

Abstract
Ligand-activated Eph tyrosine kinases regulate cellular repulsion, morphology, adhesion, and motility. EphA2 kinase is frequently up-regulated in several different types of cancers, including prostate, breast, colon, and lung carcinomas, as well as in melanoma. The existing data do not clarify whether EphA2 receptor phosphorylation or its simple overexpression, which likely leads to Eph kinase-independent responses, plays a role in the progression of malignant prostate cancer. In this study, we address the role of EphA2 tyrosine phosphorylation in prostate carcinoma cell adhesion, motility, invasion, and formation of metastases. Tumor cells expressing kinase-deficient EphA2 mutants, as well as an EphA2 variant lacking the cytoplasmic domain, are defective in ephrinA1-mediated cell rounding, retraction fiber formation, de-adhesion from the extracellular matrix, RhoA and Rac1 GTPase regulation, three-dimensional matrix invasion, and in vivo metastasis, suggesting a key role for EphA2 kinase activity. Nevertheless, EphA2 regulation of cell motility and invasion, as well as the formation of bone and visceral tumor colonies, reveals a component of both EphA2 kinase-dependent and -independent features. These results uncover a differential requirement for EphA2 kinase activity in the regulation of prostate carcinoma metastasis outcome, suggesting that although the kinase activity of EphA2 is required for the regulation of cell adhesion and cytoskeletal rearrangement, some distinct kinase-dependent and -independent pathways likely cooperate to drive cancer cell migration, invasion, and metastasis outcome.
Eph receptors, the largest subfamily of receptor tyrosine kinases (RTKs), are involved in many biological processes including angiogenesis, tissue-border formation, cell migration, axon guidance, and synaptic plasticity. Ephs\ephrins are important mediators of cell-cell communication regulating cell attachment to extracellular matrix, cell shape, and motility.1 Their frequent overexpression in human cancers and the correlation with poor prognosis and high vascularity in cancer tissues emphasize emerging roles in tumor progression.2 EphA2 has been implicated in carcinogenesis of several cancers including melanomas and prostate, breast, colon, lung, and esophageal carcinomas.3 These studies showed high levels of EphA2 in both tissue and cell explants of these diseases and especially in the more aggressive stages of progression.4 In particular, ectopic overexpression of EphA2 gives untransformed epithelial cells both tumorigenic and metastatic potential.5 Certain Ephs and their ligands are expressed and up-regulated at sites of dynamic neovascularization, eg, in endothelial cells during tumor invasion.6 EphA2-deficient mice displayed decreased tumor volume, microvasculature density, and lung metastasis.7 Actually, the role of EphA2 in the regulation of malignant transformation/progression is far from clear. Indeed recent results unveil some antitumorigenic functions of EphA2: EphA2 is localized on chromosome 1p36.13, a region frequently deleted in a number of human cancers, including prostate and brain tumor and disruption of EphA2 kinase in mice leads to increased susceptibility to skin carcinogenesis, suggesting EphA2 as a potential tumor suppressor gene in mammalian skin.8
Beside being implicated in tumor aggressiveness and vasculogenesis, class A ephrin/Eph interactions have recently been implicated in the organization of cell migration during several physiological and pathophysiological processes, including development, tissue morphogenesis, and cancer cell migration.1 As for other receptor tyrosine kinases, ligand binding of EphA receptors induces receptor clustering, activation of kinase activity, and subsequent trans-phosphorylation of the cytoplasmic domains, creating docking sites for a number of signaling proteins.9,10 The role of class A Eph receptors in regulating endothelial cell migration and assembly is strongly supported by several studies in angiogenic remodeling.11 On the contrary, a clear role of EphA2 kinase in the regulation of cancer cell motility has not been delineated. We recently reported that in prostate carcinoma cells, ephrinA1 elicits a motility response by activating a Rho- and focal adhesion kinase (FAK)-dependent cytoskeleton rearrangement, finally driving the retraction of the cell body and the inhibition of directional cell migration.12 In addition, activation of EphA2 is able to redirect motility and inhibit invasion of adenocarcinoma cells, again through a FAK-mediated pathway.13
Ephrin/Eph interaction gives rise to complex cell-cell signaling culminating in a bidirectional pathway. Cells bearing the ephrin ligand engage in reverse signaling, and cells carrying the Eph receptors undergo forward signaling.10,14,15 Although the reverse signaling of ephrin As is recognized as kinase-independent, attributable to their lack of enzymatic activity, the forward response elicited by the Eph kinase receptors is puzzling, because both kinase-dependent and -independent components have been reported. Indeed, several lines of evidence describe EphA2 signaling as mainly kinase-dependent. First, mutations of the kinase domain of EphA2 affect vascular endothelial cell growth and vascular endothelial growth factor-dependent angiogenesis.16 Second, recent data showed that EphA2 receptor phosphorylation may be vital in granting oncogenic potential.17 In agreement, the block of EphA2 receptor activation through EphA2-Fc results in a decrease in phosphorylation that was concurrent with decreased tumor volume.18 In keeping with these data, emerging evidence suggests that protein tyrosine phosphatases (PTPs) are involved in regulating Eph-mediated responses,19,20 strongly supporting a role for Eph kinase activity.
Nevertheless Eph receptors are nonclassical receptor tyrosine kinases because, beside kinase-dependent signaling, ligation of certain members of the Eph family can also trigger kinase-independent responses.21,22,23,24 First, the presence of kinase-inactive together with wild-type EphA7 within the same cell changes its ligand-induced response from repulsion to adhesion,25 indicating different functions of Ephs owing to their phosphorylation and degree of clustering. Second, EphA8 receptor localizes p110γ phosphatidylinositol 3-kinase to the plasma membrane in a tyrosine kinase-independent manner, thereby allowing access to lipid substrates to enable the signals required for integrin-mediated cell adhesion.26 Third, the simple removal of membrane-associated EphA2 through ligand-independent endocytosis reduces malignant behavior of the cells and tumor growth.13 Finally, Miao and colleagues27 recently reported that EphB3 catalytic activity is required for inhibition of integrin-mediated cell adhesion but is dispensable for directional cell migration.
In the context of this controversial literature, we investigated the role of tyrosine phosphorylation of EphA2 kinase in the regulation of prostate carcinoma cell motility and invasion. On the whole, our findings point to a kinase-dependent role of EphA2 receptor for the regulation of cell motility, adhesion, cytoskeleton rearrangements, as well as for invasion and metastasis formation in nude mice, although the invasive and prometastatic effect of EphA2 show a kinase-independent component.
Materials and Methods
Materials
Unless specified all reagents were obtained from Sigma (St. Louis, MO). PC3, DU145, LNCaP, and HEK293T cells were from ATCC, Rockville, MD; PNT1A cells were a generous gift of Rosario Notaro (the Department of Pharmacology, University of Florence, Italy), recombinant mouse Fc and ephrinA1-Fc chimera were from R&D Systems (Minneapolis, MN), antiphosphotyrosine (clone 4G10) and anti-EphA2 antibodies (clone D7) were from Upstate Biotechnology Inc. (Charlottesville, VA), anti-RhoA antibodies were from BD (New Jersey, USA).
Plasmids and Site-Directed Mutagenesis
Primers for EphA2 reverse transcriptase-polymerase chain reaction (RT-PCR) were 5′-ATGGAGCTCCAGGCAGCCCG-3′ and 5′-TCAGATGGGGATCCCCACAGT-3′. Total RNA was isolated from PC3 cells with TRIzol reagent, and cDNA obtained with SuperScript one-step RT-PCR. EphA2 was subcloned into pTargetT vector (Promega, Madison, WI). EphA2 mutants were obtained using a QuikChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). Phenylalanine replaced Tyr587 and Tyr593 in the double-mutant (DM) and arginine replaced Lys645 in the kinase dead (KD) mutant. EphA2 cytoplasmic domain truncation mutant (ΔCyto), was generated by PCR amplification of the extracellular and transmembrane domain of EphA2 (from N-ter to Lys 562).
