

Abstract
Multiple system atrophy (MSA) is a neurodegenerative disease caused by an accumulation of α-synuclein (α-syn) in oligodendrocytes. Little is known about the cellular mechanisms by which α-syn accumulation causes neuronal degeneration in MSA. Our previous research, however, revealed that in a mouse model of MSA, oligodendrocytic inclusions of α-syn induced neuronal accumulation of α-syn, as well as progressive neuronal degeneration. Here we identify the mechanisms that underlie neuronal accumulation of α-syn in a mouse MSA model. We found that the α-syn protein binds to β-III tubulin in microtubules to form an insoluble complex. The insoluble α-syn complex progressively accumulates in neurons and leads to neuronal dysfunction. Furthermore, we demonstrated that the neuronal accumulation of insoluble α-syn is suppressed by treatment with a microtubule depolymerizing agent. The underlying pathological process appeared to also be inhibited by this treatment, providing promise for future therapeutic approaches.
Important advances in hereditary neurodegenerative disorders have risen from research using molecular biology techniques. For example, identification of the genes responsible for familial Alzheimer’s disease and hereditary polyglutamine diseases is among the most significant achievements in neuroscience.1,2 In contrast, little progress has been made in research on the biology of neurodegeneration in a group of non-hereditary neurodegenerative disorders. Multiple system atrophy (MSA) is a non-hereditary neurodegenerative disease that is clinically characterized by autonomic nervous system failure as a symptom of Shy-Drager syndrome and Parkinsonism as a symptom of striatonigral degeneration.3,4 The cellular mechanisms underlying the neurodegeneration are not understood, and no prospective therapeutic target for MSA has been presented.
Three significant neuropathological features characterize MSA histologically: glial cytoplasmic inclusions (GCIs), neuronal inclusions, and neuropil threads.5 All three are composed of α-synuclein (α-syn). GCIs, the first neuropathological manifestation to be described, are oligodendrocytic inclusions.6,7,8 Previous studies on GCIs reported that filaments isolated from the central nervous system (CNS) of patients with MSA were labeled by α-syn antibodies.9 Accumulated α-syn comprises a major component of the inclusions in MSA10,11 and might be the primary lesion that eventually compromises nerve cell function and viability in MSA.12 However, the relevance of α-syn accumulation in oligodendrocytes to the neuronal degeneration in MSA was unknown.
No study had demonstrated that α-syn accumulation in oligodendrocytes leads to neuronal degeneration before the establishment of a mouse model of MSA. Three transgenic (Tg) mouse models in which human wild-type α-syn is overexpressed in CNS oligodendrocytes under the control of different promoters were generated.13,14,15 Two of the three mouse lines showed that the accumulation of α-syn as GCIs leads to neuronal degeneration in the mouse CNS.14,15
Our previous study of the Tg mouse model demonstrated that the formation of GCI-like α-syn inclusions leads finally to neuronal degeneration, as exemplified by motor impairment in the phenotype, macroscopically apparent brain atrophy, and histologically decreased numbers of neurons with gliosis.14 Thus, the accumulation of α-syn in oligodendrocytes induced the secondary neuronal degeneration, and we suggested that a similar disease process underlies MSA.
Here, we elucidated novel pathological mechanisms of neuronal accumulation of α-syn in the mouse model of MSA. We identified a protein, microtubule β-III tubulin, that interacts with α-syn and forms an insoluble protein complex. Moreover, the accumulation of α-syn is suppressed by inhibiting polymerization of microtubules. Important insights into MSA neurodegeneration and therapeutic targets have therefore emerged from this mouse model.
Materials and Methods
Primary Culture of Neurons and Glial Cells
Primary cultures of glial cells were obtained as previously described.16 Briefly, glial cells were prepared from the brains of 1- to 3-day-old (P1-3) non-Tg and Tg mice. Cerebral hemispheres were mechanically disrupted. The cell suspensions were transferred to poly-l-lysine (20 μg/ml)-coated culture flasks (4 brains/75 cm2 flask) and incubated in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum supplemented with penicillin (50 U/ml), streptomycin (50 μg/ml), glutamine (1 mmol/L), and insulin (50 μg/ml, Sigma-Aldrich). Primary cultures of neuronal and glial cells were prepared from the brains of P0-P1 non-Tg and Tg mice. Cerebral cortices were dissected from mice and treated with 0.125% trypsin for 15 minutes at 37°C as previously described.17 The dissociated cells were plated on 15 mm polyethylenimine (0.01%, Sigma-Aldrich)-coated glass coverslips (Matsunami, Japan) at a density of 2.4 × 105 cells/cm2 for immunocytochemistry or 4.0 × 106 cells/cm2 per polyethylenimine-coated 75 cm2 flask for biochemical experiments. The cells were maintained in a 1:1 mixture of Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and neurobasal medium with B27 supplement and penicillin (50 U/ml), streptomycin (50 μg/ml), glutamine (1 mmol/L), and insulin (50 μg/ml, Sigma-Aldrich). Half of the medium was replaced twice weekly. To inhibit proliferation of non-neuronal cells, after 4 days in culture, cytosine-1-β-d-arabinofuranoside (AraC, 0.5 μmol/L, Sigma-Aldrich) was added for 3 days. All chemicals for culture were purchased from Invitrogen, unless otherwise stated. To assay the effect microtubule function on α-syn, cultured cells were treated with 3 μmol/L nocodazole (Sigma-Aldrich) for 12 hours at DIV30 or 3 nmol/L nocodazole from DIV21. To enhance the spontaneous firing rate of cortical neurons, neurons cultured longer than DIV27 were treated with picrotoxin (PTX; 100 μmol/L, Wako, Japan), a blocker of gamma-aminobutyric acid A receptor/Cl channels.
