

Summary
Multidrug efflux (MDR) pumps remove a variety of compounds from the cell into the external environment. There are five different classes of MDR pumps in bacteria, and quite often a single bacterial species expresses multiple classes of pumps. Although under normal circumstances MDR pumps confer low-level intrinsic resistance to drugs, the presence of drugs and mutations in regulatory genes lead to high level expression of MDR pumps that can pose problems with therapeutic treatments. This review focuses on the resistance nodulation cell division (RND)-class of MDR pumps that assemble from three proteins. Significant recent advancement in structural aspects of the three pump components has shed new light on the mechanism by which the tripartite efflux pumps extrude drugs. This new information will be critical in developing inhibitors against MDR pumps to improve the potency of prescribed drugs.
Keywords: Drug resistance, RND-type multidrug efflux pumps, tripartite pump assembly, proton/drug antiporter, membrane fusion protein, outer membrane protein factor
Introduction
Multidrug efflux transporters provide a means by which bacteria can confer intrinsic, low-level resistance to a diverse group of antibiotics. Expression of these transporters is regulated by the antibiotics they remove from the cell; consequently, a high expression of multidrug efflux transporters confers a multidrug resistance (MDR) phenotype, thus posing a serious therapeutic problem.
Bacterial multidrug efflux transporters [1] can be divided into five major structural families: (1) resistance nodulation cell division (RND), (2) major facilitator superfamily (MFS), (3) small multidrug resistance (SMR), (4) multidrug and toxic compound extrusion (MATE), and (5) ATP-binding cassette (ABC). Energetically, RND, MFS, and SMR are H+/drug antiporters, MATE pumps are Na+/drug antiporters, while ATP hydrolysis is linked to drug transport in ABC transporter family. The first four groups are also known as secondary transporters, as they use the pre-stored energy of chemical gradients across the membrane, while the ABC transporters are directly coupled with energy generation.
In this review we will focus on RND multidrug transporters, which are the major contributors of a MDR phenotype in Gram negative bacteria.
It is thought that the functional pumps with the participation of the RND multidrug transporters typically assemble from three proteins: an inner membrane protein, which is providing the energy for the transport and hence is often referred to as the RND “pump” protein, an outer membrane protein (OMF), which connects up with IMP in the periplasm providing a continuous drug conduit to help remove drugs out of the cell into the medium, and an adapter protein, often referred to as the membrane fusion protein (MFP), which, through its direct interactions with RND and OMF, helps assemble and stabilize the tripartite drug efflux complex. All three components of the RND drug efflux complex are essential for expelling drugs from the cell.
The bulk of our present knowledge concerning the structure-function and assembly of RND multidrug transporters has come from studies carried out on two major tripartite efflux transporters, AcrAB-TolC and MexAB-OprM of Escherichia coli and Pseudomonas aeruginosa, respectively. Here, AcrA and MexA are MFPs, AcrB and MexB are RND proteins, and TolC and OprM are OMFs. (Acr and Mex are acronyms for acriflavin and multiple efflux, respectively, while Tol and Opr refer to tolerance to colicin and outer membrane protein, respectively). Though AcrAB and MexAB are primarily studied for their roles in multidrug efflux, their other less well-studied roles, which are outlined in a recent review [2], include quorum sensing [3] and metabolite disposal [4]. Similarly, TolC, besides facilitating colicin E1 import [5,6], interacts with a type I protein secretion machinery, compromised of HlyBD, to secrete hemolysin [7,8] as well as with other non-canonical ABC-transporters such as the macrolide extrusion pump MacAB [9,10], which has also recently been implicated in enterotoxin secretion [11].
During the last decade, an extraordinary amount of progress has been made concerning the structure, biochemistry and genetics of RND multidrug transporters. We will briefly review these studies and focus on the mechanism of drug extrusion through the RND-type efflux pumps. Readers are encouraged to check out several excellent recent reviews on this topic [12,13,14,15,16,17].
OMF structure
The crystal structures of TolC [18,19,20] and OprM [21] were first to be solved. In addition, a structure of a related OMF from Vibrio cholerae, known as VceC is also available [22]. Though TolC, OprM and VceC share little homology at the amino acid sequence level (in the case of TolC vs VceC, as little as 10% identity), they fold into a remarkably similar three-dimensional homotrimeric structure with an outer membrane-embedded β-barrel domain and a large α-helical domain extending 100 Å into the periplasm [Fig. 1].
Fig. 1.
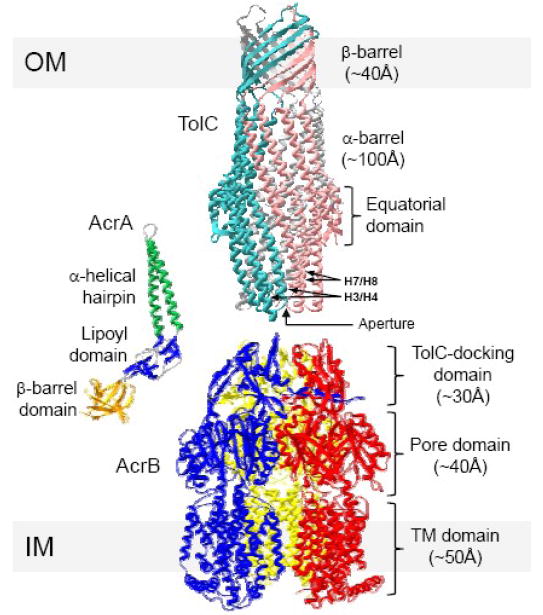
Structures of TolC (1EK9), AcrA (2F1M) and AcrB (2GIF) and their domains. Note that TolC and AcrB are shown as homotrimers, AcrA as a monomer. Locations of the static H3/H4 and mobile H7/H8 helices from two different TolC protomers are shown. OM and IM refer to the outer membrane and the inner membrane, respectively.
The β-barrel domain resembles the porin fold, however it is distinctly different in that, unlike the porins, where the barrel is formed by only one subunit, the TolC family achieves the same while all three protomers contribute 4 β-strands to form a pseudo-continuous barrel [18]. Similar to the porins however, the inclination of the barrel is to the right (at about 55° relative to the membrane plane) and it is formed by antiparallel β-strands.
Each of the TolC protomers is itself a product of internal gene duplication, manifesting itself in a structural repeat, which effectively gives the TolC assembly a pseudo-sixfold symmetry. Each “half-protomer” is formed by five helices and an antiparallel β-hairpin, which when assembled provide a full complementation of a 12-stranded β-barrel and the α-helical domain. As demonstrated clearly in the case of OprM [21] the N-terminus of the OMF presents a flexible tail, which is often lipidated and inserted in the outer membrane. The precise N-termini are either absent or disordered in the TolC and VceC structures.
A peculiar feature of the TolC family is the periplasmic domain, which is also a pseudo-continuous structure built with the participation of all three protomers. Unlike the β-barrel domain, the periplasmic domain is almost exclusively α-helical, and the helices are inclined to the left, following a sharp kink in the junction between the two domains of around 110°. Thus the orientation of the chain alters dramatically at the junction, giving the each individual protomer a sickle-like appearance [18,21]. The α-helical domain is composed of 12 helical fragments, which form a unique anti-parallel helical assembly, which was coined as “alpha-helical barrel” by Calladine et al. [23] with two long continuous pairs – H3 and H7, being complemented by four discontinuous ones, forming supercoiled pairs with the continuous ones in the following fashion: H3 + (H2/H4), and H7 + (H6/H8). The short helices H1 and H5 form a part of the so-called equatorial subdomain and connect the N-terminal tail to the first helical pair and serve as a linker between the two internal structural repeats respectively.
In order to keep the superhelical trajectories on the surface of a cylindrical barrel, some notable structural adjustments have taken place in the helical packing, causing the helices to run at an angle relative to the axis of the barrel and adjustment to the classical “knobs into holes” helical interfaces [23]. This peculiar packing puts stringent spatial restraints on the size and shape of the side chains which could occupy the knobs positions, and this is associated with a relative axial translation of the paired helices. Due to the pseudo-continuous nature of the intra- and inter-protomer interfaces, each helix packs laterally with two lateral neighbors, creating two different helical sequence patterns which have to match these unequal interfaces.