Cell Culture, Stimulation, and Protein Overexpression
PC3 human prostate carcinoma were cultured in Ham’s F12. HEK293T human embryonic kidney cells and DU-145 human prostate carcinoma cells (brain metastasis) were cultured in Dulbecco’s modified Eagle’s medium. PNT1A human postpubertal prostate normal cells and LNCaP human prostate carcinoma cells (lymph node metastasis) were cultured in RPMI 1640. All media are supplemented with 10% fetal calf serum in a 5% CO2 humidified atmosphere. HEK293T cells were transiently transfected using Lipofectamine 2000 (Invitrogen Milano, IT) using 4 μg of plasmid DNA. Forty-eight hours after transfection the cells were recovered for analysis. PC3 and DU-145 cells were stably transfected with the same procedure, except that 48 hours after transfection cells were selected with 400 mg/L G418 for neomycin resistance. For studies using soluble ephrinA1, cells were stimulated with 1 μg ml−1 Fc or ephrinA1-Fc for the indicated times.
Retraction Fiber Formation
After washing with phosphate-buffered saline (PBS), the cells were fixed with 3.7% formaldehyde solution in PBS for 20 minutes at 4°C. Then, cells were permeabilized with 0.1% Triton X-100 in PBS and stained with 50 μg/ml of phalloidin-tetramethyl-rhodamine isothiocyanate for 1 hour at room temperature, mounted with glycerol plastine, and observed under a laser-scanning confocal microscope (Leica SP5, Mannheim, Germany).
Immunoprecipitation and Western Blot Analysis
For anti-EphA2 immunoprecipitation, we used either anti-EphA2 antibodies or 1 μg ml−1 ephrinA1-Fc fusion protein with similar results. Immune complexes were collected on protein A Sepharose, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred onto nitrocellulose. For chemiluminescence detection we used a Gel Logic 2200 Kodak Imaging System (Eastman-Kodak, Rochester, NY), equipped with a charge-coupled device camera, which guarantees a high linearity, and Quantity-One software (Bio-Rad, Hercules, CA) was used to obtain quantitative analyses.
Cell Adhesion Assay
Cells were serum-starved for 24 hours before detaching with 0.25% trypsin for 1 minute. Trypsin was blocked with 0.2 mg/ml soybean trypsin inhibitor, and cells were resuspended in fresh medium, maintained in suspension for 30 minutes at 37°C, and then directly seeded onto precoated dishes treated overnight with 10 μg/ml of human fibronectin for 4 hours in the presence of either 1 μg ml−1 Fc or ephrinA1-Fc.
Wound Healing Assay
Cells were cultured in 6-cm plates until confluence. The monolayer of PC3 cells was serum-starved for 24 hours and then was scratched using a thin sterile pipette tip. Pictures were taken before and 24 hours after the addition of complete medium with either 1 μg/ml of ephrinA1-Fc or Fc using an inverted Leica microscope equipped with a Nikon digital camera (Tokyo, Japan).
In Vitro Boyden Migration Assay
The transwell system of Costar (Lowell, MA), equipped with 8-μm-pore polyvinylpyrrolidone-free polycarbonate filters was used. Cells were loaded into the upper compartment (5 × 105 cells in 500 μl) in serum-free growth medium. The upper sides of the porous polycarbonate filters were coated with 50 μg/cm2 of reconstituted Matrigel basement membrane and placed into six-well culture dishes containing 1 ml of complete growth medium together with either 1 μg/ml Fc or ephrinA1-Fc. After 24 hours of incubation at 37°C, noninvading cells were removed mechanically using cotton swabs, and the microporous membrane was stained with DiffQuick solution. Chemotaxis was evaluated by counting the cells migrated to the lower surface of the polycarbonate filters (six randomly chosen fields, mean ± SD).
Metalloproteinase Zymography
Aliquots from media conditioned by PC3 cells and human fibrosarcoma HT1080 cells, used as positive control, were electrophoresed on 8% sodium dodecyl sulfate-polyacrylamide gels co-polymerized with 0.1% (w/v) type A gelatin. After electrophoresis, the gels were washed in 2.5% v/v Triton X-100 for 30 minutes to remove sodium dodecyl sulfate. Gelatin substrate gels were then incubated in 50 mmol/L Tris-HCl, pH 7.4, 200 mmol/L NaCl, and 5 mmol/L CaCl2 for 24 hours at 37°C. After incubation, the gels were stained with 0.1% Coomassie brilliant blue in acetic acid, methanol, and distilled water at a volume ratio of 1:2:3, respectively, for 60 minutes at room temperature. After destaining, the gels were immersed in distilled water and scanned immediately with QuantityOne Image Analysis software (Bio-Rad).
RhoA or Rac1 Activity Assay
PC3 cells were directly lysed in RIPA buffer, the lysates were clarified by centrifugation, and RhoA-GTP or Rac-GTP levels were quantified. Briefly, lysates were incubated with 10 μg of Rhotekin-GST fusion protein (Becton Dickinson, Franklin Lakes, New Jersey) or p21-activated kinase (PAK)-GST fusion protein, both absorbed on glutathione Sepharose beads for 1 hour at 4°C. Immunoreactive RhoA or Rac1 were then quantified by Western blot analysis. Lysates were normalized for RhoA or Rac1 content by immunoblot.
In Vivo Experimental Model for Bone Metastasis
Male CD1 nude mice, used for in vivo bone metastasis experiments, were purchased from Charles River (Milan, Italy). Mice were maintained under the guidelines established by our institution (University of L’Aquila, Medical School and Science and Technology School Board Regulations, complying with the Italian Government Regulation n.116, 27/1/1992). The procedure of heart injection of prostate cancer cells in nude mice has been previously described.28 Briefly, 1 × 105 cells in 0.1 ml of saline solution, were injected in the left ventricle of 4-week-old nude mice previously anesthetized with a mixture of ketamine (25 mg/ml)/xylazine (5 mg/ml). The development of tumor colonies in the whole skeletal apparatus was monitored at times by radiography using a Faxitron cabinet X-ray system (Faxitron X-ray Corp., Wheeling, IL). All animals were subjected to accurate necroscopy for the evaluation of the presence of tumor colonies in other anatomical sites.
Alu PCR
The quantification of human Alu sequences was previously described.29 Briefly, mice were sacrificed by carbon dioxide inhalation and the rear limbs were accurately deprived of surrounding soft tissues and opened with a surgical blade to expose the medulla. Bone fragments were incubated with 100 mmol/L Tris-HCl, pH 8.5, 0.5 mol/L ethylenediaminetetraacetic acid, 10% sodium dodecyl sulfate, 5 mol/L NaCl, 20 mg/ml proteinase K at 37°C for 12 hours. DNA was isolated by phenol/chloroform extraction, precipitated with ethanol, and suspended in 0.1 mol/L Tris-HCl, pH 8.0, 0.5 mol/L ethylenediaminetetraacetic acid. After spectrophotometric quantification, 2 ng of genomic DNA was analyzed by real-time PCR amplification using Stratagene MX3000P personal Q-PCR (M-Medical, Milano, IT) in the presence of SYBR Green. Quantification of human DNA was based on standard curve using genomic DNA extracted from PC3 cells.
