

Introduction
Trigeminal pain disorders, such as migraine and temporomandibular disorders (TMDs), are more prevalent in women than in men (1–4), which has been attributed to oestrogen fluctuations over the course of the menstrual cycle (5). Episodes of trigeminal pain correlate temporally with the menstrual cycle, increasing at menarche (6) and declining with cessation of the menstrual cycle after menopause and during pregnancy (7). Trigeminal pain in women changes with the menstrual cycle. Migraine increases during the perimenstrual period (8), and TMJ pain increases mid-cycle (9) and at menstruation. Headache and TMDs share common features (10), and both may be accompanied by significant allodynia (11, 12). Although clinical data suggest that oestrogen increases susceptibility to trigeminal pain disorders, the fall in oestrogen at the time of menstruation may precipitate an attack, suggesting a complex relationship between oestrogen levels and trigeminal pain.
Exogenous oestrogens have been shown to have pro-nociceptive effects. Oestrogen replacement therapy decreases thermal pain thresholds (13) and may trigger migraine with aura (14). Use of exogenous oestrogens, such as hormone replacement therapy or oral contraceptives, increases the risk of TMD pain (15). Sex differences in trigeminal pain disorders affecting different cranial sites suggest that the basis for, their sex disparity lies in the trigeminal system. It has been suggested that these effects result from direct actions of oestrogen on the trigeminal system, which is rich in oestrogen receptors (16). In animal studies, cutaneous receptive fields of neurons in the spinal trigeminal nucleus expand in phase with oestrogen levels during the rodent oestrous cycle and with oestrogen replacement following ovariectomy (17, 18). Oestrogen replacement in vivo has been shown to increase the excitability of acutely dissociated primary trigeminal afferents (19). However, it has not been determined whether these changes in trigeminal excitability translate to allodynia in the awake animal.
The mechanism through which oestrogen modulates excitability of trigeminal neurons is unclear. A possible mediator is activation of extracellular signal-regulated kinase (ERK), a member of the mitogen-activated protein kinase (MAPK) family. Activation of ERK occurs through a dual phosphorylation event mediated by the upstream kinase MAPK/ERK kinase (MEK). In sensory pathways, ERK phosphorylation is a specific marker of nociceptive activation (20) and is required for thermal and mechanical hyperalgesia in several models of peripheral nerve injury and inflammation (21, 22). Oestrogen has been shown to activate ERK in cultured neurons from dorsal root ganglion (DRG) (23) and trigeminal ganglion (24), but the effects of in vivo oestrogen on ERK activation in sensory ganglion have not been examined.
Cutaneous allodynia is common in trigeminal pain disorders (11, 12, 25) and is an indicator of trigeminal sensitization that may be assessed using graded mechanical stimuli. In the current study, we used a behavioural model of trigeminal inflammatory pain to determine whether oestrogen modulates mechanical allodynia. We also investigated the in vivo effects of oestrogen treatment on ERK phosphorylation and the role of ERK activation in the development of orofacial allodynia following inflammation and oestrogen treatment.
The results of this study support the hypothesis that oestrogen-mediated ERK activation in the trigeminal ganglion modulates responses to peripheral inflammation, and suggest that facial allodynia is not a solely central phenomenon, but is also mediated, in part, by peripheral sensitization of the trigeminal ganglion.
Methods
Animals
Sixty- to 75-day-old virgin female Sprague-Dawley rats (Harlan, Indianapolis, IN) were used for all studies. Rats were maintained on a daily 12 h light, 12 h dark schedule with ad libitum access to water and food (Harlan Teklad 8604). All studies were conducted in compliance with the National Institutes of Health guidelines for the care and use of laboratory animals and the Institutional Animal Care and Use Committee of the University of Kansas Medical Center.
Ovariectomy
Rats were anaesthetized with 4% isoflurane in compressed oxygen and maintained on 1% isoflurane. Dorsal bilateral incisions were made midway between the lower ribs and the iliac crest, and both ovaries were isolated and removed. The incision was closed with suture clips and Buprenex was administered (0.1 mg/kg). Rats were allowed to regain consciousness before returning to the animal facility.
CFA treatment and hormone replacement
Two weeks following ovariectomy, rats were given a single 50 μL injection of a Complete Freund’s Adjuvant (CFA; Mycobacterium tuberculosis H37 Ra; Sigma, St Louis, MO) solution, 1:1 in 0.1 M phosphate buffer into the left masseter muscle under isoflurane anesthesia. We used 50 μL of 1:1 CFA solution, a dose that has been shown to produce molecular and behavioural effects that last for 3 days (26, 27). Control rats were injected with an equal volume of saline. Estradiol valerate (E2, Delestrogen, Monarch Pharmaceuticals, Bristol, TN), an oestradiol conjugate that provides prolonged stable oestrogen levels (28), 10 μg/kg in sesame oil (100 μL), used as a vehicle, was injected subcutaneously using a 22 gauge needle during the same surgery as masseter injection. All control rats received sesame oil vehicle. Post-mortem examination of CFA-treated rats and H and E staining of sectioned facial structures revealed granulomatous inflammation confined to the masseter muscle. We did not observe inflammatory pathology upon examination of cranial sites outside the masseter, including the whisker pad. This is supported by other studies using the CFA model that demonstrate inflammation confined to the injection site (29–31).