Due to the requirements for this specific helical packing on the surface of the alpha-barrel, one of the helices in the superhelical pair follows a roughly traditional superhelical trajectory, while the second one is severely curved, appearing to be under structural tension. As a result, the inner, tensed, helices from each protomer form triplets, which are stabilized by ion-bridges, effectively closing the periplasmic end of the channel [18,21] [Fig. 2]. Disruption of the ion bridges which maintain this structure result in relaxation of the superhelical trajectories of the corresponding helices, resembling an iris-like mechanism, leading to a dilation of the periplasmic entrance of the channel as supported by structural and functional studies [20,24] [Fig. 2].
Fig. 2.
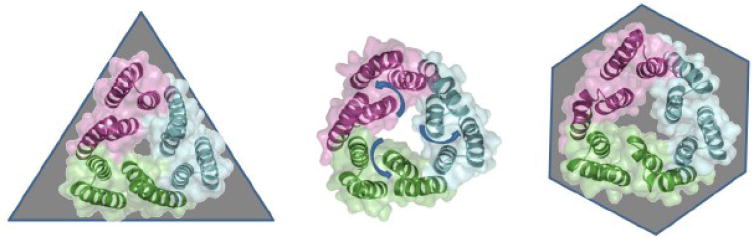
Views of the periplasmic aperture of the currently available TolC trimer structures reveal a possible transition to a fully open state. Structures of the closed state (1EK9; left), and partially open mutant structures 2VDE (center) and 2VDD (right) reveal a rearrangement of the periplasmic tip helices consistent with the proposed “aperture opening” mechanism. In the initial state (left) the aperture is maintained closed by inter-protomer salt-bridges, which are thought to become destabilized and released upon the interaction of the OMF with the RND. The following transition is also associated with a breakage of the central three-fold symmetry of the trimer and relaxation of the gating helical pairs (arrows) resulting in a roughly hexagonal arrangement (right).
MFP structure
MFPs are lipoproteins that are anchored to the inner membrane with their lipidated N-terminal ends, exposing the bulk of their structures into the periplasm [Fig. 1]. Crystal structures of AcrA and MexA, lacking the extreme N- and C-terminal information, show that both proteins fold into very similar three-dimensional structures comprised of three distinct linearly-arranged domains: a β-barrel domain comprised of six anti-parallel β-strands and a short α-helix, a central lipoyl domain assembled from two interlocking lipoyl motifs each composed of 4 β-strands, separated by an α-helical hairpin domain, which is slight larger in AcrA (105 Å) than in MexA (89 Å). The available genetic and biochemical data show that the highly flexible hairpin domain of MFPs interacts with the periplasmic helices of OMFs, while the β-barrel domain interacts with their cognate RND proteins (see below).
To date 3 crystallographic structures of the membrane fusion proteins are available – the P. aeruginosa MexA protein (residues ~20-300), which was crystallized in P21 space group independently by Akama et al. [25] and Higgins et al. [26], and the AcrA structure (residues 45-312) in P212121 orthorhombic space group with 4 copies per asymmetric unit [27]. While highly similar in appearance, the MexA features a shorter hairpin than AcrA. In both proteins the C-terminal part which is thought to form a mini-domain was not present and it is the only structural element of the tripartite assembly that awaits its discovery. This region is also thought to be involved in AcrB interaction [28,29] and the lack of structural information about it is hampering more comprehensive docking simulations. In addition to the aforementioned structures, successful crystallization of another membrane fusion protein, namely MacA, has been reported recently [30], and its crystal structure (deposited in the PDB databases as 3FPP) is going to extend our understanding of oligomerization and structural diversity in membrane fusion proteins.
RND structure
Multiple crystal structures of AcrB are available [31,32,33,34,35,36] and are described in great depth in a later section. Briefly, AcrB homotrimers fold into an α-helical transmembrane domain, and a large soluble periplasmic domain, the crown of which is thought to make a direct contact with TolC [Fig. 1].
Compositional stoichiometry of the RND efflux pump components
TolC and AcrB assemble as homo-trimers, but the oligomeric state of either complexed or uncomplexed AcrA is not fully resolved. In vivo cross-linking data indicated that AcrA can form dimers and trimers in the absence of AcrB [37], but in vitro monomers as the predominant species have also been reported for MexA [25,26]. Trimeric species have also been reported for the HlyD, a membrane fusion protein belonging to the hemolysin export system [38]. The physiological significance of uncomplexed AcrA as oligomers is unclear, but it is conceivable that self-association is induced only in the absence of interactions with AcrB and/or TolC.
The first high-resolution structures of MexA [25,26] showed a tri-decamer assembly, with a hexamer and a heptamer assembled in a head-to-head fashion, an arrangement most likely resulting from crystallographic artifacts. Recent structural resolution of AcrA’s core domains also showed unusual intermolecular association resulting from a dimer of inverted dimers [27], showing significant flexibility of the α-helical hairpin region. It is likely that these subunits arrangements are the results of anomalous crystal packing and may bear little resemblance to the stoichiometric state of free AcrA or that which exists when complexed with AcrB and TolC in vivo. It is clear that a high-resolution structure of the assembled AcrAB/TolC complex is needed to conclusively determine AcrA’s stoichiometry in the functional pump complex. In the absence of the tripartite complex structure, however, various models have been proposed to envision the possible subunit assembly of AcrA/MexA around AcrB/MexB and TolC/OprM. One such model proposes a ring of nine laterally-aligned MexA monomers completely shielding the bottom helices of OprM and the crown region of MexB [26,39]. Yet another model invokes either twelve monomers of MexA encircling MexB/OprM or six monomers, assembled as three dimers, aligned along OprM helices and MexB crown [25]. A more thorough model of tripartite pump complex stoichiometry and assembly has been put forward by Fernandez-Recio et al. [40], suggesting a 3:3:3 stoichiometry with MFP-helical hairpins bound in the inter-protomer grooves (due to the steric clashes which occur with the equatorial domain of TolC if docking is attempted to the intra-protomer grooves). However more recent docking studies and mutagenesis data such as Lobedanz et al. [41] and Bavro et al. [20] suggest that equatorial domain could undergo certain rearrangements, allowing it to accommodate the MFP-hairpin tip, in which case the intra-protomer grooves appear as more likely candidates. A similar model was put forward for the related BesA, BesB, BesC system from Borrelia burgdorferi [42]. It is noteworthy, that while the above models have all favored a 3:3:3 stoichiometry, at present there is no definitive proof of such an assembly taking place and based on the 6 grooves being available on the surface of the TolC, one can envisage also a 3:6:3 models. Indeed, each protomer in TolC trimer features an internal duplication, which results in formation of 6 structurally similar features on the surface of the trimer. Notably each two pairs of helices (H3/H4 and H7/H8, respectively) form a groove with the preceding and following pair, forming a total of 6 exposed surface grooves which could accommodate docked helical hairpins of the membrane fusion proteins. Suggestions for such a stoichiometry can be inferred from the mutagenesis work of Stegmeier et al. [43], but also from whole cell quantitative immunoblotting assays [44], which have shown that the relative molar ratio of MFPs compared to OMFs and IMPs is about 2:1.
A recent work by Mima et al. [45] on the TriABC-OpmH triclosan extrusion system in Pseudomonas suggest that for functional activity the pump requires two different MFPs, which is most easily explained with a 3:6:3 stoichiometry where the two different MFPs occupying two non-equivalent surface grooves on OpmH. Similarly, in Serratia sp. ATCC 39006 the ZrpADBC pump with two MFPs (ZrpA and ZrpD) may assemble with a 3:6:3 stoichiometry [46]. In addition, cross-linking and atomic force microscopy data (Zhang et al., manuscript in preparation) show that stable hexamers of the MtrC membrane fusion protein can be isolated, forming a ring-like structure. It is worth to remind that earlier studies of AcrA 2D crystals which belonged to P2122 space group [47] as well as the recent cryo-EM analysis by Trépout et al. [48] also reported ring-like structures distinct from the tri-decamers observed in MexA crystal structures. Sadly the low resolution in that study did not allow determining the oligomeric, but interestingly, the recent reports of Piao et al. [30] suggest a single molecule of MacA in the asymmetric unit of a crystal belonging to the P622 space group, indicative of a possible hexameric assembly. If so these crystal structures could be the first ones to land a direct structural proof that a functional assembly of membrane fusion protein is hexameric. While the above proposal is quite reasonable, it does require further experimental verification. Whatever the case may be, future structural work is bound to clarify this issue.