Cell Growth Assay
Cells (105) were plated in triplicate directly onto 24-well cell culture dishes in the presence of complete medium. Cellular growth was stopped after 4 hours (To) and after 7 days in culture, by removing the medium and 0.5% crystal violet solution in 20% methanol was added. After staining for 5 minutes the fixed cells were washed with PBS and solubilized with 200 μl/well 0.1 mol/L sodium citrate, pH 4.2. The absorbance at 595 nm was evaluated using a microplate reader.
Statistical Analysis
All experiments were done at least in triplicate. Statistical analysis of the data were performed by Student’s t-test, P values ≤0.05 were considered statistically significant. Statistical analysis of in vivo experiments was performed using SPSS 11.0 software (SPSS, Inc., Chicago, IL). All statistical tests were two-tailed. Differences in the success rate between treatments were compared with χ2 test for 2 × 2 tables or Fisher’s exact test when the tables were too sparse.
Results
EphA2 Receptor Phosphorylation in Tumor Cells Is Regulated by Cell Density
EphA2 has already been reported to be overexpressed in prostate cancers30,31 and the expression of the kinase has been correlated with tumor aggressiveness.32 The strong correlation between EphA2 expression and prostate carcinoma aggressiveness prompted us to select an experimental model to study the role of EphA2 in the regulation of cell motility and invasiveness of prostate carcinoma cells. We therefore selected PC3 prostate tumor cells as a model because of their expression of both EphA2 and ephrinA1 ligand (Figure 1A). Prior studies have shown that co-expressed EphA receptors and ephrin-A ligands mediate opposing actions on growth cone navigation from distinct membrane domains. These results raise the possibility that cis- and trans-configurations of ligand/receptor proteins allow cells to use both Ephs and ephrins as functional guidance molecules within the same moving cell, raising both reverse and forward signaling.33 We observed that the expression of endogenous ephrinA1 ligand is increased by cell density (100% or 30% confluence, respectively, dense or sparse), whereas the amount of EphA2 kinase is unaffected (Figure 1, B and C). We therefore determined whether in PC3 cells EphA2 receptors are tyrosine phosphorylated by engaging endogenous ephrinAs in a cell-cell contact-dependent manner, and whether the level of EphA2 tyrosine phosphorylation can be further elevated by exogenous ephrinA1-Fc ligand. Cells were again plated with 100% or 30% confluence and stimulated with 1 μg/ml of exogenous ephrinA1-Fc ligand for 15 minutes. As shown in Figure 1D, the basal level of EphA2 receptor phosphorylation is not influenced by increased cell concentration, suggesting that the trans-stimulation via cell-cell contacts is ineffective in PC3 cells. On the contrary, the phosphorylation of EphA2 is strongly induced by exogenous ephrinA1-Fc ligand. In particular, high cell density reduces the exogenously-induced EphA2 phosphorylation, suggesting that endogenous ephrinA1 ligands may behave as a trans-acting and cell-cell contact-dependent factor, leading to desensitize EphA2. To focus our studies on forward trans-phosphorylation of EphA2 by the exogenous ligand, thus avoiding any contamination of the forward signaling with the reverse signaling, all of the following experiments were performed in low confluence cultures (≤30%).
Figure 1.

EphA2 phosphorylation level is cell density-dependent in PC3 cells. A: Lysates of PC3 and HEK293T cells were subjected to EphA2 and ephrinA1 immunoblot analyses. B and C: Monolayers of sparse (30% confluence) and dense (100% confluence) PC3 cells were lysed and anti-ephrinA1 (B) and anti-EphA2 (C) immunoblots were performed. An anti-actin immunoblot was performed for normalization. Bar graphs obtained from densitometry analysis of triplicate experiments are shown. §P < 0.005 sparse versus dense. D: Sparse (30% confluence) and dense (100% confluence) PC3 cells were serum-deprived for 24 hours and then treated with 1 μg/ml of ephrinA1-Fc for 15 minutes. EphA2 was immunoprecipitated and divided in two aliquots used for anti-phosphotyrosine (PY99) and anti-EphA2 immunoblots. A bar graph obtained from densitometry analysis of triplicate experiments is shown. *P < 0.001 stimulated sparse versus stimulated dense.
Phosphorylation-Defective Mutants Inhibit Endogenous Epha2 Kinase in Prostate Carcinoma
To test directly the role of EphA2 receptor phosphorylation/kinase activity, we cloned the human EphA2 cDNA and by site-specific mutagenesis we generated the following mutants: Y587F/Y593F in the juxtamembrane domain (DM); K645R a kinase-dead (KD) mutant in the ATP binding site; and the ΔCyto mutant, deleted of the entire cytoplasmic region (Figure 2A). Our assumption is that all these mutants may act as dominant-negative factors by contrasting and/or eliminating the trans-phosphorylation of receptor dimers, whereas the ΔCytoEphA2 leads to abrogation to both kinase-dependent and -independent function of EphA2. Preliminary studies on the EphA2-negative HEK293T cell line as recipient (Figure 1A), demonstrated that the ΔCytoEphA2, the DM, and the KD mutants exert a dominant-negative activity on both EphA2 and FAK phosphorylation (data not shown).
Figure 2.

EphA2 kinase-deficient mutants act as dominant-negative molecules in ephrinA1-stimulated PC3 cells. A: EphA2 kinase structure and mutants generated by site-specific mutagenesis. B: PC3 cells stably overexpressing wild-type (WT) EphA2 or kinase-defective mutants were serum-deprived for 24 hours and then treated with 1 μg/ml of ephrinA1-Fc for 15 minutes. EphA2 was immunoprecipitated and divided in two aliquots used for anti-phosphotyrosine (PY99) and anti-EphA2 immunoblots. A bar graph obtained from densitometry analysis of triplicate experiments is shown. *P < 0001 stimulated versus control; §P < 0.005 stimulated versus control. C: PC3 cells were treated as in A and 25 μg of total proteins were analyzed by anti-phospho-576-577-FAK immunoblot. The blot was then stripped and an anti-FAK immunoblot was performed for normalization. The bar graph obtained from densitometry analysis of triplicate experiments is shown. *P < 0.001 stimulated versus control; #P < 0.05 stimulated versus control.
Nevertheless EphA2 has been implied in prostate carcinogenesis, a role for both ligand-dependent tyrosine phosphorylation of EphA2 or for its simple overexpression have been entailed.16,49 To investigate the role of the tyrosine kinase-dependent and -independent signaling, driven by EphA2 in prostate carcinoma, we generated stable cell lines overexpressing EphA2 mutants in PC3 human prostate carcinoma cells, derived from bone metastasis and showing high endogenous EphA2 expression. We first analyzed the phosphorylation of EphA2 in response to ligand administration. Data show that the overexpression of wild-type EphA2 leads to a strong enhancement of ligand-independent tyrosine phosphorylation of EphA2 kinase, which is further increased by ephrinA1 administration (Figure 2B). The ΔCytoEphA2 mutant behaves as a strong dominant-negative for the endogenous receptor. The ΔCytoEphA2 mutant shares the ability to inhibit the phosphorylation of endogenous EphA2 with the two other EphA2 mutants DM-EphA2 and the KD-EphA2 (Figure 2B). These data suggest that the elimination of the kinase-dependent signaling of EphA2 by blocking functional dimerization of the receptor, leads to dramatic inhibition of endogenous EphA2 phosphorylation and activation.
To investigate the ability of these EphA2 mutants to interfere with endogenous EphA2 intracellular signaling leading to a motility response, we first analyzed the activation of FAK in response to ephrinA1 (Figure 2C). The results demonstrate again that the overexpression of wild-type EphA2 leads to a ligand-independent FAK activation, further enhanced by exogenous ephrinA1. In addition, the ΔCytoEphA2, the DM-EphA2, and the KD-EphA2 mutants strongly inhibit the activation of FAK by endogenous EphA2 kinase, although with different extent. These data support the idea that the removal of the kinase-dependent signaling of EphA2, by blocking efficient trans-phosphorylation of the receptor dimers, leads to remarkable inhibition of endogenous EphA2 receptors signaling to FAK.