Orofacial sensitivity assay
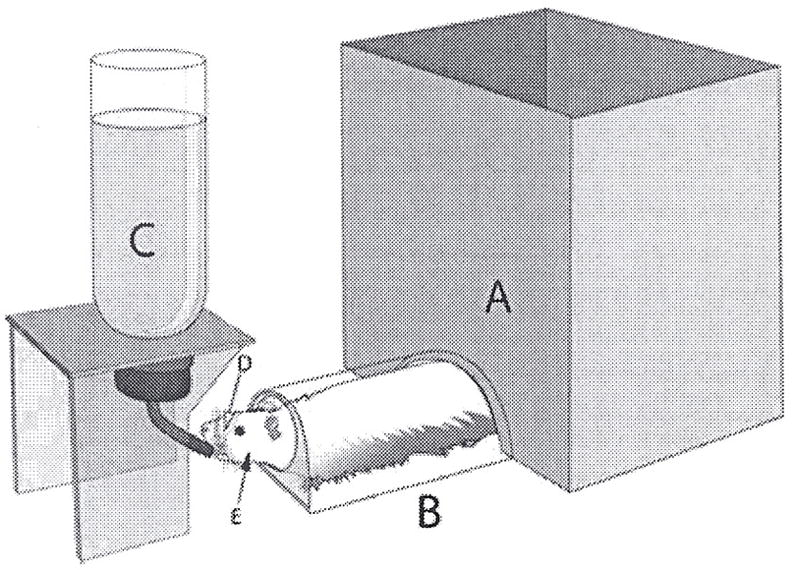
The behavior apparatus is shown in Fig. 1. Rats were conditioned to traverse a clear plastic tunnel attached to a behavioural arena (A in Fig. 1) in order to gain access to a bottle containing 0.1 M sucrose (C in Fig. 1). In this task, the rat must extend its head from a hole in the end of the tunnel in order to sample drops of sucrose solution. During the drinking task, the rat’s head was in a fixed position, allowing easy and precise access to specific areas of the face for mechanical stimulation. Rats were acclimated to the apparatus for 15-min periods once daily for 5 days. During each session, the rats consume approximately 2 mL sucrose, less than 10% of their total daily water intake. This amount of sucrose consumption is less than 1% of the amount required to produce antinociceptive effects in adult rats, approximately 1 M sucrose ad libitum over 3 to 4 weeks (32), and acute effects of sucrose on analgesia, present in neonates, disappear by P21 (33). The rats were not restrained in any way, and the tunnel is large enough for the rat to turn around to re-enter the arena at any time. After training, they spent approximately ¼ of their time in the testing position when placed in the behavioural arena. Once rats were fully trained and reliably demonstrated acquisition of target behaviour, withdrawal responses to orofacial mechanical stimulation were assessed using graded monofilaments (Stoelting, Wood Dale, IL), a well-established method for assessing mechanical sensitization in rodents. Rats were coded and experimenters blinded to experimental groups. The behavioural responses were scored as follows: no response (scored 0); flinch/orienting reflex (scored 1); partial retraction (scored 2); or complete retraction (scored 3). A flinch/orienting reflex was classified as a discontinuation in licking behaviours and orientation toward the stimulus. A partial retraction was classified as retraction of the head away from the reward. Escape back into the tunnel or arena was scored as a full retraction. A percentage withdrawal score was calculated for each rat as the sum of response scores divided by the total possible score, a score of 3 on all trials. Withdrawal was assessed in response to stimulation of the whisker pad or skin overlying the masseter using 0.16 g and 4 g monofilaments, respectively. Preliminary studies established that these filaments produced withdrawal responses at or below threshold in control rats. Mechanical withdrawal responses were measured 24, 48 and 72 h following CFA and oestradiol treatments.
Figure 1.
Hormone measurements
Blood (1.5 mL) was collected in microcentrifuge tubes by cardiac puncture at the time of perfusion. Blood was centrifuged at 7000 × g for 5 min, and the plasma transferred into fresh tubes. Serum oestradiol levels were measured by radioimmunoassay according to the method described in Terranova and Garza (34). Mean oestradiol values were 37 ± 4 pg/mL for oestrogen-replaced animals, which corresponds to a proestrus level (35) and 4 ± 2 pg/mL for ovariectomized vehicle treated animals.
