

Abstract
Injury or impaired clearance of apoptotic cells leads to the pathological accumulation of necrotic corpses, which induce an inflammatory response that initiates tissue repair1. In addition, antigens present within necrotic cells can sometimes provoke a specific immune response2-4 and it has been argued that necrosis could explain adaptive immunity in seemingly infection-free situations, such as after allograft transplantation or in spontaneous and therapy-induced tumour rejection5, 6. In the mouse, the CD8α+ subset of dendritic cells (DC) phagocytoses dead cell remnants and crossprimes CD8+ T cells against cell-associated antigens7. Here, we show that CD8α+ DC utilise CLEC9A (DNGR-1), a recently-characterised C-type lectin8-10, to recognise a preformed signal that is exposed on necrotic cells. Loss or blockade of CLEC9A does not impair uptake of necrotic cell material by CD8α+ DC but specifically reduces crosspresentation of dead cell-associated antigens in vitro and decreases the immunogenicity of necrotic cells in vivo. The function of CLEC9A requires a key tyrosine residue within its intracellular tail that allows recruitment and activation of the tyrosine kinase Syk, which is also essential for crosspresentation of dead cell-associated antigens. Thus, CLEC9A functions as a Syk-coupled C-type lectin receptor to mediate sensing of necrosis by the principal DC subset involved in regulating crosspriming to cell-associated antigens.
Keywords: DNGR-1, CLEC9A, C-type lectin, dendritic cells, necrosis, CTL, crosspriming
We and others have recently found that the clec9a gene encoding the CLEC9A protein, also known as Dendritic cell NK lectin Group Receptor-1 (DNGR-1), is selectively expressed at high levels by the CD8α+ subset of DC and its putative human equivalent8-10. To identify ligand(s) for CLEC9A, we constructed a chimera comprising the extracellular domain of CLEC9A fused to CD3ζ8 and expressed it in the T cell lines BWZ and B3Z11. Ligand binding to the chimeric receptor triggers signalling via ZAP-70, leading to activation of an NFAT reporter in those cells11. As a control, we first checked that bivalent antibodies (but not monovalent Fab fragments – see below) to CLEC9A can trigger CLEC9A-ζ, in soluble form or after immobilisation on plastic surfaces (Supplementary Fig. 1 and reference 8). Interestingly, we observed basal activation of the NFAT reporter in the reporter cell lines expressing mouse or human CLEC9A-ζ (but not expressing a control ζ chimera made with the related C-type lectin, Dectin-1), which correlated with the number of dead cells in the culture (Fig. 1a). Notably, addition of UV-irradiated mouse embryonic fibroblasts (MEFs) to the reporter cells induced signalling by mouse or human CLEC9A-ζ but not Dectin-1-ζ and this was blocked by Fab fragments of anti-mouse or anti-human CLEC9A, respectively, but not by a control Fab fragment (Fig. 1b). Although cells entered apoptosis shortly after UV treatment, they did not trigger CLEC9A-ζ signalling until 4-7.5 h post-irradiation and the signal correlated with the frequency of cells that had undergone secondary necrosis during that time period, as assessed by permeability to DNA stains such as propidium iodide and TO-PRO-3 (Fig. 1c). All cell types tested, independently of species of origin, induced mouse and human CLEC9A-ζ signalling after UV-induced secondary necrosis (data not shown), indicating that the ligand is neither species nor cell type restricted. As an independent means of measuring ligand expression, we made a recombinant version of the C-type lectin domain (CTLD) of CLEC9A, which contains the putative ligand binding site. Tetramers of CLEC9A CTLD, but not control tetramers of Dectin-1 CTLD, stained the secondary necrotic (TO-PRO-3+) fraction of MEFs that had been UV-irradiated (Fig. 1d). In contrast, Dectin-1 tetramers, but not CLEC9A tetramers, stained zymosan particles (Fig. 1d), consistent with the role of Dectin-1 as a β-glucan-specific receptor12. CLEC9A ligand exposure was seen upon treatment with additional primary or secondary necrosis-inducing protocols, including freeze-thawing, treatment with anthracyclins (mitoxantrone) or serum deprivation, and was directly proportional to the extent of TO-PRO-3 permeability induced by the treatment (Fig. 1e). Notably, cell fixation with formaldehyde, especially when followed by detergent permeabilization, rendered the cells permeable to TO-PRO-3 and instantly revealed the ligand(s) for CLEC9A (Fig. 1f-h). Further analysis revealed that the ligand(s) is predominantly cytoplasmic (Fig. 1g) and resistant to glycosidase and nuclease treatment but susceptible to the action of proteases, heat and low pH (Fig. 1h). We conclude that CLEC9A recognises a ubiquitous preformed acid-labile protein-associated ligand(s) that is normally sequestered in healthy cells but becomes exposed during necrosis, upon loss of membrane integrity.