Role of EphA2 Tyrosine Phosphorylation in Prostate Carcinoma Cell Proliferation, Adhesion, and Motility
To establish whether the expression of kinase-deficient mutants could affect cell proliferation, we first analyzed the proliferation of stably transfected PC3 cells with EphA2 mutants. As shown in Figure 3A, the different clones do not show any significant difference in their proliferation rate, suggesting that EphA2 expression/activation does not affect the regulation of cell proliferation. We have recently reported that in PC3 prostate carcinoma cells EphA2 activation elicits a motility response by activating the Src/FAK complex, leading a Rho-dependent actino/myosin contractility activation driving the retraction of cell body and cell migration.12,34 In addition, EphA2 activation inhibits integrin-mediated cell adhesion and spreading onto extracellular matrix (ECM).35,36 To investigate the role of tyrosine phosphorylation of EphA2 kinase in the adhesion and migration of ephrin-sensitive carcinomas, we again overexpressed the phosphorylation-defective mutants in PC3 cells. We first analyzed the effect of EphA2 mutants on cell adhesion onto fibronectin-coated dishes. As indicated in Figure 3B the adhering cells respond to ephrinA1 with a strong inhibition of their spreading, acquiring a round shape, and reducing their adhesive properties. The expression of the kinase defective mutants leads to the elimination of the sensitivity of the endogenous receptor to the ligand, thus supporting a kinase-dependent role of EphA2 in the regulation of ephrinA1 inhibition of ECM adhesion. We then analyzed cell de-adhesion from ECM and the formation of retraction fibers elicited by ephrinA1 (Figure 3C). The results indicate that the ΔCytoEphA2, the DM-EphA2, and the KD-EphA2 mutants strongly inhibit the retraction of cell body and the formation of retraction fibers induced by ligand, although the effect of the ΔCytoEphA2 is even evident and more marked.
Figure 3.

EphA2 phosphorylation is required for ephrinA1-mediated inhibition of ECM adhesion and spreading and for retraction fiber formation. A: Proliferation of PC3 cells transfected with wild-type (WT) and EphA2 mutants. Cells were plated in triplicate in complete medium. Cellular growth was stopped after 4 hours (To) and after 7 days in culture (T7). Cell proliferation was evaluated after crystal violet staining. B: Adhesion assay. PC3 cells expressing wild-type EphA2 or kinase-deficient mutants were kept in suspension for 30 minutes and then seeded onto fibronectin-coated dishes in the presence of 1 μg/ml of ephrinA1-Fc. After 4 hours photographs were taken. The bar graphs represent the number of adhering cells shown in six randomly chosen fields of triplicate experiments. *P < 0.001 stimulated versus control. C: Retraction fiber formation. PC3 cells overexpressing wild-type EphA2 or kinase-deficient mutants were seeded onto collagen-coated coverslips and adhesion was allowed for 24 hours. Cells were serum-starved for 24 hours before stimulating with 1 μg/ml of ephrinA1-Fc for 15 minutes. Confocal microscope analysis after phalloidin-tetramethyl-rhodamine isothiocyanate immunostaining was shown. The results are representative of at least three experiments. The bar graph represents the percentage of ephA1-stimulated cells with retraction fibers, in six randomly chosen fields of triplicate experiments. *P < 0.001 stimulated clones versus stimulated mock. Scale bar = 20 μm.
Eph ligands have been reported to influence cell migration instructing cells to hinder formerly engaged signals toward chemoattractive molecules and inducing a change of directional motility.37,38 Analysis of the motility response induced by ephrinA1 revealed that kinase-defective mutants severely impair the ability of ephrinA1 to inhibit chemotaxis toward serum both in wound healing and in Boyden assays (Figure 4, A and B). Expression of all mutants leads to a valuable ligand-independent response in both motility assays, suggesting a kinase-independent component of EphA2 effects on cell motility.
Figure 4.
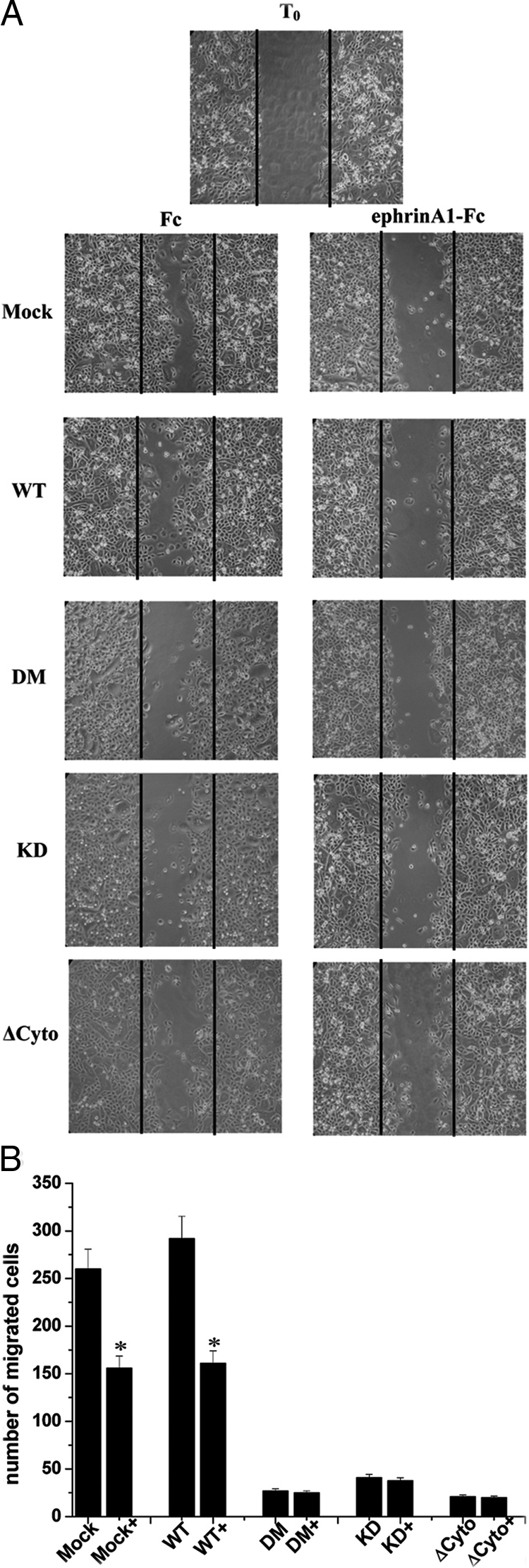
Kinase-dependent and -independent role of EphA2 in ephrinA1-mediated inhibition of directional migration. A: Wound-healing migration assay. Confluent PC3 cells, expressing wild-type (WT) EphA2 or kinase-deficient mutants were serum-starved for 24 hours, scratched with a tip, and a photograph was taken (T0). Complete medium was added to induce migration of cells in the wound, together with 1 μg/ml of ephrinA1-Fc or Fc alone, and after 24 hours photographs were taken. B: Boyden cell motility assay. PC3 cells expressing wild-type EphA2 or kinase-deficient mutants, after 24 hours of serum starvation, were seeded into the upper chamber of Boyden chambers. Cells were allowed to migrate for 24 hours toward the lower chamber filled with complete growth medium together with 1 μg/ml of ephrinA1-Fc. Cell migration was evaluated after crystal violet staining by counting cells in six randomly chosen fields. The results are representative of four experiments. *P < 0.001 stimulated versus control.