Anti-α-Synuclein Antibodies
Syn204 is a monoclonal antibody raised against recombinant human α-syn, which selectively recognizes a human α-syn epitope between amino acid residues 87 and 110.18 Anti-rat synuclein 1 (Syn-1; BD Bioscience) is a monoclonal antibody raised against rat α-syn that recognizes mouse α-syn. To generate an antibody specific to mouse α-syn, a synthetic peptide corresponding to mouse α-syn residues 115 to 125 (DMPVDPGSEAY) was conjugated with keyhole limpet hemocyanin, and used to immunize rabbits. The antisera were purified by affinity chromatography. The resulting polyclonal antibody, Syn-4469, was used for immunoreactive probing.
Immunocytochemistry
Neurons cultured on glass coverslips were fixed in 4% paraformaldehyde in PBS containing 4% sucrose for 15 minutes, and further fixed in methanol (−20°C) for 5 minutes. Neurons were then washed with PBS and blocked in PBS containing 2% bovine serum albumen and 2% goat serum for 2 hours at room temperature. Neurons were incubated with primary antibodies for 1 hour at room temperature in blocking solution. The primary antibodies used were 1:2000 Syn-1, 1:1000 mouse anti-CNPase (Lab Vision), 1:1000 mouse anti-MAP2 (Sigma-Aldrich), and 1:2000 anti-Tuj1 (Covance). After incubation with a primary antibody, the neurons were washed extensively in PBS and incubated for 20 minutes with fluorescent secondary antibody conjugates (goat anti-mouse- or goat anti-rabbit-Alexa488, 1:2000, or Alexa594, 1:2000; Molecular Probes). For cell counting, fixed cells were imaged using a microscope (Olympus, Japan) with ×10 objective lens and captured with a cooled CCD camera. Cells were manually counted in at least five fields for each series of experiments. In some of experiments, pyramidal neurons were confirmed by immunostaining with anti-MAP2 antibody. To assess cell density changes during culture, double positive cells with marker protein immunostaining and Hoechst 33258 were counted by using Metamorph software (Universal Imaging Corp.).
Biochemical Analysis of α-Synuclein Solubility
Proteins were extracted from the dissociated cultures as described above. Cultured cells were homogenized in 0.5 ml of high-salt (HS) buffer (50 mmol/L Tris, pH 7.4, 750 mmol/L NaCl, 20 mmol/L NaF, and 10 mmol/L EGTA with protease inhibitors) per flask of cells and centrifuged at 100,000 × g for 30 minutes; the supernatant was used as the HS-soluble fraction. The resulting pellet was further extracted with 10% sucrose and 50 mmol/L Tris, pH 7.6 to float and remove myelin and associated lipids. The pellets were dissolved in radioimmunoprecipitation (RIPA) buffer (50 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 5 mmol/L EGTA, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) with protease inhibitors and centrifuged at 100,000 × g for 30 minutes at 4°C. The RIPA-soluble supernatant was used as the RIPA fraction. The pellets were extracted with 70% formic acid (FA) by sonication. Protein concentrations of HS and RIPA samples were determined using a BCA protein assay kit (Pierce). FA-extracted samples were dried and resuspended in SDS sample buffer. Equal amounts of samples were separated by SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes (ATTO, Japan). Membranes were blocked with 5% solution of powdered skimmed milk-0.3% Tween 20-Tris-buffered saline (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl), incubated with primary antibodies, followed with either goat anti-mouse or goat anti-rabbit antibody conjugated to horseradish peroxidase (Zymed), detected by an ECL plus system (GE), and exposed on X-ray film (Fujifilm, Japan). The protein levels were quantified using Image J software.
Immunoprecipitation
Homogenates were extracted as described above. The lysates were pre-cleared with protein G sepharose (Santa Cruz). Agarose-coupled mouse IgG was then added and incubated at 4°C for 2 hours. For the precipitation with α-syn, the immunocomplex was incubated with protein G Sepharose at 4°C overnight, and washed three times with RIPA buffer. Proteins were eluted from the beads by boiling in SDS sample buffer, and samples were subjected to immunoblotting.
Expressing Recombinant α-Synuclein and β-III Tubulin in COS-7
The complete cDNA sequences of the Snca and Tubb3 genes were obtained by RT (reverse transcription)-PCR cloning using total RNA isolated from the brain of a 3-month-old non-Tg control mouse as a template. To fuse FLAG tag and c-Myc tag sequences to the 5′-region of Snca and Tubb3 genes, respectively, PCR was performed using primers containing the tag sequence. The PCR products were subcloned into pT7Blue (Novagene). To express recombinant Snca and Tubb3, expression vectors were changed to pcDNA3.1(+) (Invitrogen) for COS-7 cells. COS-7 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. FuGENE 6 (Roche Applied Science) was used to introduce exogenous DNA into COS-7 cells according to the manufacturer’s instructions. Briefly, 24 hours after cells were plated, each dish was transfected with Snca- or Tubb3-pcDNA3.1(+) and FuGENE, and incubated at 37°C for 48 hours. The sequences of the constructs were verified by an ABI 3100-Avant Genetic Analyzer.
