 (Biolabs, Santa Ana, CA) to generate telomere restriction fragments. The samples were resolved by electrophoresis in a 0.7% agarose gel that was hybridized to a 32P-(TTAGGG)4 probe. The signal responses were revealed using the Personal-Molecular-Imager (Bio-Rad, Hercules, CA) and analyzed by a computer-assisted system using National Institutes of Health (Bethesda, MD) Image 1.62 and ProFit software. The mean of telomere lengths was determined from three independent gels.
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Two μg of total RNA isolated using Trizol (Invitrogen) were reverse-transcribed into cDNA according to the manufacturer’s instructions. Equal amounts of the reverse transcription products were subjected to PCR amplification (ABGene, Rochester, NY). Amplification was initiated by 4 minutes of denaturation at 94°C, followed by 20 (GAPDH) or 35 cycles (p16) of amplification. Each cycle consists of 60 seconds at 94°C, 60 seconds at 55°C (GAPDH), or 62°C (p16) and 60 seconds at 72°C. A final step of extension was performed for 10 minutes at 72°C. After amplification, 15 μl of each PCR reaction product were separated on a 1% agarose gel containing ethidium bromide. The following primers, synthesized by Sigma-Proligo were used: GAPDH, forward, 5′-GATGACAAGCTTCCCGTTCTCAGCC-3′; GAPDH, reverse, 5′TGAAGGTCGGAGTCAACGGATTTGGT-3′; p16, forward, 5′-TGGAGCCTTCGGCTGACTGGCTGGC-3′; and p16, reverse, 5′-CTACGAAAGCGGGGTGGGTTGT-3′.
Western Blotting
Thirty μg of total protein extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose membranes and incubated with different antibodies directed against Rb (Pharmingen, San Diego, CA), p16 (Santa Cruz Biotechnology, Santa Cruz, CA), p53 (Santa Cruz Biotechnology), cyclin D (Cell Signaling, Beverly, MA) and Emerin (Novocastra, Newcastle, UK). Secondary antibodies coupled to horseradish peroxidase were revealed using the ECL kit (Pierce, Rockford, IL). The signal was detected on film (Fuji Film, Fuji, Tokyo, Japan) and quantified by densitometry.
Statistical Analysis
Prism software was used to calculate the statistical significance and the SEM. Significance was tested by Student’s unpaired t-test or Mann-Whitney test. For all tests, the groups were considered statistically different for P value <0.05 (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Premature Senescence of DM1 Satellite Cells
The proliferative capacity of satellite cells isolated from muscles of three congenital DM1 patients with large CTG repeats (>2000) was compared in vitro to that of satellite cells isolated from three age-matched and nonaffected individuals. The cells were grown under identical culture conditions until they ceased to respond to mitogenic stimuli for 3 weeks and entered into an irreversible cell-cycle arrest. As shown in Figure 1A, the average proliferative lifespan of the DM1 satellite cells was significantly reduced by 40% as compared with that of control cells. When they stopped dividing both DM1 and control satellite cells displayed a flattened and enlarged morphology characteristic of senescent cells (Figure 1B). Senescent-associated β-galactosidase activity was detected in growth-arrested DM1 cells, as well as high levels of cyclin D1, which is a reliable marker of replicative senescence and was detected in both DM1 and control cells at the end of their lifespan (Figure 1C). Analysis of the phosphorylation status of Rb showed that arrested DM1 and control satellite cells contained only the hypophosphorylated form, in contrast to the proliferating cells at earlier passages, which expressed in addition the hyperphosphorylated form essential to overcome the G1 checkpoint (Figure 1D). Cell-cycle fluorescence-activated cell sorting analysis confirmed that the majority of DM1 and control cells were arrested in G1 (data not shown). These results indicate that a mechanism similar to senescence is responsible for the early proliferative growth arrest of DM1 satellite cells because these cells expressed biomarkers usually observed in senescent cells, although much earlier in their replicative lifespan as compared with control cultures.
Figure 1.