Western blot
Trigeminal ganglia ipsilateral to the injection site were harvested and homogenized in cold lysis buffer (20 mM Hepes buffer, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1 mM PMSF, 5 mg/mL pepstatin A, 10 mg/mL leupeptin and 10 mg/mL aprotinin) using a Dounce homogenizer. The homogenate was centrifuged at 12 000 × g(10 min, 4°C) and the supernatant was collected. Total protein concentration was determined with a bicinchoninic acid (BCA) assay kit using bovine serum albumin as a standard (Pierce Biotechnology, Inc., Rockford, IL). Proteins were separated using SDS-PAGE on 12%TRIS-HCl gels (BioRad, Hercules, CA) and electrophoretically transferred to polyvinylidene difluoride membranes. Membranes were simultaneously probed with rabbit antibody specific for the dually-phosphorylated, active form of ERK (p-ERK) (1:200, Cell Signaling Technology, Beverly, MA) and goat anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) 1:1000 (Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were rinsed at room temperature in TBS-Tween-20 and IRDye 800 conjugated anti-goat (Rockland Immunochemicals, Gilbertsville, PA) and Alexa 680 conjugated anti-rabbit (Invitrogen, Eugene, OR) secondary antibodies were applied 1:10 000 in blocking buffer for 1 h at room temperature. Membranes were rinsed at room temperature in Tris-buffered saline containing Tween-20 (TEST) followed by TBS and visualized using an Odyssey Infrared Imaging System (LI-COR, Lincoln, NE). For total ERK, membranes were stripped for 30 min in a solution of 62.5 mM Tris HCl, 2% SDS, and 100 mM β-mercaptoethanol, and reprobed using a mouse monoclonal antibody to total ERK (1:1000, #4696, Cell Signaling Technology, Beverly, MA) followed by an Alexa 680 conjugated goat anti-mouse secondary antibody (1 : 10 000, Molecular Probes). Band intensities were determined using Odyssey software version 1.0 (LI-COR, Lincoln, NE) and expressed as a ratio of pERK/total ERK.
Immunohistochemistry
Rats were deeply anaesthetized with sodium pentobarbital (50 mg/kg i.p.) and perfused transcardially with 0.1 M PBS followed by 4% buffered paraformaldehyde. Trigeminal ganglia were removed, post-fixed overnight in 4% buffered paraformaldehyde and cryoprotected in 30% sucrose in 0.1 M phosphate buffer pH 7.2. Ganglia were frozen in Tissue Freezing Medium (Triangle Biomedical Science, Durham, NC) at −80°C. Frozen sections (20 μm) through the horizontal plane of the trigeminal ganglion were prepared using a cryostat (Carl Zeiss, Inc., Oberkochen, Germany). Horizontal sectioning of the ganglion allows visualization of the three anatomic divisions in a single section. The sections were permeabilized and blocked for 1 h in a solution of 0.3% Triton-X100 and 2% normal goat serum in 0.1 M phosphate buffered saline (PBS). Activated ERK was labelled by incubating overnight at 4°C in an antibody specific for the active form of ERK, dually-phosphorylated at Thr202/Tyr204 of ERK1 and Thrl85/Tyrl87 of ERK2 (1:200, #4376, Cell Signaling Technology, Beverly, MA), followed by 1h in AlexaFluor 568 goat anti-rabbit (1:500, Molecular Probes, Carlsbad, CA). Some slides were co-labelled using guinea pig anti-TRPV1 (1:1000, Neuromics, Edina, MN) and AlexaFluor 488 goat anti-guinea pig (Molecular Probes). Slides were coverslipped using ProLong Gold anti-fade reagent with DAPI (Molecular Probes).
Microscopy and analysis
Fluorescent digital images were obtained on a Nikon Eclipse 90i upright microscope and Nikon C1 confocal imaging system using a 20× objective and a frame size of 1024 × 1024 pixels. Sections stained with the primary antibody omitted were used to control for non-specific fluorescence. Detector gain and laser intensity were set initially using control slides, and all images were then collected under the same photomultiplier detector conditions and pinhole diameter. This ensured that all neurons considered immunoreactive in images of stained sections had fluorescence intensities above background. Individual images were montaged into a single image using Adobe Photoshop CS2 to prevent double counting on the image edges and to allow localization of immunoreactivity to subdivisions. In preliminary experiments, all neuronal profiles were immunoreactive for total ERK. Neuronal profiles positive for activated ERK and total profiles per section were counted from montaged photomicrographs in a blinded fashion. Neuronal cells were differentiated from glia by their distinctive large, round morphology and pale DAPI staining nuclei. A minimum of 2000 neurons were counted from at least two randomly selected sections from each of six animals per treatment condition.