Regional interactions among pump components
After the TolC and AcrB crystal structures were resolved, it was proposed that TolC and AcrB may directly interact with each other by virtue of their extended periplasmic domains: TolC’s α-helical domain extends 100 Å into the periplasm, while AcrB’s periplasmic domain protrudes 70 Å into the periplasm. The first experimental data supporting this notion for direct interactions between TolC and AcrB came from a study showing disulfide bond formation between engineered cysteine residues located in AcrB’s periplasmic hairpin turns and the turns of TolC’s coiled coils [49]. Unlike these synthetic, disulfide-induced interactions, however, other experiments indicated that direct interactions between TolC/OprM and AcrB/MexB are intrinsically weak but can be stabilized by AcrA/MexA [29,50]. Structural analysis of the possible extent of the interpenetration of the crown domain of AcrB and the periplasmic part of TolC indicates that such interaction is quite shallow and hence unlikely to be very stable [21,40]. Such interactions are likely restricted to the loops connecting helices H3/H4 and H7/H8 of TolC and the β-hairpins of the TolC-binding domain in AcrB [20,49].
Below we summarize studies analyzing MFP-OMF interactions. An extensive amount of research has been carried out to reveal specific residues and regions of MFP and OMF that are important for their functional contacts. An initial approach involved construction of chimeric MFPs between AcrA and YhiU (which is unable to interact with AcrB) to see which of these chimeric constructs would restore the function of the pump [28]. The results showed that the first 290 residues out of AcrA can be exchanged with YhiU without affecting the efflux function, and hence its interactions with AcrB. But a hybrid protein lacking residues 290 to 357 of AcrA failed to support efflux function, signifying the importance of this C-terminal proximal region of AcrA in AcrA-AcrB interactions. Two additional studies provided general support for this notion. In one case, isothermal calorimetry titration analyses showed interactions between an AcrA fragment, comprised of residues 172 to 397, and AcrB but not with TolC [29]. Interestingly, the N-terminal AcrA fragment expressing residues 24 to 172 showed no interactions with AcrB but interacted strongly with the C-terminal AcrA fragment (172-397); finally, the preformed complex of N- and C-terminal AcrA fragments interacted with both AcrB and TolC [29]. Not only did these findings support the notion that the C-terminal region of AcrA interacts with AcrB, they also indicated a role for the N-terminal region of AcrA in forming a complex with TolC.
Genetic studies were carried out to reveal critical residues/regions that influence the interactions among pump components also produced valuable insights. Random mutagenesis to seek efflux defective mutants produced alterations affecting the N- (P68-V129) or C-terminal (V264-G297) regions of MexA [51], with the latter being already implicated in MFP-RND interactions (see above). One of the first studies seeking revertants of a defective TolC protein produced suppressor alterations mapping primarily in the β-domain of AcrA [52], which assembles from both the N-terminal (residues 54-61 of the β1 strand) and C-terminal (residues 210-297 of a β10-α3-β11-14 fold) residues [27]. These results were somewhat surprising because the β-barrel domain of MFP is predicted to interact with RND, since lipidation of the N terminal-proximal cysteine residue places this domain in close proximity to the inner membrane and hence to RND, as demonstrated by Elkins & Nikaido [28] and Nehme et al. [51]. Curiously, these AcrA suppressor alterations elevated the mutant TolC level, indicating that the suppressors acted by stabilizing TolC-AcrA interactions. However, an AcrB-dependence of this stabilizing effect of AcrA suppressors on the mutant TolC protein indicated the suppressive effect requires a tripartite complex, therefore raising the possibility that AcrA suppressors, particularly those affecting the β-domain of AcrA, may first influence AcrA-AcrB interactions and only secondarily the AcrB-TolC or AcrA-TolC interactions [52]. However, at this point there is no experimental basis to eliminate the possibility that certain suppressor alterations in the β-domain of MFP could produce broad conformational changes to influence other domains of MFP, including the highly flexible, OMF-proximal, α-helical hairpin domain, and thus achieve suppression by directly modulating MFP-OMF interactions. Nehme and Poole [53,54] also isolated suppressor alterations in MexA’s β-barrel domain that reversed a functional defect of not only a mutant MexB protein but also a mutant OprM protein, thus pointing to a broad, perhaps long-distance, influence of alterations in MFP’s β-barrel domain.
As mentioned above, modeling predictions have postulated a direct interaction between the α-helical hairpin domain of MFP and the intra-protomer grooves formed by the bottom helices of OMF [29,40,50]. This prediction was experimentally tested indirectly through isolating compensatory mutations of a defective pump component [20,52,54,55,56] or directly through analyzing a chimeric MFP construct [43,56] and probing cysteine-specific cross-linking between MFP and OMF [41].
Compensatory alterations have been found in the α-helical hairpin domain of MFP when reversing a deficient efflux activity of either to a heterologous non-functional pump complex [56] or a defective OMF [52]. Similarly, compensatory alterations in OMF’s bottom helices have been isolated when reversing defects originating from either a heterologous non-functional pump complex [55] or a defective MFP [54]. Importantly, the work of Vidyappan et al. [55] has pointed towards the possible interaction zone of the OMF and MFP, as most of the gain-of-function mutations clustered at the region of the intra-protomer groove of the OMF, highlighting it as the likely docking site for the helical hairpin of the MFP. The importance of MFP’s α-helical hairpin of MFP in establishing functional contacts with OMF was also tested through analyzing a heterologous, non-functional pump complex [43]. Utilizing a chimeric MFP construct, the authors showed that the region corresponding to the α-helical hairpin of MFP plays a crucial role in making functional interactions with the cognate OMF. Another noteworthy work to highlight the binding determinants on TolC surface was conducted using directed evolution approach, selecting TolC variants to work with non-cognate pumps, in that case MexA/B [57]. Importantly, the mutants selected have converged to the native MexA/B partner OprM, suggesting that a limited number of “discriminator” residues is responsible for the specificity of the binding between the OMF and different MFPs. Once again the majority of the gain-of-function mutants were located at the lower end of the α-helical domain, outlining a possible interaction site in the intra-protomer groove of TolC.
Finally, Lobedanz et al. [41] carried out a comprehensive analysis of MFP-OMF interactions through probing reciprocal cysteine-mediated cross-linking. Their data supported and confirmed a model of MFP-OMF interactions in which the α-helical hairpin domain of MFP docks at the intra-protomer grooves of OMF that are observed in the crystal structures of the partially open TolC [20] [Fig. 2].
Assembly of the tripartite efflux pump
Among the tripartite drug efflux pumps, AcrAB/TolC of E. coli and MexAB/OprM of P. aeruginosa have received most attention in part due to the fact that high resolution structures of all except that of MexB have been solved. A considerable effort has been devoted to determine in vivo interactions among the components of the AcrAB/TolC and MexAB/OprM efflux systems, with the aim of establishing how these interactions lead to the assembly of a functional tripartite pump. It is clear that all three components of the pump must be present and able to interact to assemble a functional, drug extruding pump. But questions remain as to the order of assembly, the roles the individual components play in pump assembly, the mechanism allowing OMF to transition from closed to open state, and the roles that MFP and RND play in this transition.
To address these questions, scientists have taken various approaches, including isolation of the complex using affinity tags [29,37,50,51,58], analysis of chimeric or heterlogous protein constructs [28,43,55], suppressor analysis [52,53], molecular modeling [40], site-directed mutagenesis based on molecular modeling [41], biophysical studies [29] and targeted evolution approaches [56,57,61]. All of these approaches have contributed to our understanding of various interactions among the pump components, but many issues remain.
In vivo cross-linking studies have shown the existence of bipartite complexes of all three permutations in the absence of the third component of the pump [29,37,58]. Additionally, these complexes have been shown to form in the absence of a proton motive force [29,62] or externally added pump substrate [29,58], although one report suggests an enhancement in complex formation in the presence of substrates [62].