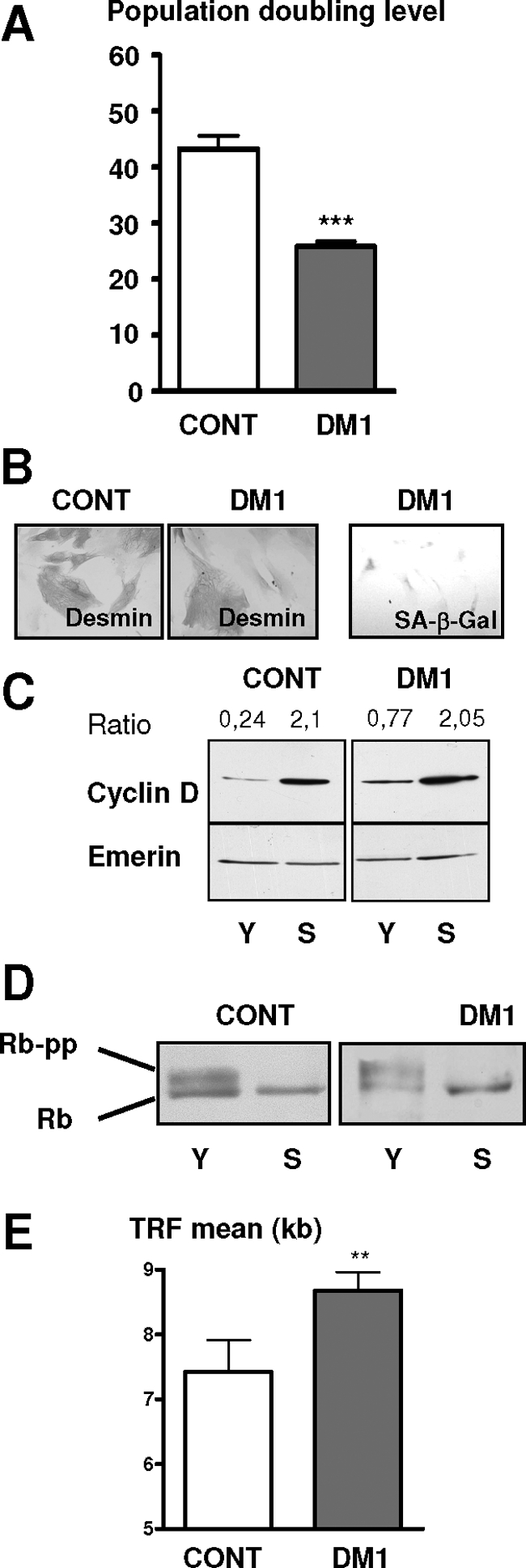
DM1 satellite cells stop dividing by a premature senescence pathway. A: Mean of the population doubling level of DM1 and control satellite cells isolated from muscles of three DM1 patients with large CTG repeats (>2000) and three age-matched and nonaffected individuals, respectively. B: Immunocytochemistry using anti-desmin antibody and revealed by peroxidase was performed on senescent control and premature growth-arrested DM1 cells. Flattened and enlarged morphologies are present in both arrested DM1 cells and control senescent cells. Senescent-associated ß-galactosidase activity (SA-ß-Gal) was observed on arrested DM1 cells. C: Western blot analyses of cyclin D in the early (Y) and late (S) stages of their proliferative lifespan. The results were normalized to the expression of the nuclear protein Emerin. Levels of cyclin D were increased in both DM1- and control-arrested cells. D: Western blot analyses of Rb in the early (Y) and late (S) stages of their proliferative lifespan. The hyperphosphorylated form of Rb is absent in both controls and DM1-arrested cells. E: Mean length of telomeric restriction fragments (TRFs) measured on DM1 and control satellite cells at proliferative arrest. DM1 satellite cells stop dividing with longer telomeres than controls. (**P < 0.01, ***P < 0.001).