Figure 1. Ligand(s) for CLEC9A are exposed upon cell death.
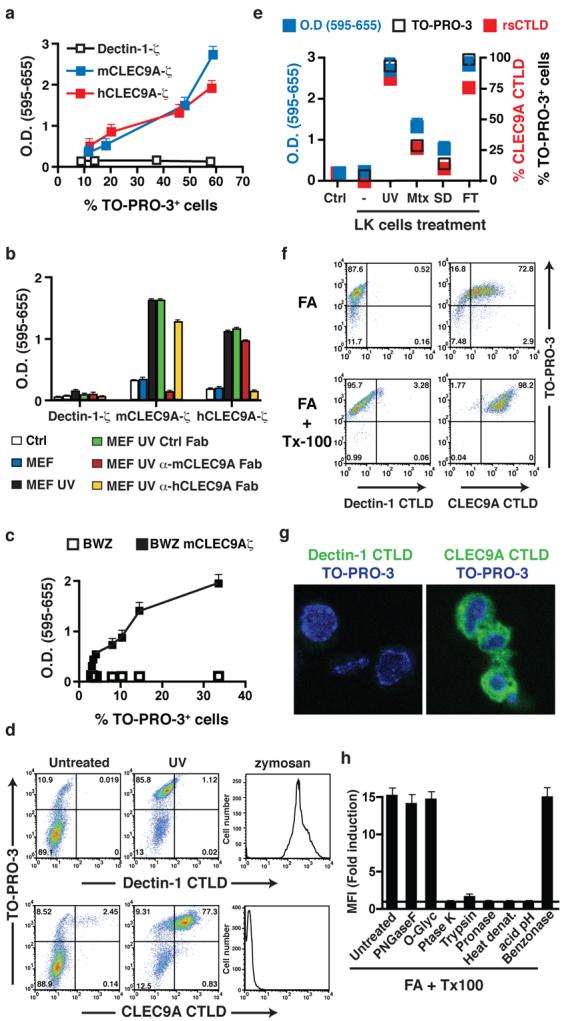
a, BWZ cells stably expressing mouse or human CLEC9A-ζ or Dectin-1-ζ were plated at different concentrations and allowed to grow for 48h. The frequency of TO-PRO-3+ cells was determined and equivalent numbers of live cells were transferred to fresh medium. After overnight culture, LacZ activity was measured using a colorimetric assay and expressed relative to the frequency of TO-PRO-3+ cells. b, Untreated or UVC-treated MEFs were cultured with BWZ cells expressing CLEC9A-ζ or Dectin-1-ζ. Where indicated, Fab fragments of control or anti-mCLEC9A or anti-hCLEC9A antibodies were added. Reporter activity in BWZ cells was measured as in (a). Ctrl., BWZ cells alone. c, LK cells were exposed to increasing doses of UVC before culturing with BWZ cells expressing, or not, mCLEC9A-ζ. LacZ activity in BWZ cells was measured as in (a) and is expressed as a function of the percentage of TO-PRO-3+ LK cells at each dose of radiation. d, PE-labelled tetramers of CLEC9A or Dectin-1 CTLD were used to stain untreated or UVC-treated MEFs or zymosan particles. e, BWZ cells expressing mouse CLEC9A were cultured overnight in medium alone (Ctrl) or together with LK cells that had been untreated (−) or treated 16h before with UVC (UV) or mitoxantrone (Mtx), or cultured overnight without serum (serum deprivation; SD) or subjected to freeze / thawing (FT). LacZ activity (left y axis) is depicted, together with the frequency of TO-PRO-3+ and CLEC9A CTLD tetramer+ LK cells at the beginning of the co-culture (right y axis). f, CLEC9A ligand is a preformed signal. LK cells were fixed with formaldehyde and permeabilized, or not, with Triton-X-100 before labelling with TO-PRO-3 and PE-tetramers of Dectin-1 or CLEC9A CTLD. g, Fixed and permeabilized MEFs were labelled with Dectin-1 or CLEC9A CTLD monomers and counterstained with TO-PRO-3 before confocal microscopy. h, Fixed and permeabilised MEFs were left untreated or were treated with acid (pH 3.5), peptido N-glycosidase F (PNGase F), O-Glycosidase (O-Glyc), proteinase K (Ptase K), trypsin, pronase-E or benzonase, or were heated for 5 min at 80°C. Cells were then labelled with Dectin-1 or CLEC9A CTLD tetramers and analysed by flow cytometry. Data are ratio between the mean fluorescence intensity (MFI) of the CLEC9A CTLD staining and the Dectin-1 CTLD staining for each of the treatments. a-h, data are from one representative experiment of at least three. All error bars are shown in a-c, e, h and represent mean ± SD of triplicate measurements.