Among the three possible bipartite complexes, the existence of a complex between OMF component and RND, in the absence of the MFP, is somewhat perplexing [29,37,49]. Molecular modeling and cysteine-specific cross-linking data [49] have suggested interactions between the bottom tip of OMF and the apex region of the antiporter, but no specific in vivo stabilizing interactions between these surfaces have been experimentally tested. Moreover, mutational analyses of the proposed interacting regions between OMF and RND have given no indication as to the existence of any side chain-specific interactions (Weeks, Husain, Bavro, & Misra, unpublished data). Finally, based on data obtained from isothermal titration calorimetry, it was concluded that OMF and RND do not stably interact in the absence of MFP, but their transient interactions, and hence close proximity, can be stabilized by chemical cross-linkers [29].
One of the major steps that must occur for successful drug extrusion is the engagement of the OMF and the RND securing the transfer of drugs into the channel of the OMF. For this to happen, the periplasmic end of the TolC channel must open [Fig. 2].
Analysis of the crystal structures of TolC (1EK9), OprM (1WP1) and VceC (1YC9) demonstrate that the periplasmic lumens of the channels are closed by complex networks of charged interactions [18,21,22]. This network has to be destabilized in order for the channel opening to occur. Indeed site-directed mutagenesis of the gating residues in vitro is demonstrated to result in a spontaneously open, leaky channel or in conditional blockage of transport [20,63,64]. In vivo however, these salt bridges are likely being destabilized by AcrB, which is demonstrated to dock at the tips of the helices that bear them [21,49]. The interaction with the AcrB alone is likely to be sufficient for the unlocking of the gates, which however on its own isn’t sufficient to fully open the channel as demonstrated by the two recent structures of the mutant TolC channels trapped in a partially open state [20] [Fig. 2]. The analysis of the TolC channel cavity, reveals however the presence of the second selectivity gate in it, which is formed by a double ring of aspartate residues [63], and which is not destroyed in the leaky mutants [20,63,65], due to its location further up the channel. For the same reason it can be argued that it is also unlikely to be unlocked via a direct interaction with AcrB, and hence for that step an interaction with the MFP is necessary [20].
An in vivo role for MFP in stabilizing the interactions between OMF and RND was examined through the isolation of the MexAB/OprM complex without the use of a chemical cross-linker so as to evaluate various interactions based on their intrinsic binding strengths [50]. The authors showed that whereas the absence of MexB made no difference in MexA’s ability to pull down OprM, MexB was unable to pull down OprM without MexA. This study also found no stable interactions between MexA and MexB in the absence of OprM. The implications of these findings are that (a) MFP plays a critical role in stabilizing OMF-RND interactions and thus central to the tripartite pump complex assembly, and (b) since only MFP-OMF complexes are stable without a cross-linker, this combination may represent a major pathway to tripartite complex assembly. In other words, formation of MFP-OMF complexes precedes MFP-OMF-RND assembly. If MFP-OMF complexes are indeed a precursor to the functional pump assembly, OMF in this bipartite complex must be in the closed state. Then, binding of RND to the preexisting MFP-OMF complex, followed by the realignment of MFP with respect to both OMF and antiporter must lead to dilation of OMF’s aperture to the open state.
An alternative model of pump assembly proposes a different order of interaction and it accommodates for the conformational transition that OMF helices must undergo to reach an open state [20,40]. It envisages a partial outward movement of OMF’s aperture-restricting helical pairs upon initial interaction with the apex region of the antiporter. This initial outward movement of helices exposes grooves in the intra-subunit regions of OMF into which the coiled-coil region of MFP can be accommodated. This then leads to further outward extension of OMF helices and thus full dilation of OMF’s aperture to establish an active pump. So the major difference in this model compared to that proposed above, is that besides giving MFP a central role in stabilizing the interaction between OMF and antiporter, and hence the pump assembly, it also considers MFP a central player in forcing the allosteric transition of OMF from a closed to open state, and suggest that the MFP is an active energy transducer from RND to the OMF. Thus the model invokes an order of assembly in which a preformed complex between MFP and RND engages OMF, followed by MFP-mediated interaction with OMF that not only completes the tripartite pump assembly but helps OMF transition from a closed to open state. Similar role of the MFP can be inferred from the MacA/B-TolC system, which however is energized by ATP [10].
Mechanism of drug extrusion
Understanding the mechanism by which drugs are removed by RND-type efflux pumps begins with a deeper knowledge of the atomic structures of all the pump components. Since drugs must be captured by the RND pump protein, we begin our discussion with AcrB. The first AcrB structure to be solved was crystallized in R32 space group [31], and displayed a crystallographic 3 fold symmetry along the centre axis of the trimer. Each of the identical protomers were revealed to be composed of three distinct domains: a transmembrane (TM) domain containing 12 TM helices; a membrane-proximal pore or porter domain which is formed from an extended loop connecting TM helices 1 and 2; and a membrane-distal TolC docking domain. Both the pore domain and the docking domain present mixed α/β-structures, with the pore domain being an α+β sandwich with antiparallel β-sheet, belonging to ferredoxin fold as described by SCOP [66], while the docking domains form a unique fold family.
A few details of the protomer structure have hinted towards the possible mechanics of the tripartite pump assembly. Indeed, the discovery of the extension of the pore- and TolC-docking domains over 70 Å into the periplasm raised the possibility of direct interaction between AcrB and TolC, thus creating an intermembrane bridge for antibiotic expulsion. Another structural peculiarity, which reminds of the TolC organization, was revealed in the structure of the AcrB protomers, which appear to also have arisen from internal gene duplication. Thus, similarly to TolC, while the AcrB assembly is a homotrimer it also shows a pseudo six-fold symmetry. Indeed, the two original copies of the gene can be split up without losing the functionality of the assembly as was elegantly demonstrated by Eda et al. [67] using a close homologue of AcrB – namely the MexB from P. aeruginosa.
The original centrally-symmetrical R32 structure also revealed a possible route by which drugs are captured and guided across the AcrB molecule and ultimately outside the cell through TolC. An initial hypothesis was raised that the drugs could move through the central inter-subunit pore from the cytoplasm to the OMF [31,68]. Since then a total of 5 new structures have been crystallized in the lower symmetry C2 and P1 space groups [32,33] and in addition an orthorhombic P212121 form was obtained by Sennhauser et al. [35] using a Designed Ankirin Repeat Protein (DARPin) adaptor, which also showed the highest resolution of 2.5 Å. The higher resolution at around 3.1 Å as well as the use of brominated-drugs for phasing also allowed unambiguous assignment of two bound drugs, minocyclin and doxorubicin respectively in two of the asymmetric structures [32]. Importantly, only one protomer was found to be drug bound, and the binding pocket appears to only form in one of the conformers. Also noteworthy is the fact that no drugs were found bound in the central cavity, thus contradicting earlier reports [69,70].
Most remarkably the drug bound forms reveal a breakdown of the 3 fold symmetry of the protomers, which appear to be in three distinct conformations designated as access (or loose), binding (or tight) and extrusion (or open) respectively [32,33]. The authors independently concluded that the asymmetric protomers trapped in the different crystal forms actually represent different stages of the expulsion cycle. These recently solved structures have led to the formulation of a novel, much more detailed model of the functioning of the RND transporters which is commonly referred to as the “peristaltic mechanism” [32,33], which we will briefly discuss below.
In analogy to rotational catalysis in the F1Fo-ATPase, Murakami et al. [32] suggested that a similar rotating mechanism, which is also likely to operate in a sequential manner, exists for the conformers of AcrB. Since it resembles the rotary action of the peristaltic pump, this mechanism was coined a “peristaltic pump mechanism” [33]. A cycle of the pump is thought to start with the access (loose) state, when the drug can access the binding site of the protomer at low affinity, after which the conformational change results in a tighter binding (tight) state, which is followed by a conformer transition to the extrusion (open) state, when the drug is released towards the outer membrane protein channel and the conformer is reset to the access (loose) state, thus completing the cycle. Readers can find a comprehensive review on the AcrB extrusion mechanisms in a previous issue of this journal [14], so we are not going to discuss the mechanistic details of the process here in full detail, however the main paradigmal shift in the new models is the emphasis on the role of the intra-protomer tunnels, instead of central cavity (and the inter-protomer vestibules leading to it) in providing the main paths of drug extrusion.