To evaluate if the premature growth arrest observed in satellite cells isolated from DM1 patients was attributable to an excessive proliferation in vivo before their isolation, we measured the size of the telomeres in these cells. Telomere length represents a predictive marker of the regenerative capacity of the human satellite cells as shown in Duchenne muscular dystrophy in which the extensive proliferation of the satellite cells after continuous cycles of degeneration and regeneration leads to reduced telomere length.23 As shown in Figure 1E, DM1 satellite cells stop dividing with telomeres significantly longer than those of control cells (respectively, 8.7 kb versus 7.4 kb), indicating that telomeres of DM1 cells had not reached their critical size and consequently that these cells had not exhausted their proliferative capacity. This confirms that a mechanism of premature senescence alters and limits in vitro the replicative lifespan of DM1 satellite cells carrying large CTG expansions.
p16 Triggers Premature Senescence in DM1 Cells
As demonstrated recently, the p16 cyclin-dependent kinase inhibitor is a key regulator of the replicative senescence of human satellite cells.17 To determine whether the p16/Rb pathway is involved in the premature senescence of DM1 cells in vitro, we have measured the expression of p16 protein at the end of their lifespan. In these experiments we found that a similar level of p16 was detected in both DM1 and control cultures once they had entered proliferative arrest (Figure 2A). Moreover, it should be noted that DM1 cells made significantly fewer divisions than control cells (Figure 2B). The accumulated p16 will bind Cdk4 thus inhibiting its activity and block cell-cycle progression. To assess the role of p16 in DM1 premature senescence, we stably overexpressed Cdk4 to neutralize p16 activity. As seen in Figure 2B, DM1-Cdk4 cells were able to bypass the premature growth arrest observed in DM1 cells. Their lifespan was extended by 59% to 51 divisions, which is very close to the 52 divisions observed in control cells. In addition, overexpression of p16 in young control cells resulted in a premature proliferative arrest, as evidenced by absence of BrdU incorporation after incubation for 72 hours (data not shown). These results demonstrate that p16 is responsible for triggering in vitro the premature senescence of DM1 satellite cells.
Figure 2.

Premature growth arrest of DM1 satellite cells is dependent on the p16 pathway. A: p16 levels in control and DM1 satellite cells at early (Y) and late (S) stages in their proliferative lifespan were determined by Western blot. Means of the p16/Emerin ratio in control and DM1 senescent cells were presented on the histogram. Control- and DM1-arrested cells present the same level of expression of p16, ns: not significant. B: Lifespan plots of control and DM1 populations transduced with or without Cdk4. Transduction of Cdk4 gene allows to bypass the arrest observed in nontransduced cells.
However, we also abolished p16 activity by overexpression of Cdk4 in control cells and observed that the DM1-Cdk4 cells still make fewer divisions than the control-Cdk4 age-matched cells (51 versus 70 divisions, respectively) before entering into replicative senescence (Figure 2B). To assess if a residual activity of p16 could be responsible for the proliferative arrest of the Cdk4-overexpressing cells, RNA interference directed to p16 was induced in these cells, but no further expansion of their lifespan was observed (data not shown). Measurements of telomere lengths showed that DM1-Cdk4 and control-Cdk4 satellite cells both stop growing with telomere lengths of 6.2 kb and 6.1 kb, respectively. These values are almost identical to the 6 kb measured in senescent human fibroblasts in which a telomere-dependent senescence is induced by short telomeres. Both the DM1- and control Cdk4-transduced cells stopped growing with short telomeres but the DM1-Cdk4 cells reached this minimum value after fewer divisions than the control-Cdk4 cells, suggesting that large CTG expansions may interfere with the telomere homeostasis machinery in DM1 cells.