Stereotactic injection
Rats were fixed in a stereotaxic apparatus (Kopf Instruments, Tujunga, CA) under isoflurane anaesthesia. The trigeminal ganglion was targeted by stereotaxic coordinates (36) and the injection carried out according to the method described by White-head et al. (37). A midline incision was made in the scalp and a 1 mm burr hole was drilled in the left calvarium at 2.5 mm lateral and 3 mm posterior to bregma. A 27-gauge Hamilton syringe was lowered slowly into the trigeminal ganglion. Because central structures of the trigeminal pathway are located contralaterally at this level, stereotactic injection ipsilateral to testing avoids structures of the tested sensory pathway. Correct placement of the needle was confirmed by observing the masseter twitch upon the needle entering the trigeminal ganglion. The MEK inhibitor U0126 or its inactive congener U0124 (Tocris Bioscience, Ellisville, MO) 100 μM in 10% DMSO (5 μL) was injected over 5 min. The scalp was closed with suture clips. Rats were administered Buprenex (0.1 mg/kg) and allowed to recover before returning to the animal facility.
Statistical analysis
Data were analysed using Graph Pad Prism version 4.0 software (Graph Pad, San Diego, CA). Behavioural data and immunohistochemical data were analysed by one-way ANOVA using a Bonferroni post hoc test. Data are presented as mean ± SEM, with a P value less than 0.05 considered significant.
Results
Oestrogen and inflammation additively increase primary and secondary allodynia
In order to determine how oestrogen influences mechanical nociception in the presence of orofacial inflammation, we used Complete Freund’s Adjuvant (CFA) to induce inflammation of the masseter. CFA injection is an established model of inflammatory pain (22, 38, 39) that can induce widespread mechanical hyperalgesia (40). In order to assess facial mechanical sensitivity, we developed a behavioural method based on operant conditioning that allows monofilament testing of the territory innervated by the trigeminal nerve (Fig. 1). Using this method, the 50% withdrawal threshold for stimulation of the masseter in preliminary experiments was 4 g. This model has advantages over other models of mechanical testing of the orofacial region that require direct contact between the subject and experimenter (41) and result in higher baseline withdrawal thresholds, perhaps due to handling (42).
We conducted a preliminary study to determine the monofilament force resulting in a 50% withdrawal threshold in ovariectomized rats. A sub-threshold hair was subsequently used for the testing of whisker pad and masseter in order to measure allodynia. In order to assess primary allodynia, we stimulated the masseter using a 4 g monofilament (Fig. 2A). Masseter inflammation, both with and without oestrogen, increased withdrawal responses to stimulation of the skin overlying the masseter 24 and 48 h following injection (P < 0.05). Oestrogen treatment in the presence of masseter inflammation increased withdrawal responses at 48 and 72 h compared with inflammation alone (P < 0.05).
Figure 2.
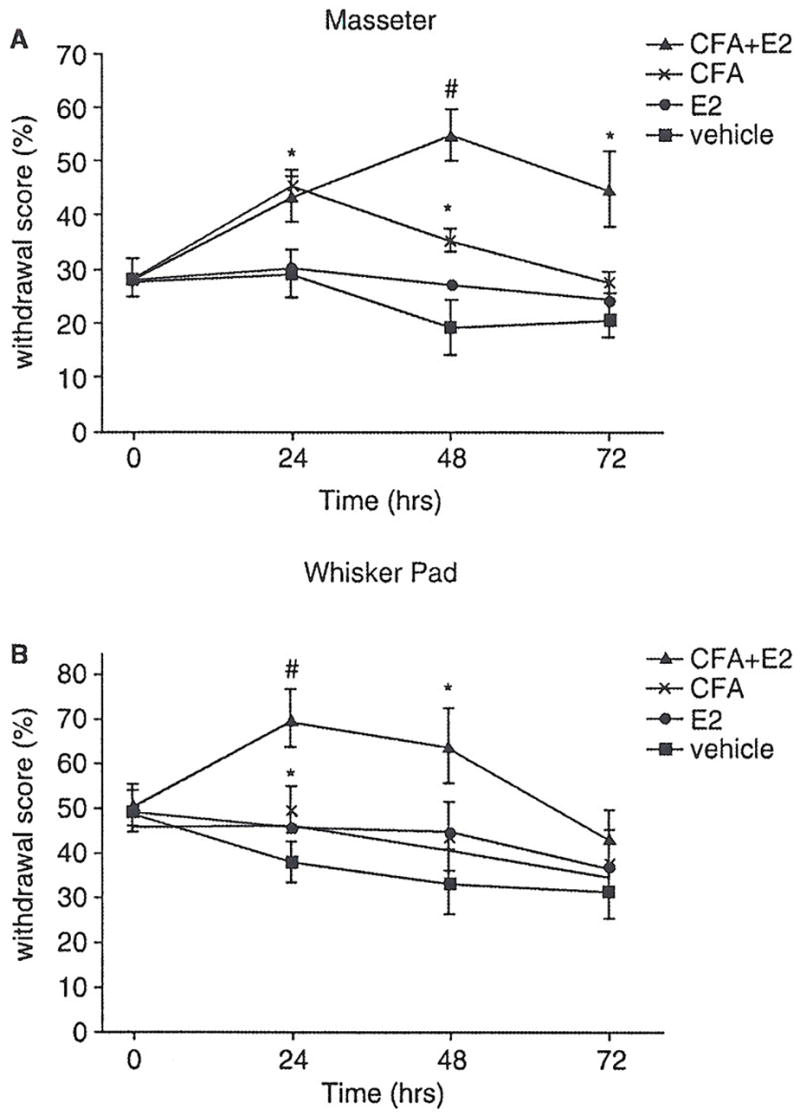
In order to assess secondary allodynia, mechanical sensitivity of the whisker pad was measured using a 0.16 g monofilament 24, 48 and 72 h following CFA injection (Fig. 2B). Masseter inflammation increased withdrawal response to whisker pad stimulation 24 h following injection (P < 0.05 compared with vehicle, one-way ANOVA). Oestrogen treatment combined with masseter inflammation increased withdrawal response to whisker pad stimulation at 24 h compared with inflammation alone (P < 0.05) and at 48 h compared with vehicle (P < 0.05).