In spite of the wealth of recent structural data, the path of the substrate through the pump is not readily clear. This is because a closer examination of the potential conduits reveals at least three different tunnels leading to/from the drug binding cavity [33]. Those tunnels are distinct from and should not be confused with the inter-protomer vestibules, which are leading to the AcrB central cavity. Importantly, availability of these intra-protomer tunnels differs between the conformers, suggesting a possible role for sequential tunnel access in the extrusion mechanism. While two of the tunnels seem to provide access to the drug binding cavity from the side of the membrane leaflet (available in loose and tight conformers), the third tunnel, which is only available in the extrusion (or open) conformation protomer leads the drug outward to the TolC. It thus appears that a single protomer is capable of conducting the drug on its own in a conformational cycling mode, or cooperatively with the others in a rotary model [32,33].
While it is currently not fully established experimentally whether all the possible states of the conformers could be occupied in a functional trimer, that is whether AAA (A for access, B for binding, E for extrusion), BBB, EEE, and all the combinations of the thereof are possible, or whether there is a cooperativity which limits the combinations and the conversion of one leads to a direct transition in the neighbor, it seems plausible that a conformational ensemble greater than the one observed in the available crystal structures does exist. These possibilities are beyond the scope of the current review, but are elaborated in recent reviews by Murakami [13] and Seeger et al. [17]. It is worth emphasizing however, that while the new C2, P1 and P212121 structures represent a functional ABE state of the molecule, there is mounting evidence that the centrally symmetric R32 spacegroup crystals probably represent a state close to the AAA (all loose), despite the earlier reports of drugs bound to it [17]. Interestingly, a recent structure of AcrB in complex with bile acid [36], which has been crystallized in a rhombohedral H32 group, appears to present an all-loose AAA state where all the vestibules are open and loaded with the substrate. It is difficult to say if this represents another crystallization artifact or a physiological state. In addition, a structure of the all-tight, fully drug bound configuration BBB is suggested to have been obtained by the group of Pos [17]. When available, this new structure will no doubt extend our understanding of the cooperativity and transition mechanisms in RND transporters.
In addition to structural cues, there have been also functional data to support the peristaltic model. Perhaps the most convincing is the recent study involving introduction of engineered disulfide bridges, which are predicted to hamper the domain movements in the periplasmic region and thus affect the drug export [71]. Indeed removal of the conditional bridges by reduction restored the activity of the pump. Conversely, drug transport also affected the disulfide bridge formation, indicative of the existence of the functional rotation in vivo [71]. The importance of inter-protomer flexibility during drug export has also been highlighted by earlier studies [72,73,74] as well as by a recent study utilizing a covalently linked AcrB trimer [75].
These results seem to undermine the earlier reports of the drugs bound in the central cavity of AcrB [69,70], however it is noteworthy, that a recent structure of the AcrB in complex with the YajC [76] has been reported with six antibiotic molecules located in the central cavity. In addition recent structural studies [34,76] also indicate that alternative drug expulsion pathway cannot be ruled out. So the role of the lateral vestibules and the central cavity might see some revision in the near future.
Still quite a few questions remain open. For instance it is not known whether the two intra-protomer tunnels are actually conduits for entry of the substrate or for release of non-selected substrates [33]. To answer that we need first to see what defines the substrate specificity in the RND transporters and where does the selection happen. Functional studies using chimeric proteins reveal the importance of the periplasmic loops in substrate selection and swapping the loops between homologous proteins resulted in reciprocal changes to their substrate specificities as demonstrated for AcrB and AcrD [59], AcrB and MexB [60] MexB and MexY [67]. Thus it is unlikely that a selection is happening in the cytoplasm and the drugs are passed through the central cavity as initially speculated. Instead the selection is happening at the level of the porter domain and most likely in the hydrophobic pocket itself.
As noted above there are two tunnels leading to the drug-binding chamber with entries from the periplasmic space, which could be hypothesized to serve as drug entry conduits, while the third one is leading outwards towards TolC. As the third tunnel is only available in extrusion (open) conformer, it appears pretty clear that it represents the end of the cycle allowing the drug to exit the cell. Situation is a bit more complicated in respect to the two “entry” tunnels. Extending the sequential mechanism, it could be hypothesized that the drugs in access (loose) protomer are guided in via one of the two laterally accessible tunnels (the entry of which is close to the membrane leaflet) and after selection in the drug-binding cavity which is formed in the binding (tight) state, if rejected are exiting via the second lateral tunnel located higher at the porter domain level [17]. Alternatively both tunnels could represent possible substrate entry points as suggested by the detected simultaneous presence of both “entry” tunnels in the 2JS8.pdb [35] in binding (tight) conformation. As both tunnels are rather hydrophilic some energy, possibly from the peristaltic mechanism itself is provided to push the compound through A different issue regarding second tunnel is that it apparently clashes with the postulated AcrA binding site on the AcrB [31,53]. This however is a rather intriguing possibility, which may suggest an active role of the MFP in delivery of the drug and/or pump activation [10], and also lends some additional support to the idea of the primal role of the tunnel originating close to the membrane leaflet as a drug entry route.
The remaining question is how the substrate export is connected with the proton gradient. Under the current model, the proton transport takes place in the transmembrane domain. A proton conduit channel is lined up with charged residues, which are conserved across the RND transporters. Mutagenesis studies have identified that just five residues are absolutely essential for the proton conductance as even their single mutants result in transport shutdown: D407, D408, K940, R971 and T978 (belonging to TM4 and TM10, respectively) [78,79]. The exact mechanisms by which proton-binding leads to the conformational switch between binding (tight) and extrusion (open) conformers as a result is still unclear [77,78,79], but changes in the TM domain seem to be transduced to the periplasmic pore domain via the TM8 which undergoes a coil-to-helix transition, causing subdomain movements in the pore domain [32,33] and also result in reorganization of the periplasmic loop between TM3 and TM4 [77]. Combined these transitions are supposed to result in a conformational switch to the extrusion state allowing the drug to leave the drug binding site.
Ultimately, drug molecules arriving at the TolC-proximal funnel region of AcrB access the TolC’s tunnel to find their way out of the cell. What then triggers TolC’s aperture opening? As discussed above, a two-step opening model envisions initially a partial aperture opening through the initial interaction with AcrB [Fig. 2], followed by AcrA-mediated full opening [20].
Despite the dramatic rearrangements between the different conformers in the asymmetric AcrB structures the TolC docking domain undergoes only limited changes. It is hence likely that the active signal for the drug bound state of AcrB is being mediated to TolC by MFP. Initial docking and the unlocking of the outer gates of the TolC channel is probably possible without AcrA participation and might be a transient, reversible event which doesn’t even require a drug-bound AcrB. Once the drug is bound however, AcrA probably becomes engaged with AcrB and upon TolC docking further transduction of conformational changes from AcrA’s α/β and lipoyl domains to its TolC-proximal coiled-coil domain allows it to properly align with the aperture-restricting helices of TolC, leading to full dilation of TolC’s aperture and a productive extrusion.
A role for YajC in AcrAB/TolC-mediated efflux
Recently Törnroth-Horsefield et al. [76] reported an X-ray structure of AcrB in complex with YajC [Fig. 3], a trans-membrane protein previously shown to interact with the SecYEG and SecDF proteins of the E. coli protein translocation machinery [80]. Although deletion of yajC caused only a slight increase in drug sensitivity, the structure revealed that the interaction of YajC with multiple trans-membrane helices of AcrB produced a significant rotation in the porter domain of AcrB that was not apparent from other AcrB crystal structures [31,32,33,35]. It was speculated that the YajC-mediated twist in AcrB’s porter domain may also induce a similar twist in TolC’s α-helical domain, presumably via AcrA, to help twist open TolC’s aperture. However, whether or not the observed YajC-mediated AcrB rotation has a biological significance could not be conclusively determined given that the absence of YajC produces only a weak phenotype. The significance of YajC may emerge in a mutant background expressing partially defective components of the AcrAB-TolC pump. In this context, it is worth noting that overexpression of YajC has been reported to compensate for the growth defect of a dominant lethal secY allele, while its disruption is reported to further impair the growth of a secY (secY39) ts mutant [81]. It is clear from the available data on YajC that more work is needed to dissect its role either as a general scaffold for many different membrane protein complexes or as a component of a specific complex dedicated to a specific function, such as drug extrusion or protein translocation.