CTG Repeat Expansion Alters Telomere Homeostasis in DM1 Cells Independently from p16 Induction
Telomere loss per division was calculated from measurements made at several time points during the lifespan of DM1 and control cells, and a significant 59% increase in the amount of telomeric DNA lost per division was measured in DM1 satellite cell cultures (171.9 ± 17 bp) as compared with control cultures (108.1 ± 10 bp) (Figure 3A). To determine whether or not the increase in the amount of telomeric DNA lost per division in DM1 satellite cells was a consequence of an increased cell death in DM1 cultures, which would result in an increase in the number of divisions made by the surviving cells and thus in an increase in mean telomere shortening, we assessed both cell death and proliferation in control and DM1 populations. No significant difference was observed in the number of proliferating cells, as demonstrated by BrdU incorporation in the middle of their lifespan: 77 ± 3% cells in control populations versus 75 ± 4% cells in DM1 populations were BrdU-positive, indicating that both populations have similar growth rates. Viability of the cells was assessed by trypan blue exclusion, and only very low levels of cell death were detected in both cultures (3.7 ± 0.8% in control and 4.6 ± 1% in DM1 cultures), with no significant difference being observed between them. These results confirm that the higher rate of telomere loss in DM1 cells is not a consequence of an increased cell death, which would result in an increase number of divisions made by the surviving cells. To determine whether a higher rate of telomere shortening may induce elevated levels of p53 and thus participate to the premature proliferative arrest, p53 protein levels were measured by Western blot in DM1 cells presenting a higher rate of telomere loss and control cells, both at the end of their lifespan. A similar level of p53 was detected in both cultures (Figure 3B), indicating that an elevation in the level of expression of p53 is not triggered by increased telomere loss.
Figure 3.
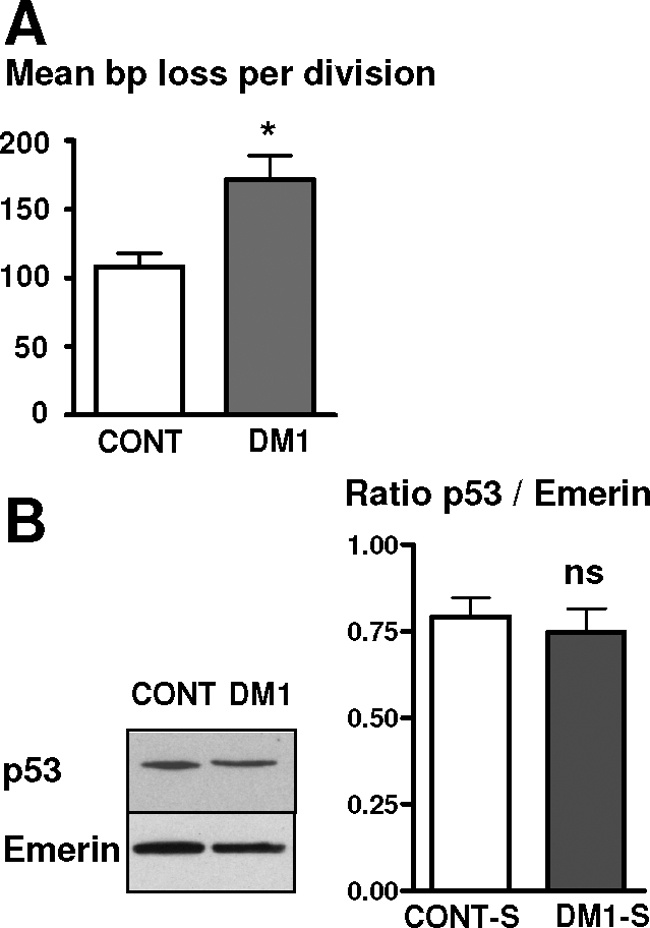
Deregulation of the telomere homeostasis in DM1 satellite cells. A: Mean length of telomeric DNA in bp lost per division in cultures of control and DM1 satellite cells. DM1 cells lost more bp per division than control cells. B: Western blot analyses of the level of p53 in control and DM1 satellite cells at the end of their lifespan. Means of the p53/Emerin ratio in control- and DM1-arrested cells are presented on the histogram. *P < 0.05, ns: not significant.