To test for a role of CLEC9A in mediating responses to necrotic cells, we generated clec9agfp/gfp mice in which CLEC9A expression is abrogated by insertion of a gene encoding a membrane anchored form of GFP into the clec9a open reading frame (Supplementary Fig. 2a). CD8α+ DC from these mice, as well as the CD8α+ DC-like (CD24hi CD11blo B220neg; reference 13) subset from Flt3L-derived bone marrow cell cultures (Flt3L-BMDC), did not stain for CLEC9A but expressed GFP (Supplementary Fig. 2b, c and Supplementary Fig. 3a). All other leucocytes were GFP-negative (Supplementary Fig. 2b and data not shown), consistent with the restricted expression of high levels of the clec9a gene to CD8α+ DC8, 10. DC phenotype and numbers were unaltered in clec9agfp/gfp mice indicating that CLEC9A is not essential for DC development (Supplementary Fig. 2b-d) and the response of DC to innate stimuli was not altered by the loss of CLEC9A (Supplementary Fig. 2f and Supplementary Fig. 3b). No alterations were found in leucocyte numbers or frequencies in spleen, lymph node or thymus (Supplementary Fig. 2b-e and data not shown) and the clec9agfp/gfp mice appeared normal and did not develop obvious symptoms of autoimmune disease (data not shown).
We first tested whether CLEC9A is necessary for engulfment of dead cells. Using a conventional flow cytometry assay, CLEC9A+ and CLEC9A-deficient CD8α+-like DC from Flt3L-BMDC cultures were seen to acquire equivalent amounts of material from dye-labelled secondary necrotic splenocytes over a similar time period (Fig. 2a and Supplementary Fig. 4). We confirmed that CLEC9A-deficient CD8α+-like DC are not impaired in their ability to phagocytose necrotic cell material in vitro using multispectral imaging flow cytometry, which differentiates between surface binding and particle internalisation (Fig. 2b). Importantly, CLEC9A deficiency also did not impair uptake of cellular material by spleen CD8α+ DC in vivo following injection of necrotic cells into mice (Fig. 2c). We therefore assessed whether CLEC9A might be necessary for steps subsequent to necrotic cell ingestion, culminating in crosspresentation of dead cell-associated antigens to CD8+ T cells. UV-treated congenic (H-2bm1) transformed MEFs untransfected (bm1 T) or transfected to express a non-secreted ovalbumin (OVA)-GFP fusion protein (bm1 T OVA) were allowed to undergo secondary necrosis and were then added to control (CLEC9A+) or clec9agfp/gfp CD8α+-like DC purified from Flt3L-BMDC cultures, together with OVA-specific OT-I TCR transgenic CD8+ T cells. Remarkably, CLEC9A-deficient CD8α+-like DC displayed a reduced ability to support OT-I expansion and differentiation into IFN-γ-producing effector cells, a readout for crosspresentation of OVA (Fig. 2d, e). In contrast, when the antigen was offered in the form of soluble protein or as a conjugate to latex beads, CLEC9A-deficient CD8α+-like DC were comparable stimulators to CLEC9A+ DC indicating that CLEC9A deficiency does not intrinsically impair crosspresentation ability (Fig. 2d, e). We conclude that CLEC9A is not required for uptake of necrotic cell material but is necessary for efficient crosspresentation of dead cell associated antigens by CD8α+ DC.