Altogether these results support the idea that tyrosine phosphorylation of EphA2 is a key determinant of the physiological response induced by ephrinA1 in prostate carcinoma cells, playing a critical function in the control of cell rounding, retraction fiber formation, inhibition of cytoskeleton spreading even if directional cell migration appears under the control of both kinase-dependent and -independent mechanisms.
Role of Epha2 Tyrosine Phosphorylation in Prostate Carcinoma Cell Invasion
EphA2 kinase has been correlated to the invasive properties of several metastatic cancers.3,39,40 We recently reported that ephrinA1 in PC3 cells induces an inhibition of Rac1 and an activation of RhoA small GTPases.12,35 These molecular events have been correlated with a proteolysis-independent style of invasion of ECM structures, known as amoeboid motility, widely used by several cancers to adjust their migratory strategies to environmental changes.41 To determine the role of EphA2 kinase activity in the regulation of RhoA and Rac1 GTPases, and ultimately in inducing an amoeboid-like motile phenotype, we again used the kinase-deficient EphA2 mutants. The results indicate that tyrosine phosphorylation of EphA2 is a key determinant of the morphological response induced by ephrinA1, correlated to both Rac1 inhibition and RhoA activation (Figure 5, A and B).
Figure 5.

Kinase-dependent and -independent role of EphA2 in ephrinA1-mediated motility response. A: PC3 cells, expressing wild-type (WT) EphA2 or kinase-deficient mutants, were serum-starved for 24 hours and then stimulated with either 1 μg/ml of ephrinA1-Fc or Fc for 15 minutes. Rac1-GTP was analyzed by a pull-down assay. The total amount of Rac-1 was then quantified by anti-Rac1 immunoblot and the bar graph obtained from densitometry analysis of triplicate experiments is shown. §P < 0.005 stimulated versus control; *P < 0.001 stimulated versus control. B: PC3 cells were treated as in A and a RhoA activity assay was performed. The total amount of RhoA was then quantified by anti-RhoA immunoblot and the bar graph obtained from densitometry analysis of triplicate experiments is shown. *P < 0.001 stimulated versus control; §P ≤ 0.005 stimulated versus control; ‡P ≤ 0.01 stimulated versus control. C: Boyden cell invasion assay. PC3 cells expressing wild-type EphA2 or kinase-deficient mutants, after 24 hours of serum starvation, were seeded into the upper chamber of Boyden chambers precoated with a Matrigel three-dimensional milieu. Cells were allowed to migrate for 24 hours toward the lower chamber filled with complete growth medium together with 1 μg/ml of ephrinA1-Fc. Cell invasion was evaluated after crystal violet staining by counting cells in six randomly chosen fields. The results are representative of four experiments. *P < 0.001 stimulated versus control. D: Analysis of MMP activity. PC3 cells expressing wild-type EphA2 or kinase-deficient mutants, were serum-deprived for 24 hours and then stimulated with 1 μg/ml of ephrinA1-Fc. After 24 hours the growth medium was collected and analyzed by gelatin zymography. The clear bands represent areas of gelatinase activity. The results are representative of four experiments.
To determine whether blocking of EphA2 kinase-dependent signaling in prostate carcinoma cells could affect metastatic invasion, PC3 cells expressing mutant forms of EphA2 proteins were analyzed for their invasion across a Matrigel barrier (Figure 5C). The results reveal that the ΔCytoEphA2, the DM-EphA2, and the KD-EphA2 mutants, even if with different extent, strongly inhibit the effect of EphA2 activation in PC3 invasion, obviously implicating that kinase-dependent EphA2 signaling is a key determinant of invasion for ephrin-sensitive prostate carcinomas. Again we observed a ligand-independent effect of invasion attributable to expression of all mutants, thereby suggesting the existence of a kinase-autonomous mechanism. In addition, the expression of all mutants does not influence MMPs regulation, as revealed by gelatin zymography (Figure 5D), confirming that regulation of invasion by EphA2 is independent from MMP expression control.
To exclude cell line-specific effects, we investigated whether the expression of EphA2 is able to influence the invasive ability of other prostate cancer cell lines. We selected two prostate carcinoma cell lines derived from lymph node (LNCaP) and brain (DU-145) metastasis and PNT1A cells from prostate normal epithelium. The results show a strong correlation between EphA2 expression and invasive properties (see Supplemental Figure S1, A and B, at http://ajp.amjpathol.org). We then selected the DU-145 cell line, which expresses the highest level of EphA2, to confirm that the overexpression of the phosphorylation-defective mutants inhibits the invasive ability of EphA2-positive cells (see Supplemental Figure S1C at http://ajp.amjpathol.org). These results underline that the ability of EphA2 kinase-deficient mutants to reduce cell invasion is a common feature of EphA2-positive cells.
EphA2 Kinase-Deficient Mutants Reduce Metastatic Potential of Prostate Carcinoma Cells
Antimetastatic potential of EphA2 mutants was evaluated using a rodent model of bone metastasis. The injection of 1 × 105 PC3 cells in the left ventricle of nude mice resulted in the formation of bone and visceral tumor colonies. In agreement with previous experiments, we selected an endpoint of 4 weeks after heart injection. Necroscopic and radiographical analyses revealed the presence of osteolytic lesions in the epiphyses of posterior limbs as prevalent sites of tumor colonies outgrowth. To evaluate the reduction in bone metastasis outcome, we obtained total body radiography of randomly selected mice, divided into five groups of eight animals each. Serial radiographies were performed from starting 36 days after cell injection and radiographical films were digitally scanned considering positive mice those with at least one osteolytic zone (Figure 6A). The experiment was repeated twice according to the same protocol. Mice treated with EphA2 mutants did not show any significant loss of weight or any distress sign. The results shown in Figure 6B reveal that expression of wild-type EphA2 causes an increase in percentage of bone tumor colonies from 50 to 75%, whereas kinase-deficient EphA2 mutants show a decrease in their development. Interestingly, the ΔCyto mutant shows the higher antimetastatic effect. Histological analyses of metastatic tibias revealed the presence of tumor masses in bone medulla proximal to the bone resorption zones (Figure 6C). To evaluate if the presence and the extension of osteolytic lesions were associated to tumor cell growth, a quantitative analysis of human Alu sequence in selected tibias was performed. An excellent correlation (R2 = 0.92) between Alu signal and the area of osteolytic lesions was observed, suggesting that the limiting factor in the formation of radiographical bone metastasis was the colonization by tumor cells (Figure 6D). In keeping with these data, we did not observe any effect of EphA2 expression/activation on in vitro cell proliferation (Figure 3A). Our results indicate that wild-type EphA2 increases, whereas kinase-deficient mutants decrease, the time of insurgence of bone tumor colonies, being again the ΔCyto mutant the most efficient (Figure 6E). When considering also lung, lymph nodes, and mediastine colonies (Figure 6F), the antimetastatic activity of EphA2 mutants results even more evident, leading ΔCyto mutant to completely abrogate extra-bone tumor colonies outgrowth, thus supporting a key role of both kinase-dependent and -independent function of EphA2.
Figure 6.

Role of EphA2 in the formation of metastasis in an experimental model for prostate cancer spreading. A: The growth of PC3 cells in bone was monitored by serial radiography (days 0, 36, 44, and 52 after cell injection). The images show a representative serial radiography of the tibia of the same mouse in which the progression of bone lytic lesions (indicated by white arrows) is evident. B: Percentage of mice revealing evidence of at least one bone osteolytic lesion. Experimental groups were obtained by injecting PC3 cells stably transfected with the kinase-defective mutants in the heart of mice. C: Representative images from the same tibia showing the area of lytic lesion and the histological localization of tumor cells. D: Correlation analysis of human DNA and area of lytic burden in tibias from mice of all experimental groups. E: The timing of the first detection of bone lytic lesions in the different experimental groups. The radiographs were performed at days 36, 44, and 52 (arrows) and values are expressed as percentages. F: Percentage of mice with evidence of at least one nonbone metastatic growth as determined by necroscopic analysis (*P < 0.05). Scale bar = 1 mm.