The increased loss of telomeric DNA per division will reduce the number of divisions that DM1 cells can make before reaching telomere-driven senescence and explains why the critical telomere size of 6 kb is reached by the DM1-Cdk4 cells after having made fewer divisions than control-Cdk4 cells. To determine whether such an accelerated telomere loss could act as a signal or could be responsible for the premature senescence of the DM1 cells, we expressed the catalytic subunit of telomerase hTERT because the activity of this enzyme should compensate for the increased telomere erosion. As seen in Figure 4A, DM1 cells expressing hTERT were not able to make more divisions than DM1 cells before entering into premature growth arrest suggesting that the higher rate of telomere erosion may not contribute to the process of premature senescence observed in vitro in DM1 satellite cells. However, among these arrested cells, a clone of proliferating cells emerged after 2 months of culture (Figure 4B), which overcame the arrested population (Figure 4B, lane S) and presented an abolition of p16 expression, as demonstrated by the decline in the level of p16 detected at two successive time points (Figure 4, B and C, lanes 1 and 2) in culture. The promoter region of the p16 gene contains a 5′CpG island that has been found to exhibit increased levels of methylation in various tumors as well as in telomerase immortalized human keratinocytes.21 The abolition of p16 expression in this clone was confirmed at the RNA level and a re-expression of p16 was measured when these cells were treated with 5-aza-2′ deoxycytidine (Figure 4D, lane 2*). This result indicates that methylation of the p16 promoter had occurred in this DM1-hTERT clone, leading to its immortalization because these cells have now made more than 200 divisions. Immortalization of the DM1 satellite cells was also obtained by the dual overexpression of hTERT and Cdk4, and these cells have been maintained for more than 150 divisions (Figure 4E). Measurement of the telomere lengths showed that the expression of hTERT in DM1-Cdk4 satellite cells stabilized the telomere length to ∼10 kb, a value similar to that observed in control hTERT/Cdk4-cells (Figure 4E, inset). We conclude that blocking both the p16 and the telomere pathways leads to the immortalization of DM1 satellite cells.
Figure 4.

Immortalization of DM1 satellite cells. A: Lifespan plots of DM1 satellite cells transduced with or without hTERT. hTERT-transduced cells were not able to bypass the premature arrest observed in DM1 satellite cells. B: Lifespan plot of a clone of cells (DM1Δ) that emerged from a DM1 satellite cell population transduced with hTERT. DM1Δ cells were immortalized. C: Western blot analyses of p16 in senescent DM1 cells (lane S), in the senescent population with emerging DM1Δ cells (lane 1), and in the DM1Δ after several divisions (lane 2). The two points (1 and 2) are also indicated on the DM1Δ lifespan curve shown above. D: Expression of p16 and GAPDH mRNA analyzed by RT-PCR: negative control (lane −), senescent DM1 satellite cells (lane S), DM1Δ (lane 2), DM1Δ treated with 5-aza-2′deoxycytidine (lane 2*). E: Lifespan plots of DM1 satellite cells transduced with or without Cdk4 and hTERT. Cdk4- and hTERT-transduced DM1 cells were immortalized. Effect of hTERT on the telomere length is presented in the inset. hTERT stabilized the telomere length of the cells ∼10 kb.
Discussion
In this report, we provide new evidence that large CTG expansions trigger in vitro a mechanism of premature senescence through a p16-dependent pathway, which reduces significantly the proliferative capacity of DM1 muscle precursor cells. Measurements of telomere lengths at the end of their lifespan indicate that DM1 cells had not exhausted their proliferative capacity confirming that a premature growth arrest, independent from an excessive in vivo turnover, occurred in satellite cells isolated from DM1 biopsies. Analysis of several biomarkers suggests that a mechanism similar to cellular senescence is responsible for the premature and irreversible cell-cycle arrest of DM1 muscle precursor cells containing large CTG expansions. Our data demonstrated that the Rb regulator p16 triggers in vitro this premature senescence because abolition of p16 activity in DM1 satellite cells reverses their precocious growth arrest and restores their proliferative capacity.
The Rb regulator p16, appears to be an important cellular sensor of stasis in response to various stresses such as DNA damage, oxidative stress, and telomere dysfunction.18,24 Even if an alteration of the telomere homeostasis, reflected by the accelerated telomere shortening, is measured in DM1 cells, the re-introduction of telomerase activity in these cells fails to extend their proliferative lifespan. Although the higher rate of telomere loss in DM1 cells may not participate to the triggering of the signal for p16 activation, as suggested by the absence of effect of the transduction by telomerase, we cannot exclude that other elements involved in telomere dysfunction could interfere with the p16 pathway. The signaling involved in the premature senescence of DM1 cells appears to be independent from the telomere-driven senescence pathway because i) the DM1 satellite cells stop dividing with telomeres significantly longer than those of control cells; ii) abolition of the dominant and irreversible p16 barrier in DM1 satellite cells prevents their premature senescence and extends their lifespan; and iii) the DM1 cells with inactive p16 stop dividing and enter into senescence with critically short telomeres of 6 kb through a telomere-dependent mechanism, like the human fibroblasts.