Results
Increase in Amount and Insolubility of Endogenous Mouse α-Synuclein in Cultured Neurons Derived from Tg Mice
A previous study revealed that GCI-like inclusions of human α-syn in oligodendrocytes led to the age-dependent accumulation of endogenous mouse α-syn in neuronal axons and nerve terminals of the CNS of Tg mice over 6 months of age.14 To elucidate the primary processes of the α-syn regulation and formation of insoluble accumulation in neurons, we prepared primary cultures of neurons and glial cells derived from non-Tg and Tg mice and compared the properties. First we confirmed the expression of human α-syn in oligodendrocytes. The cultured cells from 7 days in vitro (DIV7) to DIV35 were stained with Syn204, which specifically recognizes human α-syn. We detected Syn204-positive cells derived from only Tg mice (Figure 1A). The Syn204-positive cells increased in number after DIV14 in Tg mice (black bar) (DIV14, 0.96 ± 0.64 cells/mm2; DIV21, 2.97 ± 0.51 cells/mm2; DIV28, 3.53 ± 1.44 cells/mm2, DIV35, 4.33 ± 0.88 cells/mm2), while no Syn204-positive cells were detected in non-Tg mice (white bar) (Figure 1B). In contrast, when the cultured cells were stained with anti-CNPase antibody (CNPase) as an oligodendrocyte marker, the cell densities of oligodendrocytes showed no significant difference between Tg and non-Tg mice (see supplemental Figure 1 at http://ajp.amjpathol.org). Next, we studied the expression of mouse α-syn in neurons cultured with glial cells using immunocytochemical methods. We generated an antibody, Syn-4469, which selectively recognizes mouse α-syn (see supplemental Figure 2 at http://ajp.amjpathol.org). In the Tg mouse cultured cells, Syn-4469 and Syn-1 showed the increased staining of mouse α-syn in neurons, which was clearly distinguished from the staining of over-expressing α-syn in oligodendrocytes (see supplemental Figure 3 at http://ajp.amjpathol.org). Mouse α-syn in the cultured cells of Tg and non-Tg mice was monitored by immunofluorescence from DIV7 to DIV35 using Syn-1, an antibody that recognizes mouse α-syn (Figure 2A). Cells from non-Tg mice from DIV7 to DIV35 showed synaptic enrichment similar to that observed in other presynaptic membrane proteins such as synaptophysin, as previously demonstrated in the literature.19,20,21,22 In cells from Tg mice from DIV7 to DIV14, mouse α-syn is initially observed in neuronal cell bodies, and there was no difference in the expression level and intercellular distribution of mouse α-syn between in Tg and non-Tg mice. The protein had predominantly localized to presynaptic terminals and co-localized with synaptophysin by DIV14 (data not shown). At DIV21 and remarkably after DIV28 in Tg mice, however, a distinct increase in mouse α-syn was observed within the neuronal cell bodies and neurites in the CNS (Figure 2A, lower panels). In normal human brain extracts, α-syn was reported to be distributed almost entirely in the soluble cytosol fraction and detected in membrane fractions including those rich in vesicle and synaptic membranes, but it is also detected in the insoluble fraction in the MSA brain.14,23 We biochemically analyzed the solubility of mouse α-syn in the cultured neurons from Tg mice by sequential extraction, as described in the Materials and Methods. Neuronal and glial cells of Tg and non-Tg mice cultured more than DIV7 were sequentially extracted with HS, RIPA, and FA buffers. Immunoblotting of the extracts staining with Syn-1 and Syn-4469 detected insoluble α-syn in FA fraction of cells from Tg mice cultured more than DIV14, but not in that from non-Tg mice (Figure 2B). The immunoblotting analysis showed that insoluble mouse α-syn progressively increased with the increase in the number of days in vitro (see supplemental Figure 4 at http://ajp.amjpathol.org). To examine whether the insoluble α-syn was mouse α-syn in neurons of Tg mice, we examined pure cultures of glial cells of Tg and non-Tg mice. Syn-1 detected no immunoreactivity on the immunoblots of fractions of the pure cultures of Tg and non-Tg glial cells (Figure 2C). The results revealed that insoluble mouse α-syn was increased in the neurons but not in the oligodendrocytes of Tg mice. Thus, the histological and biochemical data together indicated that insoluble mouse α-syn was accumulated in cultured neurons derived from Tg mice.
Figure 1.

Human α-synuclein is overexpressed in cultured oligodendrocytes derived from Tg mice. A: Immunofluorescent staining of cultured oligodendrocytes for CNPase (left) and wild-type human α-syn (right) at DIV21. Cultured oligodendrocytes were derived from non-Tg and Tg mice. A Syn204-positive oligodendrocyte overexpressing human wild-type α-syn was observed in the cultured cells from Tg mice. Scale bar = 100 μm. B: Cell density of the oligodendrocytes overexpressing human wild-type α-syn was obtained by counting Syn204-positive cells over five objective fields in each series of cultures. Means data are plotted from three independent series of cultures. Data are presented as means ± SEM.
Figure 2.

Increase in amount and insolubility of endogenous mouse α-syn in cultured neurons derived from Tg mice. A: Immunofluorescent staining for endogenous mouse α-syn in cultured cells from non-Tg and Tg mice. In Tg mouse cells, the protein expression level of mouse α-syn was increased and the distribution was predominantly diffuse in cell bodies and neurites (arrows). Scale bar = 100 μm. B: Immunoblots of cultured cells were stained with Syn-1, Syn-4469, and β-actin. Cultured neuronal and glial cells of Tg mice at DIV14, DIV21, DIV28, and DIV35, and those of non-Tg mice at DIV7, DIV28, and DIV35, were sequentially extracted with HS, RIPA, and FA. Insoluble α-syn was detected in the FA fraction of Tg mouse cells. Representative immunoblots of three independent experiments. C: Sequentially extracted samples of cultured neuronal and glial cells of Tg mice at DIV35 (lane 1), pure glial cells of non-Tg mice at DIV33 (lane 2), and pure glial cells of Tg mice at DIV33 (lane 3) were immunoblotted with Syn-1. α-Syn was detected in the fractions of Tg mouse neuronal and glial cells but not in the pure glial cells of non-Tg and Tg mice. Representative immunoblots of three independent experiments.