Figure 2. CLEC9A is required for crosspresentation of dead cell-associated antigens in vitro.
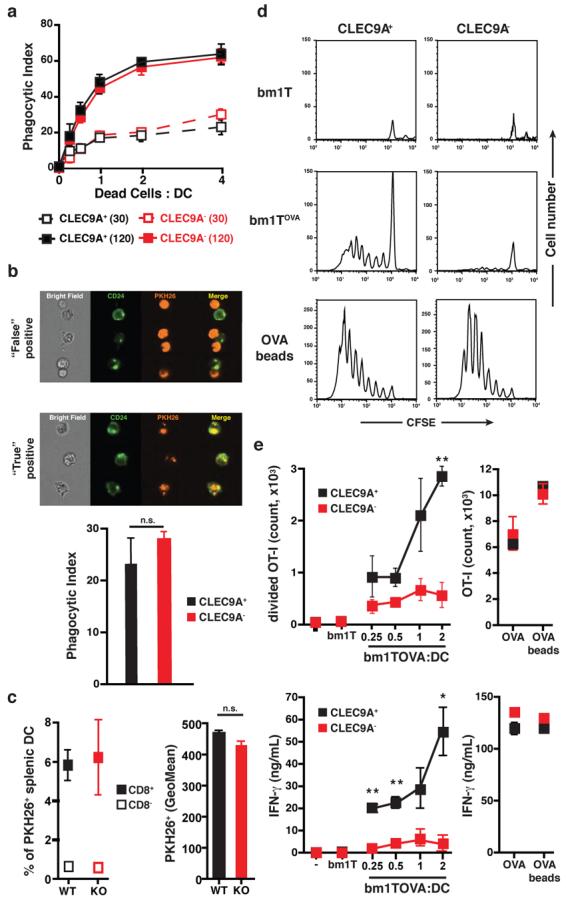
a-b, Flt3L BMDC from clec9agfp/gfp (CLEC9A−) or control (CLEC9A+) mice were incubated at 4°C or 37°C for 0, 30 or 120 min with PKH26-labelled and UVC-treated H-2bm1 splenocytes at different ratios. (a) Acquisition of PKH26 label by the CD8α+-like DC subset was quantified by flow cytometry and normalised by subtracting the signal obtained at 4°C from the one obtained at 37°C. Data are mean ± SD of two biological replicates. (b) Uptake of dead cell material by CD8α+-like DC was determined using multispectral imaging flow cytometry to discriminate between “true” (uptake) and “false” (binding) positive events. Results are mean ± SD of two independent biological replicates from one experiment and are representative of two independent experiments. c, clec9agfp/gfp (KO) or control clec9a+/+ or clec9aegfp/+ (WT) mice were injected with PKH26-labelled UVC-treated H-2bm1 splenocytes. 2h later, the frequency (left) and mean fluorescence (right) of CD8α− and CD8α+ splenic DC that acquired dead cell material was determined by flow cytometry (left panel). Results are the mean ± SD of three mice. Differences between WT and KO in (a-c) are not statistically significant. d-e, Purified CD8α+-like Flt3L BMDC from clec9aegfp/egfp (CLEC9A−) or CLEC9A+ littermates were cultured with CFSE-labelled OVA-specific OT-I cells and UVC-treated H-2bm1 MEFs expressing (bm1 T OVA), or not (bm1 T), OVA. OT-I proliferation (d and e, top panel) and IFN-γ in the supernatant were quantified (e, bottom panel). Soluble OVA and OVA-coated beads were used as a control source of antigen. Results are mean ± SEM of three independent biological replicates from one experiment and are representative of three independent experiments. *, p<0.05, **, p<0.01 and ***, p<0.001 (Student's t test).