However, the question as to how the CTG mutation interferes with telomere homeostasis remains to be solved. The expanded CTG repeat is unstable and we have shown, in a previous study, that the size of the CTG repeat increases during the lifespan of the DM1 satellite cells.12 The amplified CTG expansion adopts several unusual structures that disturb DNA metabolism and require the involvement of the DNA repair machinery.25 Because proteins involved in the regulation of telomere homeostasis could also participate to the DNA damage response,26 the possibility that the large CTG expanded repeats may participate to accelerated telomere shortening in DM1 cells, eg, by modifying activities or by titrating such dual proteins, is still open. It should also be noted that accelerated telomere shortening has been associated with higher oxidative stress.27,28 Interestingly, increased levels of free radicals and lipid peroxidation have also been measured in DM1 patients suggesting that oxidative stress could be involved in the disease progression of DM1.29 In addition, the cellular susceptibility to oxidative stress was positively correlated with the number of CTG repeats.30 In the present study, a 59% increase in the amount of telomeric DNA lost per division was measured in vitro in DM1 cells when compared with control cells, although both control and DM1 cells were grown under identical culture conditions. Although results observed in vitro cannot be directly applied to the in vivo situation in adult DM1 patients, an increased susceptibility to reactive oxygen species of DM1 cells carrying large CTG as well as the potential implication of oxidative stress in the increased telomere loss measured in DM1 cells deserve further investigation.
Our study has revealed a new and novel feature of the DM1 mutation: the large CTG and/or CUG expansions trigger in vitro stress-signaling events, which are responsible for the early activation of p16 and subsequently, the premature senescence of DM1 satellite cells. Numerous noxious stresses including reactive oxygen species and DNA-damaging stimuli have been reported to induce p16 both in vitro and in vivo, and the induction of p16 in response to various stresses is a slow process that takes several weeks.18 Interestingly, an inappropriate activation of PKCs by a large CUG repeat RNA that causes hyperphosphorylation and stabilization of CUGBP1 was demonstrated in DM1.31 PKCs are stress-sensing kinases that can also be activated by several types of physiological and oxidative stresses32,33 suggesting a potential role for the nuclear aggregates of mutant mRNA with CUG expansions as a primary event in this stress-signaling cascade. The pathway involved in the abnormal activation of p16 in DM1 satellite cells has not yet been defined, however the consequences for these muscle precursors cells result in vitro in their premature senescence and replicative arrest. Recently, p16 was shown to contribute to the decline in the number of stem cells during aging and consequently, to the reduced tissue regenerative capacities.34,35,36 We propose that expanded CTG repeats may also interfere and activate the p16 senescence pathway in muscle precursor cells in vivo. Interestingly, a premature senescence has also been observed in satellite cells isolated from severely DM1-affected adults (unpublished data). The physiological role of the myogenic precursor cells is to maintain the muscle mass and repair skeletal muscle. After injury or damage, the quiescent satellite cells located under the basal lamina of muscle fibers are activated, proliferate, differentiate, and fuse to form newly regenerated fibers or be incorporated into growing fibers. A proliferation step is required to amplify the number of myogenic precursor cells that will regenerate the damaged fibers and a decrease in their number attributable to a reduced proliferative capacity could limit both the regenerative capacity and the nuclear turnover of muscle fibers in DM1. If confirmed in vivo, the premature senescence of the DM1 satellite cells would most probably result in the slowing down or alteration of the regenerative process or the maintenance of muscle mass because the differentiation program of senescent myogenic precursor cells is defective as characterized by impaired myogenesis and down-regulation of the myogenic regulatory factors.37 Interestingly, it was suggested that abnormal regeneration could be involved in the myopathic appearance of the muscle fibers in DM1.38 More recently, a report suggests that in DM1 muscle fibers containing sarcoplasmic masses, a characteristic feature of DM1 disease, the myogenic differentiation program is interrupted at a late stage leading to incomplete maturation of these fibers.39 p16 as well as other cell-cycle regulators have been detected in the DM1 fibers containing these sarcoplasmic masses supporting the implication of the p16 pathway in the lack of regeneration observed in DM1. Altogether our results suggest that the large CTG mutation induces in vitro a p16-dependent premature senescence of the DM1 satellite cells resulting in an important reduction in their proliferative capacity when compared with the entire proliferative lifespan of normal satellite cells. The premature senescence of the DM1 myogenic precursor cells caused by the CTG expansion, if confirmed in vivo, could alter the mechanism of muscle mass maintenance and participate to the muscular atrophy that takes place in DM1.