Discussion
Typical neoplastic transformation by RTKs involves increased receptor autophosphorylation and tyrosine kinase activity. The abnormally high level of tyrosine kinase activity of ErbB2/Neu, causally linked to its oncogenic potential, is only a prototypical example.42,43 Nevertheless EphAs are nonclassical receptors and for many of them, including EphA2, the available literature is controversial. They can elicit a reverse signaling in cells bearing the ligand ephrin As, which is independent from tyrosine phosphorylation. Nevertheless the forward signaling, elicited in cells conveying the Eph As, is performed by transmembrane kinase receptors, some indications suggest that some EphA functions may be transduced independently from their kinase activity. Indeed, several proofs argue for a phosphorylation-independent role of EphA2 in oncogenic transformation.21,22,23,24 For example, EphA6, EphA7, and EphB1 have kinase-deficient alternative splicing variant forms,44 whereas EphB6 and EphA10 have kinase domains with defective catalytic activity.45,46 These receptors are able to elicit a biological response, thus supporting the idea that some Eph functions do not need receptor kinase activity. EphA8 regulates adhesion to the ECM in a kinase-independent manner,47 and EphB3 elicits a kinase-dependent de-adhesive response although its effect on cell migration is kinase-independent.27 siRNA knockdown of EphA2 in pancreatic cancer cells inhibits tumor growth and metastasis,48 supporting the idea of a key role for is simple overexpression in cancer progression and malignancy. Moreover, EphA2 kinase receptor is under-phosphorylated in some human breast cancer cell lines, although the unphosphorylated receptor still retains its kinase activity49 and its overexpression induces transformation in mammary epithelium.5
In addition to these data presenting Eph as nonclassical RTK, evidence for a strict dependence from their kinase activity are spread over the whole Eph family. High levels of EphA2 phosphorylation have been reported in xenografts of ASPC-1, U87MG, and SVP cell lines and in human mammary carcinoma cells.11,18 In addition, studies using the 4T1 model of tumorigenesis have found that blocking EphA2 receptor activation through EphA2-Fc results in a decrease in phosphorylation that was concurrent with decreased tumor volume.50 In agreement, emerging evidence suggests that PTPs are involved in regulating Eph-mediated responses of nontransformed and tumor cells, strongly supporting a role for Eph kinase activity. Indeed, low molecular weight-PTP (LMW-PTP) dephosphorylation regulates EphB2-mediated endothelial capillary-like assembly and adhesion51 as well as the motile response of carcinoma cells.34 Src homology phosphatase-2 is rapidly and transiently recruited to ephrinA1-activated EphA2 and is implicated in the loss of integrin-mediated cell adhesion19 and both EphB2 and EphA4 are negatively controlled by protein tyrosine phosphatase receptor type O.21
Our data contribute to the idea that several features of prostate cancer cell motility and invasion are mainly dependent from the integrity of kinase activity of EphA2. Indeed, we herein report that disruption of phosphorylation/activation of EphA2 leads to: i) abrogation of ephrinA1-mediated cell rounding, retraction fiber formation, and de-adhesion from ECM; ii) severe inhibition of the FAK-mediated motility response and ability to seal injuries; iii) inhibition of EphA2-mediated invasion, through a Rho-dependent and a MMP-independent mechanism; and iv) inhibition of bone and visceral tumor outgrowth. We used as tools two independent kinase-deficient mutants: the KD, which maintains all of the characteristic of wild-type EphA2, but on overexpression, is unable to trans-phosphorylate the dimerized endogenous wild-type receptor; and the DM, lacking the two juxtamembane tyrosines, serving as a negative control for several RTKs against ligand-independent activation.9,52 As a consequence both these mutants are unable to elicit the trans-phosphorylation step required to achieve the full activation state of RTKs. In addition, we used a mutant deleted in the whole intracellular portion of EphA2, the ΔCytoEphA2. On overexpression, the ΔCytoEphA2 mutant is likely to dimerize with endogenous wild-type EphA2s, inhibiting both kinase-dependent and -independent responses. The deletion of the intracellular portion of RTKs has been indicated as serving as a dominant-negative mutation for several RTKs, including Ephs, in cell culture models.11,53 KD mutants may behave as dominant-negative as indicated by c-Src dominant-negative, mutated in the ATP binding site.54 Our data indicate that both kinase-defective mutants (KD and DM) and the ΔCyto mutant behave as dominant-negative molecules, although at different extent.
Here we report a complete dependence from kinase activity of the ability of EphA2 to inhibit integrin-mediated cell adhesion and cytoskeleton spreading, which is similarly inhibited by kinase-deficient and ΔCyto mutants. These data are in keeping with the observation of a key role of PTP-mediated dephosphorylation in the control of de-adhesive properties of EphA2, implicating both SHP2 and LMW-PTP phosphatases in this feature.19,34 Conversely EphA2-mediated directional cell motility is both kinase-dependent and -independent, in agreement with the available literature on Ephs. Indeed, Parri and colleagues34 reported a role of LMW-PTP-mediated dephosphorylation of EphA2 in the control of wound healing and retraction fiber formation, although Miao and colleagues27 found that the directional control of cell migration exerted by EphB3 is kinase-independent. Our observation that the regulation of prostate carcinoma cell invasion and metastasis formation by EphA2 is dependent on both kinase and nonkinase mechanisms, is a novel finding. Although LMW-PTP is able to regulate tumor progression by regulating EphA2 dephosphorylation,55 further data on the role of dephosphorylation by PTPs in the control of EphA2-mediated invasion and metastasis spread are still lacking and are therefore highly warranted to confirm our observation.
Prostate carcinomas have a strong propensity to metastasize to bone sites, although they may give rise to lung and lymph node metastases. Although expression of Ephs has been correlated with bone homeostasis,56 data supporting a specific targeting of EphA2-expressing prostate cancers to bone are lacking. Our data indicate that intracardiac injection of EphA2-kinase defective mutants produces a decrease in both bone and visceral tumor colonies. The effect of ΔCyto mutant is more pronounced than kinase-deficient EphA2s, leading to a stronger decrease of tumor colonies and to complete abrogation of visceral ones. These results propose a role of EphA2 in bone targeting, but suggest that the kinase-independent mechanisms elicited by EphA2 particularly specify for visceral targeting.
The role of Ephs overexpression in invasive tumors is far to be understood. One open question is whether Eph implication in carcinogenesis is really linked to its nature of motility factor. The recent key advances that have challenged the view of cancer cell motility and invasion indicate as a key milestone that cancer cells display a particular plasticity in motility style, shifting on different opportunities from mesenchymal migration, based on the generation of a path through proteolysis of barriers, to amoeboid motility, based on the capacity to squeeze into gaps in the ECM.41 In contrast to mesenchymal migration, amoeboid-like migration across a three-dimensional environment involves a fast squeezing movement across ECM proteins, without their MMP-based proteolytic degradation, the inhibition of both cell-cell and cell-ECM interactions, the inhibition of Rac-induced cytoplasmic protrusion and the activation of a Rho-dependent cell body contraction to permit rounded cells to squeeze forward.41,57 In prostate carcinoma cells, ligand-induced activation of EphA2 leads to a strong induction of a motility style resembling the amoeboid-like. Indeed, EphA2 activation induces an inhibition of Rac1, an activation of RhoA, the release of integrin-mediated constraints and de-adhesion, the contraction of cell body, and the achievement of a rounded shape. Finally EphA2 activation does not affect MMP regulation. Interestingly, both kinase-dependent and -independent mechanisms are determinant for EphA2-mediated invasive and metastatic phenotype. In conclusion our data suggest that ephrin-sensitive carcinomas may achieve an invasive advantage through the activation of EphA2 signaling. This event causes a shift toward amoeboid-like motility, allowing these cancer cells to adapt to environmental changes adjusting their invasive strategy.