The CLEC9A cytoplasmic tail contains a hemITAM motif14 with a tyrosine at position 7, which allows binding of Syk kinase upon phosphorylation (reference 9 and Supplementary Fig. 5a). Consistent with those data, mAb-mediated crosslinking of CLEC9A induced phosphorylation of endogenous Syk in a CLEC9A-transfected B cell line (Supplementary Fig. 5b) and promoted activation of NFAT in B3Z cells, which was dependent on the co-expression of Syk and the presence of Tyr7 in CLEC9A (Supplementary Fig. 5c and Supplementary Fig. 6). Similarly, necrotic cells triggered CLEC9A signalling in a Syk and Tyr7-dependent fashion (Supplementary Fig. 7a, b) independently of phagocytosis (data not shown). To address the role of signalling in DC, we first assessed if Syk is activated in response to necrotic cells. CD8α+-like DC incubated with dead cells showed phosphorylated Syk at the interface between the DC and the corpse, which was significantly decreased when using CLEC9A deficient cells (Fig. 3a). We then reconstituted DC from clec9agfp/gfp mice with wild type CLEC9A or the Y7F signalling-deficient mutant by retroviral transduction. Ectopic expression of the wild type receptor restored the ability of CD8α+-like DC to efficiently crosspresent dead cell-associated OVA, confirming that the deficiency observed in the CLEC9A-deficient cells is not due to a secondary defect unrelated to CLEC9A ablation (Fig. 3b, c). In contrast, cells transduced with the signalling-deficient receptor behaved as CLEC9A-negative control cells transduced with a virus lacking the insert and showed a profound deficiency in crosspresentation of dead cell-associated OVA (Fig. 3b, c). In all cases, DC cross-presented bead-associated OVA with equal efficiency (Fig. 3b, c), again indicating the selectivity of CLEC9A for dead cell-associated antigens. We therefore assessed the ability of Syk-deficient DC to crosspresent such antigens. As for CLEC9A-deficiency, loss of Syk did not impair uptake of dead cells by CD8α+-like DC (Supplementary Fig. 8a) but blocked crosspresentation of dead-cell associated OVA, but not soluble OVA, to OT-I T cells (Supplementary Fig. 8b and data not shown – see reference 15). We conclude that CLEC9A signalling via Syk kinase is required for crosspresentation of dead cell-associated antigens by CD8α+ DC.
Figure 3. CLEC9A signalling is necessary for efficient cross-presentation of dead cell-associated antigens.
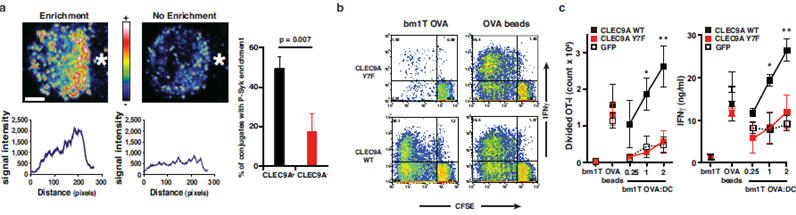
a, CLEC9A-dependent enrichment for phospho-Syk at the contact area between DC and dead cells. Quantification of conjugates showing phospho-Syk enrichment at the contact area between CD8α+-like DC and dead cell is shown as mean± SEM of three independent experiments. b-c, CLEC9A signalling is required for efficient cross-presentation of dead cell-associated material to CD8+ T cells. Bone marrow cells from CLEC9A− mice were retrovirally transduced with a vector encoding only for GFP (GFP) or encoding additionally for the wild-type (CLEC9A WT) or a mutant form (CLEC9A Y7F) of CLEC9A. After culture in the presence of Flt3L, GFP+ CD8α+-like Flt3L BMDC were purified by cell sorting and co-cultured with UVC-treated H-2bm1 MEFs expressing (bm1T OVA), or not (bm1T), OVA in the presence of CFSE-labelled OVA-specific OT-I cells. OT-I proliferation and IFN-γ production were measured. OVA-coated beads were used as a control source of antigen. b, one representative experiment out of three. c, Data are mean ± SEM of three independent experiments. *, p<0.05 and **, p<0.01 two way ANOVA.