Oligodendrocytic Inclusions Induce Neuronal Accumulation of Insoluble Mouse α-Synuclein
To confirm that the oligodendrocytes in which human α-syn was overexpressed caused the increase of mouse α-syn in the neurons of Tg mice, we treated the neuronal and glial cell cultures with cytosine-1-β-d-arabinofuranoside (AraC; 0.5 μmol/L) for three days after DIV7 to inhibit proliferation of glial cells. When examined microscopically, the number of glial cells, including oligodendrocytes, was reduced in the culture. The immunoblotting analysis showed that the mouse α-syn expression was decreased by the treatment with AraC (HS, 52.8 ± 12.43%; RIPA, 66.5 ± 12.65%; FA, 48.0 ± 16.1%), although 160-kDa neurofilament and synaptophisin expressions showed no change due to the treatment (Figure 3A). The cell density of MAP2-positive neurons showed no significant difference between cultured neurons with and without AraC. The CNPase-positive and Syn204-positive oligodendrocytes decreased in number by the treatment with AraC (Figure 3B). Next, we investigated how oligodendrocytic accumulation of human α-syn causes neuronal accumulation of insoluble mouse α-syn. Non-Tg mouse cells after DIV14 were treated with conditioned media derived from Tg mouse cell cultures and vice versa. The immunoblots of sequentially extracted cultured cells showed that the insoluble α-syn was detected in the FA fraction of non-Tg mouse treated with Tg mouse conditioned media. No remarkable difference was detected between the FA fractions of Tg mouse cell cultured with and without non-Tg conditioned media (non-Tg mice: FA, 404.1 ± 172.3%; Tg mice: FA, 138.2 ± 54.1%) (Figure 3C). The result indicates that the conditioned media of Tg mouse cell cultures contained oligodendrocyte-derived signals that induced neuronal α-syn accumulation. Thus, it was the oligodendrocytic accumulation of α-syn that induced accumulation of insoluble α-syn in the Tg mouse neurons.
Figure 3.

Effects of oligodendrocytes overexpressing human α-syn on neurons. A: Cells of Tg mice at DIV30 cultured with (+) and without (−) AraC were sequentially extracted. The immunoblots were stained with Syn-1 and antibodies to 160 kDa-neurofilament (NF160) and anti-synaptophisin (SVP38), and only the immunoblots probed with Syn-1 showed that expression of α-syn was decreased by the AraC treatment. α-Syn expression on the immunoblots of Tg mouse cultures with (closed bars) and without AraC (open bars) was quantitated. All α-syn signals were normalized to the signal for NF160 to account for the amount of neuronal protein loaded per lane. HS, 52.8± 12.4%; RIPA, 66.5± 12.7%; FA, 48.0± 16.1%, five series of cultures. *P < 0.01. Data are presented as means ± SEM. B: Bar graphs comparing the number of MAP2 positive cells (neurons), CNPase-positive cells (oligodendrocytes), and syn204-positive cells of Tg mouse cultures with (closed bars) and without AraC (open bars) (n = 3). Data are presented as means ± SEM. C: At DIV30, cultured cells of non-Tg mice treated with conditioned media (CM) from Tg mouse cell cultures and those of Tg mice treated with conditioned media derived from non-Tg mouse cell cultures were sequentially extracted, and the immunoblots were stained with Syn-1 and NF160. The FA fractions of α-syn were quantified on the immunoblots of non-Tg and Tg mouse cultures with (closed bars) and without (open bars) conditioned media (n = 3). Note that insoluble α-syn was detected in FA fraction of non-Tg mouse treated with the conditioned media of Tg mouse cells. Data are presented as means ± SEM. *P < 0.05 (Mann-Whitney U-test).
α-Synuclein Binds to β-III Tubulin and Forms Insoluble Protein Complex
We next addressed the mechanism by which the accumulation of insoluble α-syn causes neuronal degeneration in Tg mice. We investigated which proteins interact with α-syn in Tg mice. First, we tried to identify the proteins that change the soluble fraction to an insoluble one on the immunoblots of the sequentially extracted samples from Tg mice. We found that neuron-specific β-III tubulin antibody (Tuj1) labeled a band in the insoluble fractions on the immunoblots of the Tg mouse cells, but showed no immunoreactivity to the cell fractions of non-Tg mice (Figure 4A). The other neuronal proteins showed no difference in solubility between Tg and non-Tg mice. Second, we evaluated the interaction between α-syn and β-III tubulin by immunoprecipitation analysis. The analysis showed that β-III tubulin was co-immunoprecipitated with mouse α-syn in the RIPA fraction of Tg mouse cells (Figure 4B). No other β-tubulin isoforms was co-immunoprecipitated. Third, double-labeling immunofluorescent staining of Tg mouse cells showed that mouse α-syn was colocalized with β-III tubulin in the neurites and neuronal cell bodies (Figure 4C). The cell density of Tuj1-positive neurons showed no significant difference between Tg and non-Tg mice (see supplemental Figure 5 at http://ajp.amjpathol.org). These data demonstrate that the increase of mouse α-syn expression in neurites and cell bodies caused binding of the α-syn with β-III tubulin to form an insoluble complex. To verify that direct binding of the α-syn with β-III tubulin causes formation of the insoluble complex, COS-7 cells were transfected with Snca, Tubb3, or cotransfected with Snca and Tubb3 respectively. The recombinant proteins expressed in COS-7 cells were sequentially extracted. The insoluble α-syn was detected in the FA fraction only in the COS-7 cells cotransfected with Snca and Tubb3 (Figure 5A). Then, we evaluated the direct interaction between the recombinant α-syn and β-III tubulin by immunoprecipitation analysis (Figure 5B). We concluded that the direct binding of α-syn with β-III tubulin caused formation of the insoluble complex.
Figure 4.

Protein-protein interaction of α-syn and β-III tubulin in Tg mice. A: Cultured neuronal and glial cells of Tg and non-Tg mice at DIV33 were sequentially extracted with HS, RIPA, and FA. The immunoblots were stained by Syn-1, Tuj1, β-actin as a protein marker, and SVP38 (anti-synaptophisin antibody) as a neuronal protein marker. Insoluble β-III tubulin was detected in the same FA fraction of Tg mice as the insoluble α-syn on the immunoblots of Tg mouse cells. Representative immunoblots of three independent experiments. B: Association of α-syn and β-III tubulin in RIPA fraction of Tg mouse cells was examined by immunoprecipitation with Syn-1 or mouse IgG as a negative control antibody, followed by immunoblotting with Syn-1, Tuj1, β-IV tubulin, or β-actin. β-III tubulin was co-immunoprecipitated with mouse α-syn. C: Double-labeling immunofluorescent staining using antibodies Syn-1 to mouse α-syn (green) and Tuj1 to β-III tubulin (red) shows that α-syn colocalizes with β-III tubulin in the neurites (arrows) and neuronal cell bodies (arrowhead) derived from Tg mice (yellow). Scale bar = 100 μm.
Figure 5.