Acknowledgments
We thank Woodring Wright and Geron Corporation for the kind gifts of cdk-4 and hTERT vectors; Rob Hoeben and Françoise Carlotti for p16 vector; the Human Cell Culture Platform of The Myology Institute; and Alban Vignaud, Kamel Mamchaoui, Lidia Dollé, Virginie François, Stéphane Vasseur, and Maud Chapart for technical support.
Footnotes
Address reprint requests to Dr. Denis Furling, UPMC-Paris VI/ Inserm/CNRS, UMRS 974, 105 Bld de l’ Hopital, Paris, 75013, France. E-mail: denis.furling@upmc.fr.
Supported by the Association Française contre les Myopathies (AFM), University Paris 6, Institut National de la santé et de la recherche médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), and MYOAMP (grants EC, FP6, 037479).
References
- Harper PS. London: W.B. Saunders,; Myotonic Dystrophy. (ed 3) 2004:pp. 17–45. [Google Scholar]
- Aslanidis C, Jansen G, Amemiya C, Shutler G, Mahadevan M, Tsilfidis C, Chen C, Alleman J, Wormskamp NG, Vooijs M, Buxton J, Johnson K, Smeets JM, Lennon G, Carrano AV, Korneluk R, Wieringa B, de Jong P. Cloning of the essential myotonic dystrophy region and mapping of the putative defect. Nature. 1992;355:548–551. doi: 10.1038/355548a0. [DOI] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, Sohn R, Zemelman B, Snell RG, Rundle SA, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper P, Shaw DJ, Housman D. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- Fu YH, Pizzuti A, Fenwick RG, Jr, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, Wieringa B, Korneluk R, Perryman MB, Epstein HF, Caskey CT. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O'Hoy K, Leblond S, Earl-Macdonald J, de Jong P, Wieringa B, Korneluk R. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- Hunter A, Tsilfidis C, Mettler G, Jacob P, Mahadevan M, Surh L, Korneluk R. The correlation of age of onset with CTG trinucleotide repeat amplification in myotonic dystrophy. J Med Genet. 1992;29:774–779. doi: 10.1136/jmg.29.11.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsilfidis C, MacKenzie AE, Mettler G, Barcelo J, Korneluk RG. Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat Genet. 1992;1:192–195. doi: 10.1038/ng0692-192. [DOI] [PubMed] [Google Scholar]
- Paul S, Dansithong W, Kim D, Rossi J, Webster NJ, Comai L, Reddy S. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 2006;25:4271–4283. doi: 10.1038/sj.emboj.7601296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- Anvret M, Ahlberg G, Grandell U, Hedberg B, Johnson K, Edstrom L. Larger expansions of the CTG repeat in muscle compared with lymphocytes from patients with myotonic dystrophy. Hum Mol Genet. 1993;2:1397–1400. doi: 10.1093/hmg/2.9.1397. [DOI] [PubMed] [Google Scholar]
- Thornton CA, Johnson K, Moxley RT., III Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann Neurol. 1994;35:104–107. doi: 10.1002/ana.410350116. [DOI] [PubMed] [Google Scholar]
- Furling D, Coiffier L, Mouly V, Barbet JP, St Guily JL, Taneja K, Gourdon G, Junien C, Butler-Browne GS. Defective satellite cells in congenital myotonic dystrophy. Hum Mol Genet. 2001;10:2079–2087. doi: 10.1093/hmg/10.19.2079. [DOI] [PubMed] [Google Scholar]
- Vaziri H, West MD, Allsopp RC, Davison TS, Wu YS, Arrowsmith CH, Poirier GG, Benchimol S. ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J. 1997;16:6018–6033. doi: 10.1093/emboj/16.19.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Di Donna S, Mamchaoui K, Cooper RN, Seigneurin-Venin S, Tremblay J, Butler-Browne GS, Mouly V. Telomerase can extend the proliferative capacity of human myoblasts, but does not lead to their immortalization. Mol Cancer Res. 2003;1:643–653. [PubMed] [Google Scholar]