Finally, to assess the importance of CLEC9A in crosspresentation of necrotic cell-associated antigens in vivo, we used a crosspriming model that relies on the intrinsic immunogenicity of dead cells to induce CTL responses to associated antigens2-4, 16-18. We immunised clec9agfp/gfp mice and littermate controls with UV-treated bm1 T OVA cells undergoing secondary necrosis and measured anti-OVA responses six days later. In CLEC9A+ mice, these dead cells induced robust CD8+ T cell crosspriming as measured by OVA /H-2Kb tetramer staining or by the production of IFN-γ in response to ex vivo spleen cell restimulation with OVA peptide (Fig. 4a-c). Notably, crosspriming was significantly diminished in clec9agfp/gfp mice when compared to littermate controls across all litters tested (Fig. 4a-c). Finally, to exclude the possibility that CLEC9A-deficient mice have an altered T cell repertoire, we sought to confirm these results in wild type animals. We found that soluble anti-CLEC9A mAbs block the ability of dead cells to trigger mouse CLEC9A signalling (Supplementary Fig. 7) and, therefore, treated C57BL/6 mice with anti-CLEC9A mAb and measured crosspriming to dead cells expressing OVA. Anti-CLEC9A but not an isotype-matched control antibody reduced crosspriming to bm1 T OVA (Fig. 4d, e and Supplementary Fig. 9). The effect of antibody blockade was consistently more marked than that of genetic ablation (Fig. 4 and data not shown), perhaps because of redundancy in CLEC9A deficient mice, incomplete backcrossing to the C57BL/6 strain and/or because the antibody promotes clearance of a complex of CLEC9A and other receptors involved in crosspriming to dead cell-associated antigens. We conclude that CLEC9A is necessary in vivo for efficient crosspriming to antigens associated with dead cells.
Figure 4. CLEC9A is necessary for crosspriming to dead cell-associated antigens in vivo.
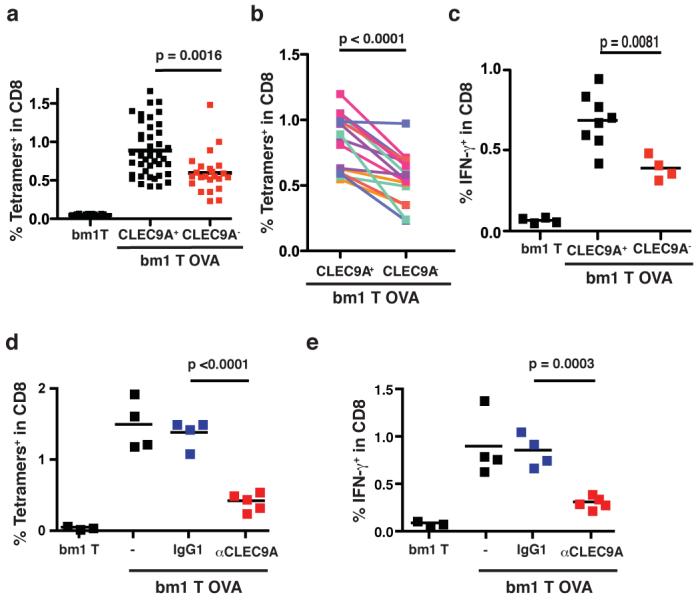
a-c, clec9agfp/gfp (CLEC9A−) or control clec9a+/+ or clec9agfp/+ (CLEC9A+) littermates were immunised i.v. with 7.5×105 UVC-treated bm1T OVA MEFs. Six days later, the frequency of OVA-specific CD8+ T cells was determined in the spleen (a-b). a, each dot corresponds to an individual mouse; the data are pooled from seven independent experiments. b, the average response in each litter for clec9agfp/gfp or control CLEC9A+ littermates. Lines link sex-matched (female) littermates. c, IFN-γ production in response to OVA peptide restimulation ex vivo. Data indicate frequency of IFN-γ+ cells within the CD8+ T cell population. Individual mice and average for one representative experiment (out of three) is shown. d-e, C57BL/6 mice untreated or treated with anti-CLEC9A antibody or an isotype-matched control (IgG1) were immunised i.v. with 7.5×105 UVC-treated bm1T OVA MEFs. The frequency of OVA-specific CD8+ T cells (d) or of IFN-γ producing CD8+ T cells in response to OVA peptide restimulation (e) was measured six days later. Data show individual mice and average response from three different litters. (a-e). p values were determined using Student's t test (paired for b)