ERK is activated in trigeminal ganglion by inflammation and oestrogen
Because ERK, an established mediator of inflammatory nociception, is activated by oestrogen in trigeminal ganglion neurons maintained in culture (24), we explored whether ERK activation contributed to the development of allodynia in our behavioural model. We first examined ERK activation at 24 h, when CFA-treated rats demonstrated mechanical allodynia in the behavioural assay.
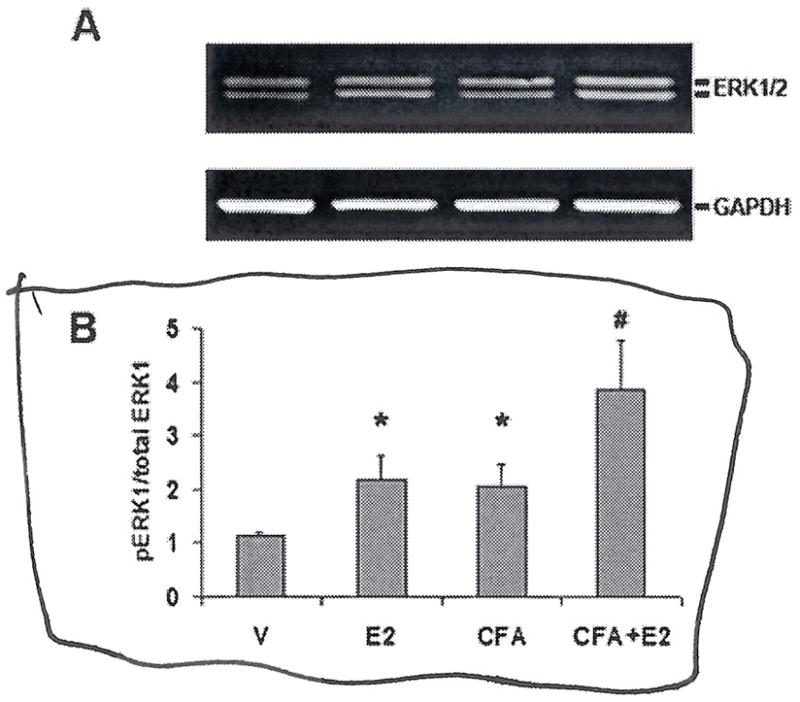
We used Western blots to analyse ERK activation in trigeminal ganglion samples from rats treated with CFA+E2, CFA, E2, and vehicle-treated rats. Membranes labelled with an antibody specific for the dually-phosphorylated, activated form of ERK showed two bands of molecular weight 44 and 42 kD, corresponding to the phosphorylated forms of ERK1 and ERK2, respectively. A representative blot of trigeminal ganglion pERK is shown in Fig. 3A. Both masseter inflammation and oestrogen treatment increased ERK activation compared with vehicle (Fig. 3B). Masseter inflammation in conjunction with oestrogen treatment increased ERK activation compared to inflammation alone (P < 0.05).
Figure 3.
In order to examine the anatomical distribution of ERK activation, immunohistochemically labelled frozen sections of trigeminal ganglion from each treatment group, CFA+E2, CFA, E2, and vehicle were compared to determine the percentage of neuronal profiles containing activated ERK relative to total neuronal profiles per ganglion. In preliminary experiments, ganglion sections labelled with an antibody to total ERK showed immunoreactivity in all neurons. Activated ERK was present in the nuclei and cytoplasm of neuronal soma.