Direct binding of α-syn with β-III tubulin causes to form insoluble complex. A: COS-7 cells were transfected with the mock (lane 1), Snca (lane 2), Tubb3 (lane 3), or cotransfected with Snca and Tubb3 (lane 4). The recombinant proteins expressed by COS-7 were sequentially extracted. The immunoblots stained with Syn-1 showed that insoluble α-syn in the FA fraction was detected only in the COS-7 cells cotransfected with Snca and Tubb3 (arrowhead). B: COS-7 cells were transfected with FLAG-Snca, or cotransfected with FLAG-Snca and cMyc-Tubb3 for immunoprecipitation analysis. The fusion proteins expressed were immunoprecipitated with anti-FLAG antibody using anti-FLAG-agarose affinity gels, and the immunoblots of the immunoprecipitates were stained with Syn-1, anti-cMyc antibody, and Tuj1. The recombinant α-syn directly bound to the recombinant β-III tubulin.
Accumulation of Insoluble α-Synuclein Is Suppressed by Depolymerization of Microtubules
Because microtubule β-III tubulin binds α-syn and the insoluble protein complex thus formed accumulates in neurons, it was hypothesized that the accumulation of insoluble α-syn might be controlled by blocking microtubule polymerization in Tg mice. We investigated the effect of treatment with nocodazole, a microtubule-depolymerizing agent (3 μmol/L), on the accumulation of α-syn. Primary cultured cells of Tg and non-Tg mice were treated with nocodazole for 12 hours at DIV30 and sequentially extracted for immunoblotting (Figure 6A). Although soluble α-syn decreased in non-Tg mouse cells after nocodazole treatment, the soluble and insoluble α-syn in Tg mouse cells showed no difference after this treatment (non-Tg mice: HS, 41.6 ± 16.3%; RIPA, 65.0 ± 25.5%; Tg mice: HS, 89.9 ± 9.32%; RIPA, 100.2 ± 5.1%, FA, 96.8 ± 17.6%) (Figure 6B). The data indicate that microtubules play an important role in the regulation of α-syn in non-Tg mice. The accumulation of α-syn in Tg mice was not inhibited by nocodazole when it had already accumulated and formed the insoluble protein complex. Then we investigated whether nocodazole treatment prevented the accumulation of α-syn in Tg mice before soluble α-syn starts to form an insoluble protein complex. Primary cultured cells from Tg mice were treated with nocodazole (3 nmol/L) from DIV20 to DIV30 and then sequentially extracted at DIV30 (Figure 6A). The immunoblotting analysis showed that the accumulation of α-syn decreased in Tg mouse cells with nocodazole pretreatment (non-Tg mice: HS, 7.89 ± 3.47%; RIPA, 9.52 ± 1.56%; Tg mice: HS, 109.2 ± 42.5%; RIPA, 90.6 ± 20.7%, FA, 34.5 ± 12.1%) (Figure 6C). The cell density of MAP2-positive neurons showed no significant difference between cultured neurons with and without nocodazole (Figure 6D). The data demonstrated that microtubule depolymerization suppressed the accumulation of insoluble α-syn in Tg mice. Therefore, the binding of α-syn to microtubules and formation of an insoluble protein complex induced the accumulation of α-syn, and it could be suppressed before the insoluble complex was formed.
Figure 6.

Effect of microtubule-depolymerizing agent on accumulation of insoluble α-syn. A: Cultured neuronal and glial cells from non-Tg and Tg mice were treated with (+) and without (−) nocodazole, and they were sequentially extracted at DIV30 (n = 3). The cells were treated with 3 μmol/L nocodazole at DIV30 for 12 hours (3 μM 12 h) and with 3 nmol/L nocodazole from DIV21 to DIV30 for 10 days (3nM pretreatment). Representative immunoblots of three independent experiments. B: Quantitative analyses of α-syn on the immunoblots of non-Tg and Tg mouse cultures with (closed bars) and without (open bars) treatment of 3 μmol/L nocodazole for 12 hours at DIV30. Soluble α-syn decreased in non-Tg mouse cells, whereas soluble and insoluble α-syn showed no difference in Tg mouse cells with the treatment. All α-syn signals are normalized to the signals for synaptophisin (SVP38; anti-synaptophisin antibody) to account for the amount of neuronal protein loaded per lane. *P < 0.05. Data are presented as means ±SEM. C: Quantitative analyses of α-syn on the immunoblots of non-Tg and Tg mouse cultures with (closed bars) and without (open bars) pretreatment with 3 μmol/L nocodazole for 10 days. Insoluble α-syn was significantly reduced in the FA fraction of Tg mice by the pretreatment with nocodazole. All α-syn signals were normalized to the signal for synaptophisin (SVP38) to account for the amount of neuronal protein loaded per lane. **P < 0.01. Data are shown as means ± SEM. D: Neuronal densities of non-Tg and Tg mouse cultures with (closed bars) and without (open bars) pretreatment with 3 μmol/L nocodazole for 12 hours and 3 nmol/L for 10 days were obtained by counting Hoechst 33258 and MAP2 double-positive cells over five objective fields in each series of cultures. Means data are plotted from three independent series of cultures.
Effect of Neuronal Function on Insoluble α-Synuclein Accumulation
Despite the accumulation and insolubilization of α-syn in neurons, we didn’t find that the accumulation has a direct effect on whether or not the neurons will degenerate during cultured period. However we previously reported that progressive neuronal degeneration was accompanied in Tg mice after the sixth month. It is possible that the accumulation and insolubilization of α-syn in neurons affect the neuronal function and cause neuronal dysfunction. Because the localization of multiple synaptic proteins, including α-syn was modulated by activity dependent manner,24 we examined whether neuronal activity affects the accumulation of α-syn in neuronal and glial cell cultures. The cultured cells were treated with picrotoxin (PTX; 100 μmol/L), a gamma-aminobutyric acid A receptor/Cl channel blocker, for 3 days from DIV27 to DIV30, and the proteins were sequentially extracted. Although non-Tg mouse cells showed a decrease in soluble α-syn with PTX treatment (Figure 7A), Tg mouse cells showed no difference in either soluble or insoluble α-syn with PTX treatment (Figure 7B). The cell density of MAP2-positive neurons showed no significant difference between cultured neurons with and without PTX (Figure 7C). The data showed that the modulation of α-syn by neuronal activity was disturbed in Tg mice and that the disturbance may accelerate the accumulation of α-syn.