Here we describe CLEC9A as a CD8α+ DC-expressed receptor for necrotic cells that regulates crosspresentation of dead cell-associated antigens through signalling via Syk kinase. Despite the fact that CLEC9A is an endocytic receptor8, 9, it does not appear to function in particle phagocytosis9 and, like Syk, is dispensable for uptake of necrotic cell fragments. Rather, CLEC9A and Syk play a non-redundant role at a subsequent step(s) required for crosspresentation of dead cell-associated antigens. We find that antibodies against CLEC9A are not delivered to lysosomal compartments upon endocytosis, unlike antibodies to DEC-205 (ref. 19 and Supplementary Fig. 10a), and that CLEC9A co-localises exclusively with the non-lysosomal fraction of necrotic cell material internalised by CD8α+ DC (Supplementary Fig. 10b). Thus, we favour a model in which CLEC9A, like the mannose receptor20, diverts cargo away from lysosomal compartments to allow retrieval of antigens for cross-presentation. As CLEC9A appears to be an activating receptor9, 10, similar to Dectin-121, the CLEC9A/Syk pathway may additionally act to activate DC in response to dead cells, either autonomously22 or in collaboration with toll-like receptors18, 23, 24. Notably, as predicted by the “danger” model5, CLEC9A is involved in translating innate recognition of dead cells into adaptive immunity following recognition of a preformed cellular ligand(s) that becomes exposed upon necrosis. Nevertheless, we cannot exclude the possibility that CLEC9A is also involved in crosstolerance to dead cell-associated antigens. Whichever the case, downstream of CLEC9A, our data additionally implicate the Src family kinase / Syk axis as a mediator of dead cell recognition by the innate immune system. Notably, orthologues of Src and Syk mediate clearance of apoptotic and necrotic cells by Drosophila glial cells25 and another Syk-coupled C-type lectin was recently shown to promote inflammatory responses to necrotic cells26. Thus, CLEC9A may be one of a family of Syk-coupled receptors that tap into an evolutionarily ancient mechanism for recognising cell death.
METHODS SUMMARY
Detailed descriptions of the methods are provided as Supplementary Information
Mice
Generation of CLEC9A knock-out mice is described in the Supplementary Methods. clec9a+ and clec9agfp/gfp mice were on a mixed 129/Sv × C57BL/6 genetic background at the third generation of backcrossing to C57BL/6. All experiments and analyses were carried out using littermate comparisons.