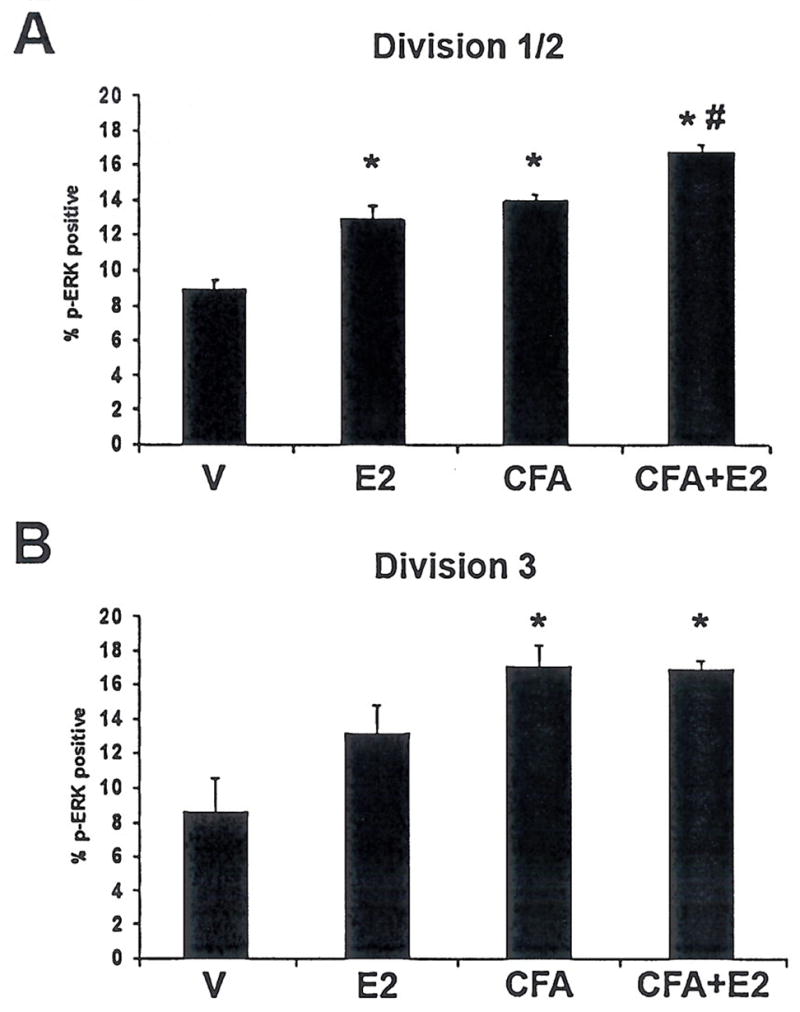
The somatotopic arrangement of the trigeminal ganglion has long been established, but recent work has clarified the anatomy of the ganglion to allow identification of cell bodies projecting to each of the three branches (43). Neurons innervating division 3 can easily be distinguished from those innervating divisions 1 and 2 based on anatomic markers (Fig. 4). The neurons with axons innervating the masseter, that are exposed to local inflammation following CFA treatment, are located in division 3, while the whisker pad is innervated by division 2. The allodynia of the whisker pad following inflammation of the masseter suggests that neurons located in division 2 may have increased excitability. In order to determine whether there is increased ERK activation in each division, we compared the change in proportion of neurons containing activated ERK in division 1/2 and division 3. In division 1/2, both masseter inflammation and oestrogen increased the percentage pf pERK immunoreactive neurons relative to vehicle-treated OVX controls (P < 0.05), and concurrent masseter inflammation and oestrogen treatment increased pERK immunoreactivity in V1/2 compared with inflammation or oestrogen alone (Fig. 6). In division 3, inflammation, both with and without oestrogen, increased the percentage of pERK immunoreactive neurons compared with vehicle (P < 0.05, Fig. 6). Oestrogen treatment increased pERK immunoreactivity relative to vehicle (P < 0.05).
Figure 4.
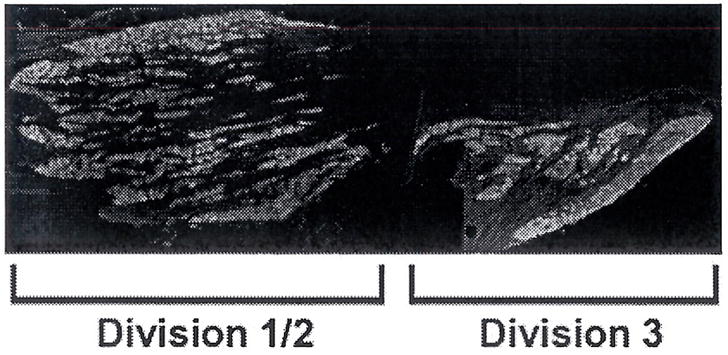
Figure 6.