Fig. 3.
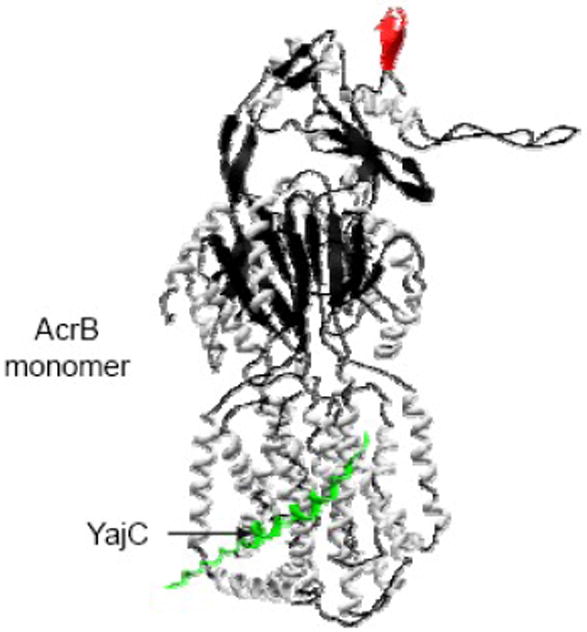
A view of AcrB monomer (2RDD) bound to the YajC helix (green).
Inhibition of MDR efflux pumps
Continuing bacterial drug resistance in society has kept up a demand for developing novel and more effective antibacterial drugs and smarter treatment strategies. MDR pumps provide attractive therapeutic targets not only to enhance the effectiveness of other drugs but because often clinical drug resistance is associated with a high level expression of MDR pumps and thus requiring a higher dosage and prolonged usage of the prescribed drugs, which further manifest ill side effects and can lead to the development of even an higher level of drug resistance. The best recognized strategy for using MDR efflux pump inhibitors (EPIs) is in conjunction with the prescribed antibacterial drugs so as to enhance the potency of these drugs. Readers are encouraged to consult an excellent recent reviews that outlines our current standing and strategies for developing EPIs [12,82,83,84].
Central to developing EPIs is the attainment of structural knowledge of the components of the MDR pumps and comprehending the mechanism of drug selection and extrusion by them. Recent structural papers on AcrB, with and without a bound ligand, have given us new and exciting insights on the mechanism of drug extrusion, but more work is needed to fully comprehend the process of drug selection and disposal. Though we have come a long way in understanding the possible pathway of the tripartite pump assembly, full understanding will no doubt require continuing collaborations among geneticists, biochemists, and structural and theoretical biologists.
Acknowledgments
We would like to thank Jon Weeks for reading the manuscript and assistance in making figures 1 and 3. This work was supported in part by a grant R01 GM66988 from the National Institutes of Health to RM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Piddock LJ. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbio Rev. 2006;19:382–402. doi: 10.1128/CMR.19.2.382-402.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poole K. Bacterial multidrug efflux pumps serve other functions. Microbe. 2008;3:179–185. [Google Scholar]
- 3.Evans K, Passador L, Srikumar R, Tsang E, Nezezon J, Poole K. Influence of the MexAB-OprM Multidrug efflux system on quorum sensing in Pseudomonas aeruginosa. J Bacteriol. 1998;180:5443–5447. doi: 10.1128/jb.180.20.5443-5447.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Helling RB, Janes BK, Kimball H, Tran T, Bundesmann M, Check P, Phelan D, Miller C. Toxin disposal in Escherichia coli. J Bacteriol. 2002;182:3699–3703. doi: 10.1128/JB.184.13.3699-3703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zakharov SD, Eroukova VY, Rokitskaya TI, Zhalnina MV, Sharma O, Loll PJ, Zgurskaya HI, Antonenko YN, Cramer WA. Colicin occlusion of OmpF and TolC channels: outer membrane translocons for colicin import. Biophys J. 2004;87:3901–3911. doi: 10.1529/biophysj.104.046151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masi M, Vuong P, Humbard M, Malone K, Misra R. Initial steps of colicin E1 import across the outer membrane of Escherichia coli. J Bacteriol. 2007;189:2667–2676. doi: 10.1128/JB.01448-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wandersman C, Delepelaire P. TolC, an Escherichia coli outer membrane protein required for hemolysin secretion. Proc Natl Acad Sci USA. 1990;87:4776–4780. doi: 10.1073/pnas.87.12.4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delepelaire P. Type I secretion in gram-negative bacteria. Biochim Biophys Acta. 2004;1694:149–161. doi: 10.1016/j.bbamcr.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Tikhonova EB, Devroy VK, Lau SY, Zgurskaya HI. Reconstitution of the Escherichia coli macrolide transporter: the periplasmic membrane fusion protein MacA stimulates the ATPase activity of MacB. Mol Microbiol. 2007;63:895–910. doi: 10.1111/j.1365-2958.2006.05549.x. [DOI] [PubMed] [Google Scholar]
- 10.Lin HT, Bavro VN, Barrera NP, Frankish HM, Velamakanni S, van Veen HW, Robinson CV, Borges-Walmsley MI, Walmsley AR. The MacB ABC transporter is a dimer whose ATPase activity and macrolide-binding capacity are regulated by the membrane fusion protein MacA. J Biol Chem. 2008 doi: 10.1074/jbc.M806964200. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamanaka H, Kobayashi H, Takahashi E, Okamoto K. MacAB is involved in the secretion of Escherichia coli heat-stable enterotoxin II. J Bacteriol. 2008;190:7693–7698. doi: 10.1128/JB.00853-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lomovskaya O, Zgurskaya HI, Totrov M, Watkins WJ. Waltzing transporters and ‘the dance macabre’ between humans and bacteria. Nature Rev. 2007;6:56–65. doi: 10.1038/nrd2200. [DOI] [PubMed] [Google Scholar]
- 13.Murakai S. Multidrug transporters, AcrB–the pumping mechanism. Curr Opin Struct Biol. 2008;18:459–465. doi: 10.1016/j.sbi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Nikaido H, Takatsuka Y. Mechanisms of RND multidrug efflux pumps. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbapap.2008.10.004. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pietras Z, Bavro VN, Furnham N, Pellegrini-Calace M, Milner-White EJ, Luisi BF. Structure and mechanism of drug efflux machinery in Gram negative bacteria. Curr Drug Targets. 2008;9:719–728. doi: 10.2174/138945008785747743. [DOI] [PubMed] [Google Scholar]
- 16.Poole K. Efflux pumps as antimicrobial resistance mechanisms. Ann Med. 2007;39:162–176. doi: 10.1080/07853890701195262. [DOI] [PubMed] [Google Scholar]
- 17.Seeger MA, Diederichs K, Eicher T, Brandstätter L, Schiefner A, Verrey F, Pos KM. The AcrB efflux pump: conformational cycling and peristalsis lead to multidrug resistance. Curr Drug Targets. 2008;9:729–749. doi: 10.2174/138945008785747789. [DOI] [PubMed] [Google Scholar]
- 18.Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- 19.Higgins MK, Eswaran J, Edwards P, Schertler GF, Hughes C, Koronakis V. Structure of the ligand-blocked periplasmic entrance of the bacterial multidrug efflux protein TolC. J Mol Biol. 2004;342:697–702. doi: 10.1016/j.jmb.2004.07.088. [DOI] [PubMed] [Google Scholar]
- 20.Bavro VN, Pietras Z, Furnham N, Pérez-Cano L, Fernández-Recio J, Pei XY, Misra R, Luisi B. Assembly and channel opening in a bacterial drug efflux machine. Mol Cell. 2008;30:114–121. doi: 10.1016/j.molcel.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akama H, Kanemaki M, Yoshimura M, Tsukihara T, Kashiwagi T, Yoneyama H, Narita S, Nakagawa A, Nakae T. Crystal structure of the drug discharge outer membrane protein, OprM, of Pseudomonas aeruginosa: dual modes of membrane anchoring and occluded cavity end. J Biol Chem. 2004;279:52816–52819. doi: 10.1074/jbc.C400445200. [DOI] [PubMed] [Google Scholar]
- 22.Federici L, Du D, Walas F, Matsumura H, Fernandez-Recio J, McKeegan KS, Borges-Walmsley MI, Luisi BF, Walmsley AR. The crystal structure of the outer membrane protein VceC from the bacterial pathogen Vibrio cholerae at 1.8 A resolution. J Biol Chem. 2005;280:5307–5314. doi: 10.1074/jbc.M500401200. [DOI] [PubMed] [Google Scholar]
- 23.Calladine CR, Sharff A, Luisi B. How to untwist an alpha-helix: structural principles of an alpha-helical barrel. J Mol Biol. 2001;305:603–618. doi: 10.1006/jmbi.2000.4320. [DOI] [PubMed] [Google Scholar]