Figure 7.
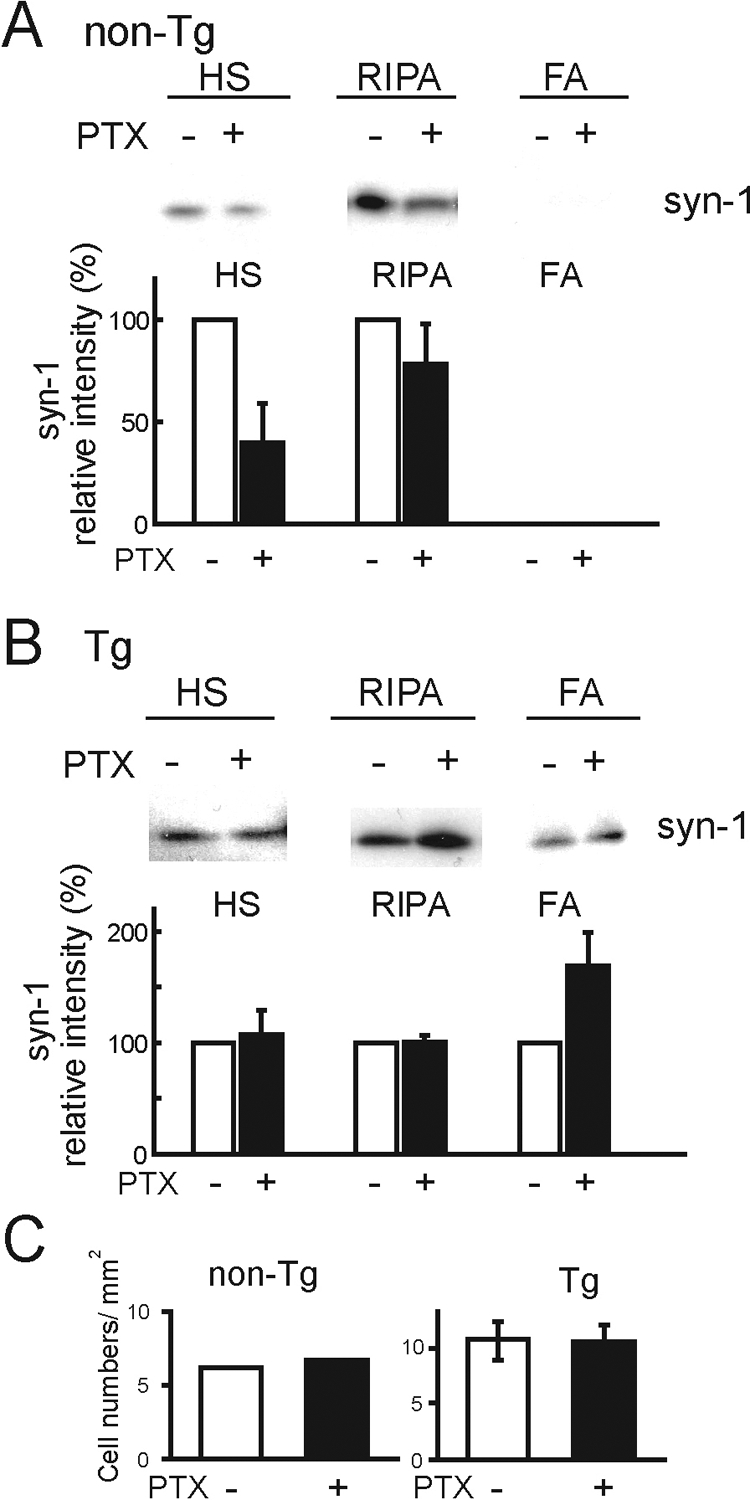
Neuronal activity controls accumulation of α-syn. Cells of non-Tg (A) and Tg (B) mice at DIV30 cultured with (+) and without (−) PTX were sequentially extracted. Although soluble α-syn decreased in non-Tg mouse cells after PTX treatment, the accumulation of α-syn showed no difference in Tg mouse cells with PTX treatment (n = 5). Bar graphs show quantitative analyses of α-syn expression on the immunoblots with (closed bars) and without (open bars) PTX in non-Tg (A) and Tg (B) mice. All α-syn signals were normalized to the signal for NF160 to account for the amount of neuronal protein loaded per lane (non-Tg mice: HS, 39.63± 19.34%; RIPA, 78.35± 19.50%; Tg mice: HS, 106.95± 22.24%; RIPA, 100.60± 6.09%; FA, 168.73± 30.61%, two series of cultures). C: Neuronal cell densities were obtained by counting Hoechst 33258 and MAP2 double positive cells over five objective fields in each series of cultures. Means data are plotted from two independent series of cultures.
Discussion
We previously reported that progressive neuronal degeneration was accompanied by neuronal accumulation of mouse α-syn in Tg mice overexpressing human α-syn in oligodendrocytes.14 Although the accumulation of α-syn in neurons plays an important role in neuronal degeneration, the primary cellular process underlying the neuronal degeneration was not understood. The present study provides novel evidence that endogenous mouse α-syn, which is increased by the formation of oligodendrocytic inclusions, binds to β-III tubulin in microtubules. The insoluble complex is progressively accumulated in neurons, leading to neuronal dysfunction in the Tg mouse model of MSA. Moreover, we demonstrated that the accumulation of the insoluble α-syn complex is suppressed by treatment with a microtubule depolymerizing agent. The results indicate that the binding of α-syn to β-III tubulin is a key process in the development of neuronal degeneration in the MSA mouse model.