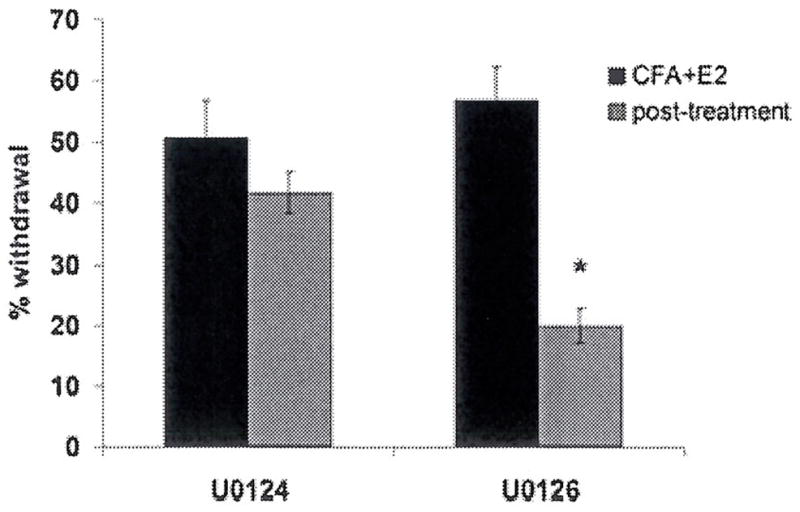
Blockade of ERK activation reduces secondary allodynia
In order to determine whether ERK activation within the ganglion is required for trigeminal sensitization, the effect of peripheral administration of an inhibitor of ERK activation on allodynia was determined using our behavioural model. We administered U0126 using stereotactic injection, an established method for targeting agents to the trigeminal ganglion (37, 44). Twenty-four hours after treatment with CFA and E2, a timepoint following the development of allodynia, we stereotactically injected the MEK inhibitor U0126, or its inactive congener U0124, into the trigeminal ganglion ipsilateral to CFA treatment. We assessed withdrawal responses 24 h following administration of U0126/4. U0126 treatment reduced the withdrawal response to whisker pad stimulation compared with U0124-treatment (P < 0.05, Fig. 7).
Figure 7.
Discussion
The results of this study provide evidence that oestrogen increases the duration of primary allodynia and both the duration and magnitude of secondary allodynia in the region innervated by the trigeminal nerve in a model of trigeminal inflammatory pain. Data also show that inflammation of the masseter increases neuronal ERK activation in all three divisions of the trigeminal ganglion, and that oestrogen increases ERK activation in the trigeminal ganglion in both the presence and absence of peripheral inflammation. Furthermore, the results demonstrate that locally blocking ERK activation in the trigeminal ganglion reduces secondary allodynia as shown by increased mechanical sensitivity of whisker pad after CFA induced inflammation of the masseter. The results of this study strongly suggest that ERK activation in the trigeminal ganglion is important for maintenance of facial allodynia, and that oestrogen amplifies nociceptive activation of ERK.
Oestrogen exacerbates inflammatory trigeminal nociception
The behavioural results show that oestrogen increases the duration of primary inflammatory allodynia of the facial region. This is the first study to demonstrate enhanced sensitization of the trigeminal territory by oestrogen in a behavioral model of allodynia. Oestrogen has previously been shown to alter the physiological properties of trigeminal neurons. High oestrogen states, in both normal cycling and oestrogen replacement, increase receptive field sizes in the spinal trigeminal nucleus (17, 18). The current results are consistent with the data of Flake et al. (45), who demonstrated increased excitability of dissociated trigeminal afferents 3 days following oestrogen treatment and inflammation of the temporomandibular joint. By extending this work to a behavioural model, the results of this study suggest that previously described increases in excitability and receptive field size result in hyperalgesia in the awake animal.
The effect of oestrogen on secondary allodynia was tested using mechanical stimulation of the whisker pad following inflammation of the masseter, a measure of sensitization extending across dermatomal divisions. Results show that inflammation of the masseter increases withdrawal responses to whisker pad stimulation, indicating that inflammation of the mandibular branch can sensitize the maxillary branch. Secondary hyperalgesia following peripheral inflammation has been demonstrated previously. Widespread sensitization occurs following CFA inflammation (40), and inflammation of the TMJ decreases escape threshold and increases excitability of whisker pad afferents in response to mechanical stimulation (31).