Supplementary Material
Footnotes
Address reprint requests to Paola Chiarugi, Department of Biochemical Sciences, University of Florence, viale Morgagni 50, 50134 Florence, Italy. E-mail: paola.chiarugi@unifi.it.
Supported by the Italian Association for Cancer Research, the Fondazione Italiana Per La Ricerca Sul Cancro (fellowship to M.L.T.), the Interuniversity Biotecnology Consortium, the Ente Cassa di Risparmio di Firenze, and the Tuscany Regional Project TRESOR.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- Kiyokawa E, Takai S, Tanaka M, Iwase T, Suzuki M, Xiang YY, Naito Y, Yamada K, Sugimura H, Kino I. Overexpression of ERK, an EPH family receptor protein tyrosine kinase, in various human tumors. Cancer Res. 1994;54:3645–3650. [PubMed] [Google Scholar]
- Kinch MS, Carles-Kinch K. Overexpression and functional alterations of the EphA2 tyrosine kinase in cancer. Clin Exp Metastasis. 2003;20:59–68. doi: 10.1023/a:1022546620495. [DOI] [PubMed] [Google Scholar]
- Dodelet VC, Pasquale EB. Eph receptors and ephrin ligands: embryogenesis to tumorigenesis. Oncogene. 2000;19:5614–5619. doi: 10.1038/sj.onc.1203856. [DOI] [PubMed] [Google Scholar]
- Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61:2301–2306. [PubMed] [Google Scholar]
- Shin D, Garcia-Cardena G, Hayashi S, Gerety S, Asahara T, Stavrakis G, Isner J, Folkman J, Gimbrone MA, Jr, Anderson DJ. Expression of ephrinB2 identifies a stable genetic difference between arterial and venous vascular smooth muscle as well as endothelial cells, and marks subsets of microvessels at sites of adult neovascularization. Dev Biol. 2001;230:139–150. doi: 10.1006/dbio.2000.9957. [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders DM, Fang WB, Hicks DJ, Zhuang G, Shyr Y, Chen J. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. FASEB J. 2005;19:1884–1886. doi: 10.1096/fj.05-4038fje. [DOI] [PubMed] [Google Scholar]
- Guo H, Miao H, Gerber L, Singh J, Denning MF, Gilliam AC, Wang B. Disruption of EphA2 receptor tyrosine kinase leads to increased susceptibility to carcinogenesis in mouse skin. Cancer Res. 2006;66:7050–7058. doi: 10.1158/0008-5472.CAN-06-0004. [DOI] [PubMed] [Google Scholar]
- Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:464–471. doi: 10.1038/nrm1399. [DOI] [PubMed] [Google Scholar]
- Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Pasqualini R, Lindberg RA, Kain R, Freeman AL, Pasquale EB. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 2000;19:6043–6052. doi: 10.1038/sj.onc.1204004. [DOI] [PubMed] [Google Scholar]
- Parri M, Buricchi F, Giannoni E, Grimaldi G, Mello T, Raugei G, Ramponi G, Chiarugi P. EphrinA1 activates a Src/focal adhesion kinase-mediated motility response leading to rho-dependent actino/myosin contractility. J Biol Chem. 2007;282:19619–19628. doi: 10.1074/jbc.M701319200. [DOI] [PubMed] [Google Scholar]
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Ligation of EphA2 by ephrin A1-Fc inhibits pancreatic adenocarcinoma cellular invasiveness. Biochem Biophys Res Commun. 2004;320:1096–1102. doi: 10.1016/j.bbrc.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Holland SJ, Gale NW, Mbamalu G, Yancopoulos GD, Henkemeyer M, Pawson T. Bidirectional signalling through the EPH-family receptor Nuk and its transmembrane ligands. Nature. 1996;383:722–725. doi: 10.1038/383722a0. [DOI] [PubMed] [Google Scholar]
- Henkemeyer M, Orioli D, Henderson JT, Saxton TM, Roder J, Pawson T, Klein R. Nuk controls pathfinding of commissural axons in the mammalian central nervous system. Cell. 1996;86:35–46. doi: 10.1016/s0092-8674(00)80075-6. [DOI] [PubMed] [Google Scholar]
- Cheng N, Brantley DM, Liu H, Lin Q, Enriquez M, Gale N, Yancopoulos G, Cerretti DP, Daniel TO, Chen J. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol Cancer Res. 2002;1:2–11. [PubMed] [Google Scholar]
- Fang WB, Brantley-Sieders DM, Parker MA, Reith AD, Chen J. A kinase-dependent role for EphA2 receptor in promoting tumor growth and metastasis. Oncogene. 2005;24:7859–7868. doi: 10.1038/sj.onc.1208937. [DOI] [PubMed] [Google Scholar]
- Dobrzanski P, Hunter K, Jones-Bolin S, Chang H, Robinson C, Pritchard S, Zhao H, Ruggeri B. Antiangiogenic and antitumor efficacy of EphA2 receptor antagonist. Cancer Res. 2004;64:910–919. doi: 10.1158/0008-5472.can-3430-2. [DOI] [PubMed] [Google Scholar]
- Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- Shintani T, Ihara M, Sakuta H, Takahashi H, Watakabe I, Noda M. Eph receptors are negatively controlled by protein tyrosine phosphatase receptor type O. Nat Neurosci. 2006;9:761–769. doi: 10.1038/nn1697. [DOI] [PubMed] [Google Scholar]
- Birgbauer E, Cowan CA, Sretavan DW, Henkemeyer M. Kinase independent function of EphB receptors in retinal axon pathfinding to the optic disc from dorsal but not ventral retina. Development. 2000;127:1231–1241. doi: 10.1242/dev.127.6.1231. [DOI] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- Grunwald IC, Korte M, Wolfer D, Wilkinson GA, Unsicker K, Lipp HP, Bonhoeffer T, Klein R. Kinase-independent requirement of EphB2 receptors in hippocampal synaptic plasticity. Neuron. 2001;32:1027–1040. doi: 10.1016/s0896-6273(01)00550-5. [DOI] [PubMed] [Google Scholar]
- Kullander K, Mather NK, Diella F, Dottori M, Boyd AW, Klein R. Kinase-dependent and kinase-independent functions of EphA4 receptors in major axon tract formation in vivo. Neuron. 2001;29:73–84. doi: 10.1016/s0896-6273(01)00181-7. [DOI] [PubMed] [Google Scholar]
- Holmberg J, Clarke DL, Frisen J. Regulation of repulsion versus adhesion by different splice forms of an Eph receptor. Nature. 2000;408:203–206. doi: 10.1038/35041577. [DOI] [PubMed] [Google Scholar]
- Gu C, Park S. The p110 gamma PI-3 kinase is required for EphA8-stimulated cell migration. FEBS Lett. 2003;540:65–70. doi: 10.1016/s0014-5793(03)00223-0. [DOI] [PubMed] [Google Scholar]
- Miao H, Strebhardt K, Pasquale EB, Shen TL, Guan JL, Wang B. Inhibition of integrin-mediated cell adhesion but not directional cell migration requires catalytic activity of EphB3 receptor tyrosine kinase. Role of Rho family small GTPases. J Biol Chem. 2005;280:923–932. doi: 10.1074/jbc.M411383200. [DOI] [PubMed] [Google Scholar]
- Angelucci A, Gravina GL, Rucci N, Festuccia C, Muzi P, Vicentini C, Teti A, Bologna M. Evaluation of metastatic potential in prostate carcinoma: an in vivo model. Int J Oncol. 2004;25:1713–1720. [PubMed] [Google Scholar]