One pathological process underlying the accumulation of α-syn in MSA is the accumulation of an insoluble protein complex in neurons induced by the binding of α-syn with β-III tubulin. We identified the microtubule component β-III tubulin as the protein that specifically interacts with α-syn in the Tg mice. Previous studies showed that β-tubulin is located in GCIs in the brain tissue of patients with MSA.8,25,26 Ultrastructural studies demonstrated that GCIs were composed of oligodendrocytic microtubules with diameters of 20 to 30 nm.25 Immunoelectron microscopy showed that α-syn positive inclusions in neuronal nuclei and oligodendrocytes of MSA were composed by bundles of filaments with a diameter of 10 to 20 nm.27 Our previous observation of aged Tg mice with motor impairment demonstrated the distinct axonal accumulation of α-syn and the formation of α-syn inclusions in the degenerating axons and axonal terminals.14 The axonal terminal inclusions were ultrastructurally composed of disordered tubular structures with diameters of approximately 20 to 35 nm.14 The present data reveal that the formation and accumulation of insoluble complexes of α-syn and β-III tubulin is an important disease process in the neuronal dysfunction of an MSA mouse model. β-tubulin was reported to be co-immunoprecipitated with α-syn in normal human brain extracts.27,28 We revealed no evidence of directly binding to the other β-tubulin isoforms. Although the physiological role of the interaction between α-syn and β-tubulin is not completely clear, previous reports showed that α-syn interacted with heterodimeric tubulin28 and that tubulin initiated and promoted α-syn fibril formation under physiological conditions in vitro.29 Indeed, in the MSA Tg mice, once the insoluble complex was formed, microtubule depolymerization agent was ineffective in blocking of the α-syn accumulation. However, microtubule depolymerization agent suppressed the accumulation of insoluble α-syn complex before the neuronal α-syn expression was developed by the oligodendrocytes overexpressing human α-syn. We plan to test the hypothesis that the regulation of insoluble α-syn complexes by the treatment is a therapeutic target for MSA. Because the microtubule depolymerizing agent blocks microtubule conformation and prevents microtubule function, the potential use can be toxic. There may be several approaches to inhibition of the accumulation of α-syn. Although the pathological mechanisms are not fully understood in MSA, this study could contribute to development of a therapeutic strategy against MSA neuronal degeneration.
Another pathological process that underlies the neuronal degeneration in MSA is the increase in the expression of α-syn in neurons due to the formation of GCIs in oligodendrocytes. We here demonstrated that insoluble α-syn accumulated in neuronal cell bodies and neurites after DIV21 in Tg mice. DIV21 is the crucial time at which the GCI-like inclusions start to develop in oligodendrocytes. Inhibition of glial cell proliferation by treatment with AraC prevented the accumulation of insoluble α-syn in Tg mouse neurons. Because reciprocal communication between neurons and oligodendrocytes is essential for the development of the CNS,30 our study suggested that degenerated oligodendrocytes might regulate α-syn protein expression in neurons. Moreover, we demonstrated that conditioned media derived from Tg mouse cell cultures induced insoluble α-syn in non-Tg mouse cell cultures, suggesting abnormal regulation of α-syn due to the formation of GCI-like inclusions. We speculate that abnormal regulation of neuronal α-syn expression by degenerated oligodendrocytes may trigger the onset of MSA.
Modulation and physiological function of α-syn by neuronal activity is disturbed in an MSA mouse model. Our present finding reveals that α-syn decreased in non-Tg mouse cells in response to neuronal activity due to PTX treatment, whereas the α-syn accumulation in response to neuronal activity due to PTX treatment showed no difference in Tg mouse cells. Neuronal activity controls the synaptic accumulation of α-syn, and α-syn disperses from the nerve terminal in response to neuronal activity.24 Although steady-state levels of α-syn in non-Tg mouse cells are balance between production and clearance, the regulation of α-syn is disturbed in Tg mouse neurons due to the binding with β-III tubulin and formation of the insoluble complex. Because α-syn localizes to nerve terminals,20,21,31 when expressed at physiological levels, it functions as a regulator of synaptic vesicle fusion and neurotransmitter release at the synapse.32 The total number of vesicle fusion events in cell α-syn-overexpressing mice was significantly decreased.33 Because endogenous mouse α-syn expression was increased in the presynaptic terminals of Tg mouse neurons,14 the total number of synaptic vesicle fusion events may be significantly decreased. Therefore, it is hypothesized that the insolubility of α-syn causes abnormal α-syn synaptic accumulation, resulting to disturb the normal function of α-syn and age-dependent progressive neuronal degeneration. We expect to evaluate the synaptic function in Tg mice by electrophysiological approach.
Supplementary Material
Acknowledgments
We thank Virginia Lee and John Trojanowski for reading the manuscript and providing the MSA Tg mice, and Ryogen Sasaki for technical assistance.
Footnotes
Address reprint requests to Ikuru Yazawa, MD, Ph.D., Laboratory of Research Resources, National Institute for Longevity Sciences, National Center for Geriatrics and Gerontology, 36-3 Gengo, Morioka-cho, Obu-shi, Aichi 474-7522, Japan. E-mail: yazawa@nils.go.jp.