- 24.Andersen C, Koronakis E, Bokma E, Eswaran J, Humphreys D, Hughes C, Koronakis V. Transition to the open state of the TolC periplasmic tunnel entrance. Proc Natl Acad Sci USA. 2002;99:11103–11108. doi: 10.1073/pnas.162039399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akama H, Matsuura T, Kashiwagi S, Yoneyama H, Narita S, Tsukihara T, Nakagawa A, Nakae T. Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J Biol Chem. 2004;279:25939–25942. doi: 10.1074/jbc.C400164200. [DOI] [PubMed] [Google Scholar]
- 26.Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V. Structure of the periplasmic component of a bacterial drug efflux pump. Proc Natl Acad Sci USA. 2004;101:9994–9999. doi: 10.1073/pnas.0400375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikolosko JK, Bobyk K, Zgurskaya HI, Ghosh P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure. 2006;14:577–587. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elkins CA, Nikaido H. Chimeric analysis of AcrA function reveals the importance of its C-terminal domain in its interaction with the AcrB multidrug efflux pump. J Bacteriol. 2003;185:5349–5356. doi: 10.1128/JB.185.18.5349-5356.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Touze T, Eswaran J, Bokma E, Koronakis E, Hughes C, Koronakis V. Interactions underlying assembly of the Escherichia coli AcrAB-TolC multidrug efflux system. Mol Microbiol. 2004;53:697–706. doi: 10.1111/j.1365-2958.2004.04158.x. [DOI] [PubMed] [Google Scholar]
- 30.Piao S, Xu Y, Ha NC. Crystallization and preliminary X-ray crystallographic analysis of MacA from Actinobacillus actinomycetemcomitans. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:391–393. doi: 10.1107/S1744309108008701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- 32.Murakami S, Nakashima1 R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:73–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- 33.Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- 34.Das D, Xu QS, Lee JY, Ankoudinova I, Huang C, Lou Y, DeGiovanni A, Kim R, Kim SH. Crystal structure of the multidrug efflux transporter AcrB at 3.1 Å resolution reveals the N-terminal region with conserved amino acids. Struct Biol. 2007;158:494–502. doi: 10.1016/j.jsb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PloS Biol. 2007;5:106–113. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drew D, Klepsch MM, Newstead S, Flaig R, de Gier JW, Iwata S, Beis K. The structure of the efflux pump AcrB in complex with bile acid. Mol Membr Biol. 2008;25:677–682. doi: 10.1080/09687680802552257. [DOI] [PubMed] [Google Scholar]
- 37.Zgurskaya HI, Nikaodo H. Cross-linked complex between oligomeric periplasmic lipoprotein AcrA and the inner-membrane-associated multidrug efflux pump AcrB from Escherichia coli. J Bacteriol. 2000;182:4264–4267. doi: 10.1128/jb.182.15.4264-4267.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thanabalu T, Koronakis E, Hughes C, Koronakis V. Substrate-induced assembly of a contiguous channel for protein export from E. coli: reversible bridging of an inner-membrane translocase to an outer membrane exit pore. EMBO J. 1998;17:6487–6496. doi: 10.1093/emboj/17.22.6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eswaran J, Koronakis E, Higgins MK, Hughes C, Koronakis V. Three’s company: component structures bring a closer view of tripartite drug efflux pumps. Curr Opin Struct Biol. 2004;14:741–747. doi: 10.1016/j.sbi.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Recio J, Walas F, Federici L, Venkatesh Pratap J, Bavro VN, Miguel RN, Mizuguchi K, Luisi B. A model of a transmembrane drug-efflux pump from Gram-negative bacteria. FEBS Lett. 2004;578:5–9. doi: 10.1016/j.febslet.2004.10.097. [DOI] [PubMed] [Google Scholar]
- 41.Lobedanz S, Bokma E, Symmons MF, Koronakis E, Hughes C, Koronakis V. A periplasmic coiled-coil interface underlying TolC recruitment and the assembly of bacterial drug efflux pumps. Proc Natl Acad Sci USA. 2007;104:4612–4617. doi: 10.1073/pnas.0610160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bunikis I, Denker K, Ostberg Y, Andersen C, Benz R, Bergström S. An RND-type efflux system in Borrelia burgdorferi is involved in virulence and resistance to antimicrobial compounds. PLoS Pathog. 2008;4:1–11. doi: 10.1371/journal.ppat.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stegmeier JF, Polleichtner G, Brandes N, Hotz C, Andersen C. Importance of the adaptor (membrane fusion) protein hairpin domain for the functionality of multidrug efflux pumps. Biochemistry. 2006;45:10303–10312. doi: 10.1021/bi060320g. [DOI] [PubMed] [Google Scholar]
- 44.Narita S-I, Eda S, Yoshihara E, Nakae T. Linkage of the efflux-pump expression level with substrate extrusion rate in the MexAB-OprM efflux pump of Pseudomonas aeruginosa. Biochem Biophys Res Commun. 2003;308:922–926. doi: 10.1016/s0006-291x(03)01512-2. [DOI] [PubMed] [Google Scholar]
- 45.Mima T, Joshi S, Gomez-Escalada M, Schweizer HP. Identification and characterization of TriABC-OpmH, a triclosan efflux pump of Pseudomonas aeruginosa requiring two membrane fusion proteins. J Bacteriol. 2007;189:76007609. doi: 10.1128/JB.00850-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gristwood T, Fineran PC, Everson L, Salmond GP. PigZ, a TetR/AcrR family repressor, modulates secondary metabolism via the expression of a putative four-component resistance-nodulation-cell-division efflux pump, ZrpADBC, in Serratia sp. ATCC 39006. Mol Microbiol. 2008;69:418–435. doi: 10.1111/j.1365-2958.2008.06291.x. [DOI] [PubMed] [Google Scholar]
- 47.Avila-Sakar AJ, Misaghi S, Wilson-Kubalek EM, Downing KH, Zgurskaya HI, Nikaido H, Nogales E. 2D crystals of AcrA appear to accommodate a central cavity. J Struct Biol. 2001;136:81–88. doi: 10.1006/jsbi.2001.4418. [DOI] [PubMed] [Google Scholar]
- 48.Trépout S, Taveau JC, Mornet S, Benabdelhak H, Ducruix A, Lambert O. Organization of reconstituted lipoprotein MexA onto supported lipid membrane. Eur Biophys J. 2007;36:1029–1037. doi: 10.1007/s00249-007-0208-5. [DOI] [PubMed] [Google Scholar]
- 49.Tamura N, Murakami S, Oyama Y, Ishiguro M, Yamaguchi A. Direct interaction of multidrug efflux transporter AcrB and outer membrane channel TolC detected via site-directed disulfide cross-linking. Biochemistry. 2005;44:11115–11121. doi: 10.1021/bi050452u. [DOI] [PubMed] [Google Scholar]
- 50.Mokhonov VV, Akama A, Nakae T. Role of the membrane fusion protein in the assembly of resistance-nodulation-cell division multidrug efflux pump in Pseudomonas aeruginosa. Biochem Biophys Res Commun. 2004;322:483–489. doi: 10.1016/j.bbrc.2004.07.140. [DOI] [PubMed] [Google Scholar]
- 51.Nehme D, Li XZ, Elliot R, Poole K. Assembly of the MexAB-OprM multidrug efflux system of Pseudomonas aeruginosa: identification and characterization of mutations in mexA compromising MexA multimerization and interaction with MexB. J Bacteriol. 2004;186:2973–2983. doi: 10.1128/JB.186.10.2973-2983.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerken H, Misra R. Genetic evidence for functional interactions between TolC and AcrA proteins of a major antibiotic efflux pump of Escherichia coli. Mol Microbial. 2004;54:620–631. doi: 10.1111/j.1365-2958.2004.04301.x. [DOI] [PubMed] [Google Scholar]