- Schneider T, Osl F, Friess T, Stockinger H, Scheuer WV. Quantification of human Alu sequences by real-time PCR—an improved method to measure therapeutic efficacy of anti-metastatic drugs in human xenotransplants. Clin Exp Metastasis. 2002;19:571–582. doi: 10.1023/a:1020992411420. [DOI] [PubMed] [Google Scholar]
- Walker-Daniels J, Coffman K, Azimi M, Rhim JS, Bostwick DG, Snyder P, Kerns BJ, Waters DJ, Kinch MS. Overexpression of the EphA2 tyrosine kinase in prostate cancer. Prostate. 1999;41:275–280. doi: 10.1002/(sici)1097-0045(19991201)41:4<275::aid-pros8>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Wang XD, Reeves K, Luo FR, Xu LA, Lee F, Clark E, Huang F. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: rationale for patient selection and efficacy monitoring. Genome Biol. 2007;8:R255. doi: 10.1186/gb-2007-8-11-r255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng G, Hu Z, Kinch MS, Pan CX, Flockhart DA, Kao C, Gardner TA, Zhang S, Li L, Baldridge LA, Koch MO, Ulbright TM, Eble JN, Cheng L. High-level expression of EphA2 receptor tyrosine kinase in prostatic intraepithelial neoplasia. Am J Pathol. 2003;163:2271–2276. doi: 10.1016/S0002-9440(10)63584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt T, Shirasaki R, Ghosh S, Andrews SE, Carter N, Hunter T, Pfaff SL. Coexpressed EphA receptors and ephrin-A ligands mediate opposing actions on growth cone navigation from distinct membrane domains. Cell. 2005;121:127–139. doi: 10.1016/j.cell.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Parri M, Buricchi F, Taddei ML, Giannoni E, Raugei G, Ramponi G, Chiarugi P. EphrinA1 repulsive response is regulated by an EphA2 tyrosine phosphatase. J Biol Chem. 2005;280:34008–34018. doi: 10.1074/jbc.M502879200. [DOI] [PubMed] [Google Scholar]
- Buricchi F, Giannoni E, Grimaldi G, Parri M, Raugei G, Ramponi G, Fiaschi T, Chiarugi P. Redox regulation of ephrin/integrin cross talk. Cell Adhes Migr. 2007;1:33–42. [PMC free article] [PubMed] [Google Scholar]
- Deroanne C, Vouret-Craviari V, Wang B, Pouyssegur J. EphrinA1 inactivates integrin-mediated vascular smooth muscle cell spreading via the Rac/PAK pathway. J Cell Sci. 2003;116:1367–1376. doi: 10.1242/jcs.00308. [DOI] [PubMed] [Google Scholar]
- Davy A, Soriano P. Ephrin signaling in vivo: look both ways. Dev Dyn. 2005;232:1–10. doi: 10.1002/dvdy.20200. [DOI] [PubMed] [Google Scholar]
- Holmberg J, Frisen J. Ephrins are not only unattractive. Trends Neurosci. 2002;25:239–243. doi: 10.1016/s0166-2236(02)02149-5. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Kato H, Fukuchi M, Nakajima M, Kuwano H. EphA2 overexpression correlates with poor prognosis in esophageal squamous cell carcinoma. Int J Cancer. 2003;103:657–663. doi: 10.1002/ijc.10860. [DOI] [PubMed] [Google Scholar]
- Saito T, Masuda N, Miyazaki T, Kanoh K, Suzuki H, Shimura T, Asao T, Kuwano H. Expression of EphA2 and E-cadherin in colorectal cancer: correlation with cancer metastasis. Oncol Rep. 2004;11:605–611. [PubMed] [Google Scholar]
- Friedl P. Prespecification and plasticity: shifting mechanisms of cell migration. Curr Opin Cell Biol. 2004;16:14–23. doi: 10.1016/j.ceb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Madhusudan S, Ganesan TS. Tyrosine kinase inhibitors in cancer therapy. Clin Biochem. 2004;37:618–635. doi: 10.1016/j.clinbiochem.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Segatto O, King CR, Pierce JH, Di Fiore PP, Aaronson SA. Different structural alterations upregulate in vitro tyrosine kinase activity and transforming potency of the erbB-2 gene. Mol Cell Biol. 1988;8:5570–5574. doi: 10.1128/mcb.8.12.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisch AH, Pasquale EB. The Eph family: a multitude of receptors that mediate cell recognition signals. Cell Tissue Res. 1997;290:217–226. doi: 10.1007/s004410050926. [DOI] [PubMed] [Google Scholar]
- Gurniak CB, Berg LJ. A new member of the Eph family of receptors that lacks protein tyrosine kinase activity. Oncogene. 1996;13:777–786. [PubMed] [Google Scholar]
- Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 2002;27:514–520. doi: 10.1016/s0968-0004(02)02179-5. [DOI] [PubMed] [Google Scholar]
- Gu C, Park S. The EphA8 receptor regulates integrin activity through p110gamma phosphatidylinositol-3 kinase in a tyrosine kinase activity-independent manner. Mol Cell Biol. 2001;21:4579–4597. doi: 10.1128/MCB.21.14.4579-4597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. EphA2: a determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene. 2004;23:1448–1456. doi: 10.1038/sj.onc.1207247. [DOI] [PubMed] [Google Scholar]
- Zantek ND, Azimi M, Fedor-Chaiken M, Wang B, Brackenbury R, Kinch MS. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. 1999;10:629–638. [PubMed] [Google Scholar]
- Brantley DM, Cheng N, Thompson EJ, Lin Q, Brekken RA, Thorpe PE, Muraoka RS, Cerretti DP, Pozzi A, Jackson D, Lin C, Chen J. Soluble Eph A receptors inhibit tumor angiogenesis and progression in vivo. Oncogene. 2002;21:7011–7026. doi: 10.1038/sj.onc.1205679. [DOI] [PubMed] [Google Scholar]
- Stein E, Lane AA, Cerretti DP, Schoecklmann HO, Schroff AD, Van Etten RL, Daniel TO. Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genes Dev. 1998;12:667–678. doi: 10.1101/gad.12.5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner S, Wybenga-Groot LE, Warner N, Lin H, Pawson T, Forman-Kay JD, Sicheri F. A change in conformational dynamics underlies the activation of Eph receptor tyrosine kinases. EMBO J. 2006;25:4686–4696. doi: 10.1038/sj.emboj.7601315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren NK, Lu M, Freeman AL, Koolpe M, Pasquale EB. Interplay between EphB4 on tumor cells and vascular ephrin-B2 regulates tumor growth. Proc Natl Acad Sci USA. 2004;101:5583–5588. doi: 10.1073/pnas.0401381101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroiher M, Miller MA, Steele RE. Deceiving appearances: signaling by “dead” and “fractured” receptor protein-tyrosine kinases. Bioessays. 2001;23:69–76. doi: 10.1002/1521-1878(200101)23:1<69::AID-BIES1009>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Chiarugi P, Taddei ML, Schiavone N, Papucci L, Giannoni E, Fiaschi T, Capaccioli S, Raugei G, Ramponi G. LMW-PTP is a positive regulator of tumor onset and growth. Oncogene. 2004;23:3905–3914. doi: 10.1038/sj.onc.1207508. [DOI] [PubMed] [Google Scholar]
- Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4:111–121. doi: 10.1016/j.cmet.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Brocker EB, Friedl P. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.