Supported by the Ministry of Health, Labor and Welfare, Japan (the Research Grant for Longevity Sciences (18A-4) and a Grant-in-Aid for “the Research Committee for Ataxic Diseases” of the Research on Measures for Intractable Diseases), Grants-in-Aid (20700327) for Scientific Research from the Japan Society for the Promotion of Science, Japan Foundation for Aging and Health, Okinaka Memorial Institute for Medical Research, and the Life Science Foundation of Japan.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Yazawa I. Pathological mechanism of neurodegeneration in polyglutamine diseases. Pandalai SG, editor. India: Research Signpost,; 2003:pp 21–28. [Google Scholar]
- Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry. 1969;32:28–34. doi: 10.1136/jnnp.32.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ, Kaufmann H, Klockgether T, Lang AE, Lantos PL, Litvan I, Mathias CJ, Oliver E, Robertson D, Schatz I, Wenning GK. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci. 1999;163:94–98. doi: 10.1016/s0022-510x(98)00304-9. [DOI] [PubMed] [Google Scholar]
- Lowe JS, Leigh N. Disorders of movement and system degeneration. Graham DI, Lantos PL, editors. London: Hodder Arnold; 2002:pp 343–346. [Google Scholar]
- Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci. 1989;94:79–100. doi: 10.1016/0022-510x(89)90219-0. [DOI] [PubMed] [Google Scholar]
- Arima K, Ueda K, Sunohara N, Arakawa K, Hirai S, Nakamura M, Tonozuka-Uehara H, Kawai M. NACP/ α-synuclein immunoreactivity in fibrillary components of neuronal and oligodendroglial cytoplasmic inclusions in the pontine nuclei in multiple system atrophy. Acta Neuropathol (Berl) 1998;96:439–444. doi: 10.1007/s004010050917. [DOI] [PubMed] [Google Scholar]
- Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee VM-Y. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α–synuclein. Ann Neurol. 1998;44:415–422. doi: 10.1002/ana.410440324. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. The α-synucleinopathies: parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann NY Acad Sci. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- Duda JE, Lee VM-Y, Trojanowski JQ. Neuropathology of synuclein aggregates. J Neurosci Res. 2000;61:121–127. doi: 10.1002/1097-4547(20000715)61:2<121::AID-JNR1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Spooren W, Fuss B, Mallon B, Macklin WB, Fujiwara H, Hasegawa M, Iwatsubo T, Kretzschmar HA, Haass C. Hyperphosphorylation and insolubility of α-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 2002;3:583–588. doi: 10.1093/embo-reports/kvf109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VM-Y. Mouse model of multiple system atrophy: α-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45:847–859. doi: 10.1016/j.neuron.2005.01.032. [DOI] [PubMed] [Google Scholar]
- Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, Hashimoto M, Song D, Iwatsubo T, Tsuboi K, Masliah E. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human α-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci. 2005;25:10689–10699. doi: 10.1523/JNEUROSCI.3527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Landsberg C, Heinrich M. OLN-93: a new permanent oligodendroglia cell line derived from primary rat brain glial cultures. J Neurosci Res. 1996;45:161–173. doi: 10.1002/(SICI)1097-4547(19960715)45:2<161::AID-JNR8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Kiyosue K, Taguchi T. Diminished neuronal activity increases neuron-neuron connectivity underlying silent synapse formation and the rapid conversion of silent to functional synapses. J Neurosci. 2005;25:4040–4051. doi: 10.1523/JNEUROSCI.4115-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Jakes R, Goedret M, Duda JE, Leight S, Trojanowski JQ, Lee VM-Y. A panel of epitope-specific antibodies detects protein domains distributed throughout human α-synuclein in Lewy bodies of Parkinson’s disease. J Neurosci Res. 2000;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res. 1991;11:335–343. doi: 10.1016/0169-328x(91)90043-w. [DOI] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Hsu LJ, Mallory M, Xia Y, Veinbergs I, Hashimoto M, Yoshimoto M, Thal LJ, Saitoh T, Masliah E. Expression pattern of synucleins (non-A beta component of Alzheimer’s disease amyloid precursor protein/ α-synuclein) during murine brain development. J Neurochem. 1998;71:338–344. doi: 10.1046/j.1471-4159.1998.71010338.x. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Liu W, Hardy J, Farrer M, Mehta N, Uitti R, Mark M, Zimmerman T, Golbe L, Sage J, Sima A, D'Amato C, Albin R, Gilman S, Yen SH. Widespread alterations of α-synuclein in multiple system atrophy. Am J Pathol. 1999;155:1241–1251. doi: 10.1016/s0002-9440(10)65226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. Neural activity controls the synaptic accumulation of α-synuclein. J Neurosci. 2005;25:10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazato Y, Yamazaki H, Hirato J, Ishida Y, Yamaguchi H. Oligodendroglial microtubular tangles in olivopontocerebellar atrophy. J Neuropathol Exp Neurol. 1990;49:521–530. doi: 10.1097/00005072-199009000-00007. [DOI] [PubMed] [Google Scholar]
- Abe H, Yagishita S, Amano N, Iwabuchi K, Hasegawa K, Kowa K. Argyrophilic glial intracytoplasmic inclusions in multiple system atrophy: immunocytochemical and ultrastructural study. Acta Neuropathol (Berl) 1992;84:273–277. doi: 10.1007/BF00227820. [DOI] [PubMed] [Google Scholar]
- Lin WL, DeLucia MW, Dickson DW. α-Synuclein immunoreactivity in neuronal nuclear inclusions and neurites in multiple system atrophy. Neurosci Lett. 2004;354:99–102. doi: 10.1016/j.neulet.2003.09.075. [DOI] [PubMed] [Google Scholar]
- Payton JE, Perrin RJ, Clayton DF, George JM. Protein-protein interactions of alpha-synuclein in brain homogenates and transfected cells. Brain Res Mol Brain Res. 2001;95:138–145. doi: 10.1016/s0169-328x(01)00257-1. [DOI] [PubMed] [Google Scholar]
- Alim MA, Hossain MS, Arima K, Takeda K, Izumiyama Y, Nakamura M, Kaji H, Shinoda T, Hisanaga S, Ueda K. Tubulin seeds α-synuclein fibril formation. J Biol Chem. 2002;277:2112–2117. doi: 10.1074/jbc.M102981200. [DOI] [PubMed] [Google Scholar]
- Simons M, Trajkovic K. Neuron-glia communication in the control of oligodendrocyte function and myelin biogenesis. J Cell Sci. 2006;119:4381–4389. doi: 10.1242/jcs.03242. [DOI] [PubMed] [Google Scholar]
- Withers GS, George JM, Banker GA, Clayton DF. Delayed localization of synelfin (synuclein. NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res. 1997;99:87–94. doi: 10.1016/s0165-3806(96)00210-6. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Shorter J. Prime time for α-synuclein. J Neurosci. 2007;27:2433–2434. doi: 10.1523/JNEUROSCI.0094-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.