- 53.Nehme D, Poole K. Interaction of the MexA and MexB components of the MexAB-OprM multidrug efflux system of Pseudomonas aeruginosa: identification of MexA extragenic suppressors of a T578I mutation in MexB. Antibicrob Agents Chemother. 2005;49:4375–4378. doi: 10.1128/AAC.49.10.4375-4378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nehme D, Poole K. Assembly of the MexAB-OprM multidrug pump of Pseudomonas aeruginosa: component interactions defined by the study of pump mutant suppressors. J Bacteriol. 2007;189:6118–6127. doi: 10.1128/JB.00718-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vediyappan G, Borisova T, Fralick JA. Isolation and characterization of VceC gain-of-function mutants that can function with the AcrAB multiple-drug-resistant efflux pump of Escherichia coli. J Bacteriol. 2006;188:3757–3762. doi: 10.1128/JB.00038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krishnamoorthy G, Tikhonova EB, Zgurskaya HI. Fitting periplasmic membrane fusion proteins to inner membrane transporters: mutations that enable Escherichia coli AcrA to function with Pseudomonas aeruginosa MexB. J Bacteriol. 2008;190:691–698. doi: 10.1128/JB.01276-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bokma E, Koronakis E, Lobedanz S, Hughes C, Koronakis V. Directed evolution of a bacterial efflux pump: adaptation of the E. coli TolC exit duct to the Pseudomonas MexAB translocase. FEBS Lett. 2006;580:5339–5343. doi: 10.1016/j.febslet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 58.Husain F, Humbard M, Misra R. Interaction between the TolC and AcrA proteins of a multidrug efflux system of Escherichia coli. J Bacteriol. 2004;186:8533–8536. doi: 10.1128/JB.186.24.8533-8536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elkins CA, Nikaido H. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominately by two large periplasmic loops. J Bacteriol. 2002;184:6490–6498. doi: 10.1128/JB.184.23.6490-6498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tikhonova EB, Wang Q, Zgurskaya HI. Chimeric analysis of the multicomponent multidrug efflux transporters from gram negative bacteria. J Bacteriol. 2002;184:6499–6507. doi: 10.1128/JB.184.23.6499-6507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eda S, Maseda H, Yoshihara E, Nakae T. Assignment of the outer-membrane-subunit-selective domain of the membrane fusion protein in the tripartite xenobiotic efflux pump of Pseudomonas aeruginosa. FEMS Microbiol Lett. 2006;254:101–107. doi: 10.1111/j.1574-6968.2005.00010.x. [DOI] [PubMed] [Google Scholar]
- 62.Tikhonova EB, Zgurskaya HI. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J Biol Chem. 2004;279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- 63.Andersen C, Koronakis E, Hughes C, Koronakis V. An aspartate ring at the TolC tunnel entrance determines ion selectivity and presents a target for blocking by large cations. Mol Microbiol. 2002;44:1131–1139. doi: 10.1046/j.1365-2958.2002.02898.x. [DOI] [PubMed] [Google Scholar]
- 64.Eswaran J, Hughes C, Koronakis V. Locking TolC entrance helices to prevent protein translocation by the bacterial type I export apparatus. J Mol Biol. 2003;327:309–315. doi: 10.1016/s0022-2836(03)00116-5. [DOI] [PubMed] [Google Scholar]
- 65.Augustus AM, Celaya T, Husain F, Humbard M, Misra R. Antibiotic-sensitive TolC mutants and their suppressors. J Bacteriol. 2004;186:1851–1860. doi: 10.1128/JB.186.6.1851-1860.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313:903–919. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- 67.Eda S, Yoneyama H, Nakae T. Function of the MexB efflux-transporter divided into two halves. Biochemistry. 2003;42:7238–7244. doi: 10.1021/bi0300074. [DOI] [PubMed] [Google Scholar]
- 68.Murakami S, Yamaguchi A. Multidrug-exporting secondary transporters. Curr Opin Struct Biol. 2003;13:443–452. doi: 10.1016/s0959-440x(03)00109-x. [DOI] [PubMed] [Google Scholar]
- 69.Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE., Jr Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- 70.Yu EW, Aires JR, McDermott G, Nikaido H. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J Bacteriol. 2005;187:6804–6815. doi: 10.1128/JB.187.19.6804-6815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seeger MA, von Ballmoos C, Eicher T, Brandstätter L, Verrey F, Diederichs K, Pos KM. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nature Struct Mol Biol. 2008;15:199–205. doi: 10.1038/nsmb.1379. [DOI] [PubMed] [Google Scholar]
- 72.Dastidar V, Mao W, Lomovskaya O, Zgurskaya HI. Drug-induced conformational changes in multidrug efflux transporter AcrB from Haemophilus influenzae. J Bacteriol. 2007;189:5550–5558. doi: 10.1128/JB.00471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takatsuka Y, Nikaido H. Site-directed disulfide cross-linking shows that cleft flexibility in the periplasmic domain is needed for the multidrug efflux pump AcrB of Escherichia coli. J Bacteriol. 2007;189:8677–8684. doi: 10.1128/JB.01127-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Murakami S, Tamura N, Saito A, Hirata T, Yamaguchi A. Extramembrane central pore of multidrug exporter AcrB in Escherichia coli plays an important role in drug transport. J Biol Chem. 2004;279:3743–3748. doi: 10.1074/jbc.M308893200. [DOI] [PubMed] [Google Scholar]
- 75.Takasuka Y, Nikaido H. Covalently linked trimer of the AcrB multidrug efflux pump provides support for the functional rotating mechanism. J Bacteriol. Dec 5; doi: 10.1128/JB.01441-08. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Törnroth-Horsefield ST, Gourdon P, Horsefield R, Brive L, Yamamoto N, Mori H, Snijder A, Neutze1 R. Crystal structure of AcrB in complex with a single transmembrane subunit reveals another twist. Structure. 2007;15:1663–1673. doi: 10.1016/j.str.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 77.Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J Bacteriol. 2006;188:7290–7296. doi: 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guan L, Nakae T. Identification of essential charged residues in transmembrane segments of the multidrug transporter MexB of Pseudomonas aeruginosa. J Bacteriol. 2001;183:1734–1739. doi: 10.1128/JB.183.5.1734-1739.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takatsuka Y, Nikaido H. Threonine-978 in the transmembrane segment of the multidrug efflux pump AcrB of Escherichia coli is crucial for drug transport as a probable component of the proton relay network. J Bacteriol. 2006;188:7284–7289. doi: 10.1128/JB.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Doung F, Wickner W. Distinct catalytic roles of the SecYE, SecG and SecDFyajC subunits of preprotein translocase holoenzyme. EMBO J. 1997;16:2756–2768. doi: 10.1093/emboj/16.10.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taura T, Akiyama Y, Ito K. Genetic analysis of SecY: additional export-defective mutations and factors affecting their phenotypes. Mol Gen Genet. 1994;243:261–269. doi: 10.1007/BF00301061. [DOI] [PubMed] [Google Scholar]
- 82.Mahamoud A, Chevalier J, Alibert-Franco S, Kern WV, Pagès J-M. Antibiotic efflux pumps in Gram-negative bacteria: the inhibitor response strategy. J Antimicrob Chemother. 2007;59:1223–1229. doi: 10.1093/jac/dkl493. [DOI] [PubMed] [Google Scholar]
- 83.Pagès J-M, Amaral L. Mechanisms of drug efflux and strategies to combat them: challenging the efflux pump of Gram negative bacteria. Biochim Biophys Acta. 2008 Dec 25; doi: 10.1016/j.bbapap.2008.12.011. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 84.Alibert-Franco S, Pradines B, Mahamoud A, Davin-Regli A, Pagès JM. Efflux mechanism, an attractive target to combat multidrug resistant plasmodium falciparum and Pseudomonas aeruginosa. Curr Med Chem. 2009;16:301–317. doi: 10.2174/092986709787002619. [DOI] [PubMed] [Google Scholar]