Cross-presentation in vitro and in vivo
H-2bm1 MEFs were immortalized with SV40 T large antigen (bm1 T MEFs) and transduced to express a truncated and non-secreted OVA-GFP fusion protein. Before the assay, MEFs were UVC-irradiated and cultured overnight in complete medium to induce secondary necrosis and are referred to as UVC-treated cells in the figure legends. For cross-presentation assays in vitro, UVC-treated bm1 T OVA MEFs were cultured with purified CD8α+-like Flt3L BMDC and CFSE-labelled OVA-specific OT-I T cells. OT-I responses were quantified four days later by analysing IFN-γ staining and CFSE-dilution profiles by flow cytometry and by monitoring IFN-γ in supernatants. For in vivo experiments, UVC-treated bm1 T OVA MEFs were injected i.v. into clec9agfp/gfp or control CLEC9A+ littermates or, alternatively, C57BL/6 mice pre-treated with an i.p. injection of PBS, 400 μg isotype control (rat IgG1) or 1F6 anti-CLEC9A. CD8+ T cell responses were measured six days later by quantitating the number of H-2Kb-OVA tetramer positive cells and by IFN-γ production in response to OVA peptide restimulation.
Supplementary Material
Acknowledgements
This work was funded by Cancer Research UK. D.S. was supported by an EMBO long-term fellowship (ALTF 336-2004) and by a Marie Curie Intra-European Fellowship within the 6th European Community Framework Programme (MEIF-CT-2005-009205). We thank Edina Schweighoffer and Victor Tybulewicz for fetal liver from syk−/− embryos. We are grateful to members of the Immunobiology Laboratory, Cancer Research UK, for advice and discussions and the Biological Resources staff for animal care and assistance with mouse experiments.
Footnotes
Supplementary Information accompanies the paper on Nature's website (http://www.nature.com/nature).
The authors declare no competing financial interests.
References
- 1.Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 3.Shi Y, Zheng W, Rock KL. Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc. Natl. Acad. Sci. U.S.A. 2000;97:14590–14595. doi: 10.1073/pnas.260497597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sauter B, et al. Consequences of Cell Death. Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 6.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 7.Villadangos J, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7:543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 8.Sancho D, et al. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest. 2008;118:2098–2110. doi: 10.1172/JCI34584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huysamen C, Willment JA, Dennehy KM, Brown GD. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J. Biol. Chem. 2008;283:16693–16701. doi: 10.1074/jbc.M709923200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caminschi I, et al. The dendritic cell subtype restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood. 2008 doi: 10.1182/blood-2008-05-155176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc. Natl. Acad. Sci. U.S.A. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 13.Naik S, et al. Cutting edge: generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J. Immunol. 2005;174:6592–6597. doi: 10.4049/jimmunol.174.11.6592. [DOI] [PubMed] [Google Scholar]
- 14.Robinson M, Sancho D, Slack E, LeibundGut-Landmann S, Reis e Sousa C. Myeloid C-type lectins in innate immunity. Nat Immunol. 2006;7:1258–1265. doi: 10.1038/ni1417. [DOI] [PubMed] [Google Scholar]
- 15.LeibundGut-Landmann S, Osorio F, Brown GD, Reis e Sousa C. Dendritic cell stimulation via the Dectin-1 / Syk pathway allows priming of cytotoxic T cell responses. Blood. 2008;112:4971–4980. doi: 10.1182/blood-2008-05-158469. [DOI] [PubMed] [Google Scholar]
- 16.Casares N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obeid M, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2006;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 18.Apetoh L, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 19.Mahnke K, et al. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J Cell Biol. 2000;151:673–684. doi: 10.1083/jcb.151.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burgdorf S, Kautz A, Böhnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. 2007;316:612–616. doi: 10.1126/science.1137971. [DOI] [PubMed] [Google Scholar]
- 21.Leibundgut-Landmann S, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 22.Janssen E, et al. Efficient T cell activation via a Toll-Interleukin 1 Receptor-independent pathway. Immunity. 2006;24:787–799. doi: 10.1016/j.immuni.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 23.Kim H, et al. Toll-like Receptor 2 Senses beta-Cell Death and Contributes to the Initiation of Autoimmune Diabetes. Immunity. 2007;27:321–333. doi: 10.1016/j.immuni.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Cavassani KA, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziegenfuss JS, et al. Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature. 2008;453:935–939. doi: 10.1038/nature06901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamasaki S, et al. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. 2008;9:1179–1188. doi: 10.1038/ni.1651. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.