- Ramirez RD, Herbert BS, Vaughan MB, Zou Y, Gandia K, Morales CP, Wright WE, Shay JW. Bypass of telomere-dependent replicative senescence (M1) upon overexpression of Cdk4 in normal human epithelial cells. Oncogene. 2003;22:433–444. doi: 10.1038/sj.onc.1206046. [DOI] [PubMed] [Google Scholar]
- Zhu CH, Mouly V, Cooper RN, Mamchaoui K, Bigot A, Shay JW, Di Santo JP, Butler-Browne GS, Wright WE. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell. 2007;6:515–523. doi: 10.1111/j.1474-9726.2007.00306.x. [DOI] [PubMed] [Google Scholar]
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, de Lange T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr Biol. 2004;14:2302–2308. doi: 10.1016/j.cub.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Darbro BW, Lee KM, Nguyen NK, Domann FE, Klingelhutz AJ. Methylation of the p16(INK4a) promoter region in telomerase immortalized human keratinocytes co-cultured with feeder cells. Oncogene. 2006;25:7421–7433. doi: 10.1038/sj.onc.1209729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decary S, Hamida CB, Mouly V, Barbet JP, Hentati F, Butler-Browne GS. Shorter telomeres in dystrophic muscle consistent with extensive regeneration in young children. Neuromuscul Disord. 2000;10:113–120. doi: 10.1016/s0960-8966(99)00093-0. [DOI] [PubMed] [Google Scholar]
- Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998;16:1113–1123. doi: 10.1038/sj.onc.1201862. [DOI] [PubMed] [Google Scholar]
- Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- Longhese MP. DNA damage response at functional and dysfunctional telomeres. Genes Dev. 2008;22:125–140. doi: 10.1101/gad.1626908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- Oikawa S, Kawanishi S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999;453:365–368. doi: 10.1016/s0014-5793(99)00748-6. [DOI] [PubMed] [Google Scholar]
- Toscano A, Messina S, Campo GM, Di Leo R, Musumeci O, Rodolico C, Aguennouz M, Annesi G, Messina C, Vita G. Oxidative stress in myotonic dystrophy type 1. Free Radic Res. 2005;39:771–776. doi: 10.1080/10715760500138932. [DOI] [PubMed] [Google Scholar]
- Usuki F, Ishiura S. Expanded CTG repeats in myotonin protein kinase increase susceptibility to oxidative stress. Neuroreport. 1998;9:2291–2296. doi: 10.1097/00001756-199807130-00027. [DOI] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, Nishizuka Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci USA. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigot A, Jacquemin V, Debacq-Chainiaux F, Butler-Browne GS, Toussaint O, Furling D, Mouly V. Replicative aging down-regulates the myogenic regulatory factors in human myoblasts. Biol Cell. 2008;100:189–199. doi: 10.1042/BC20070085. [DOI] [PubMed] [Google Scholar]
- Borg J, Edstrom L, Butler-Browne GS, Thornell LE. Muscle fibre type composition, motoneuron firing properties, axonal conduction velocity and refractory period for foot extensor motor units in dystrophia myotonica. J Neurol Neurosurg Psychiatry. 1987;50:1036–1044. doi: 10.1136/jnnp.50.8.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattemi G, Tomelleri G, Filosto M, Savio C, Rizzuto N, Tonin P. Expression of late myogenic differentiation markers in sarcoplasmic masses of patients with myotonic dystrophy. Neuropathol Appl Neurobiol. 2005;31:45–52. doi: 10.1111/j.1365-2990.2004.00602.x. [DOI] [PubMed] [Google Scholar]