Results show that inflammatory secondary sensitization is increased by the addition of oestrogen. While oestrogen increased the duration of allodynia to primary site stimulation, it increased both the duration and magnitude of sensitization of the whisker pad. Oestrogen thus appears to spatially broaden the area of allodynia. These results may be relevant to cutaneous allodynia, a common clinical manifestation of secondary hyperalgesia that occurs in a large proportion of migraineurs and TMD patients (11, 46). Cutaneous allodynia varies with the menstrual cycle, and the allodynic area is greater in women during both the luteal and menstrual phases of the cycle compared with men (47). The current findings suggest mechanisms by which oestrogen could exacerbate cutaneous allodynia during episodes of migraine and TMD.
ERK is activated by oestrogen in the trigeminal ganglion
Results show that inflammation of the masseter increases ERK activation in neurons of the trigeminal ganglion. ERK is specifically activated by nociceptive input (20), and neuronal ERK activation mediates hyperalgesia (48). ERK is activated by peripheral inflammation in dorsal root ganglion and the spinal cord dorsal horn (49). In the trigeminal system, ERK is activated in the nucleus caudalis by passive jaw movement in the presence of TMJ inflammation (50) and by capsaicin injection of the tooth pulp (51).
Our data provide evidence that oestrogen treatment alone can activate ERK in sensory ganglia of female rats. Oestrogen can have direct effects on trigeminal ganglion neurons, as oestrogen receptor α is prevalent throughout the trigeminal pathway, including neurons of the trigeminal ganglion (16, 24). ERK activation in the presence of increased oestrogen in vivo is potentially significant in light of the demonstrated role of ERK in mechanisms of pain and provides a potential mechanism linking high oestrogen states and increases in excitability of trigeminal ganglion neurons.
Furthermore, these data show that oestrogen administered in the presence of masseter inflammation produces a larger increase in ERK activation than inflammation alone. An additive effect of oestrogen on ERK activation in the context of inflammation may have important implications for trigeminal pain. Neurogenic inflammation mediated by peripheral terminals is implicated as an important mechanism of migraine and other disorders of trigeminal pain (52). Our results suggest that, during trigeminal pain episodes, ERK activation in the trigeminal ganglion induced by peripheral inflammation would be increased by a surge in oestrogen. Additional ERK activation induced by oestrogen may sensitize trigeminal neurons to the degree that a subacute inflammatory stimulus becomes painful.
Analysis of ERK activation in the maxillary/ophthalmic division (1/2) shows that masseter inflammation increases ERK activation outside the third division of the trigeminal ganglion. This is an intriguing finding, as it shows that peripheral inflammation can activate ERK in soma that have no direct contact with the inflamed tissue. These results suggest that ERK activation can occur through intercellular communication within the ganglion. It is possible that neurons containing activated ERK can activate the soma of neighbouring cells through paracrine signalling. Neuropeptides, including CGRP (53), are released within the trigeminal ganglion from neuronal soma, and may signal to neighbouring cell bodies (54). TMJ inflammation has been shown to increase excitability of Aβ neurons projecting to the whisker pad via an NK1 mediated mechanism, presumably through local release of substance P (55). TNFα administration into the TMJ increases p38 MAPK activation throughout the trigeminal ganglion by neuron-glia communication through gap junctions (56). Because the trigeminal ganglion, unlike the dorsal root ganglion, contains neurons that project to three separate dermatomes in close proximity, intracellular communication may lead to activation across divisions, contributing to trans-dermatomal allodynia, as seen in this study.
Peripheral ERK activation in trigeminal sensitization
Results of this study show an additive effect of oestrogen and inflammation on ERK activation within the maxillary/ophthalmic division of the trigeminal ganglion. Because of the role of ERK in mediating somatic pain (57) and the current behavioural results showing that inflammation and oestrogen sensitize the orofacial region in an additive manner, we asked whether ganglionic ERK activation is involved in changes in facial sensitivity. The data from these experiments provide evidence that delivery of an inhibitor of ERK activation to the trigeminal ganglion reduces facial sensitivity, and ERK activation mediates increased peripheral sensitization of trigeminal neurons. Whereas many studies have employed intrathecal infusion of MEK inhibitors, in this study, a MEK inhibitor was delivered directly to the ganglion in order to examine the role of peripheral activation in the primary afferents. The current results suggest that peripheral ERK activation within trigeminal ganglion neurons is an important component of sensitization of the orofacial region.
Cutaneous allodynia has been proposed to arise when input from sensitized first-order trigeminal afferents sensitizes second-order neurons in the trigeminal nucleus caudalis, thereby lowering the activation threshold of convergent first-order neurons, a phenomenon known as central sensitization. Chemical stimulation of dural afferents enhances the response of second order sensory neurons in the trigeminal nucleus caudalis to stimulation of facial skin, taken to indicate central sensitization in the trigeminal system (58). The current observation that secondary allodynia is ameliorated by administration of MEK inhibitor to the trigeminal ganglion suggests that ERK activation in neurons of the trigeminal ganglion contributes to the maintenance of cutaneous allodynia. These results are potentially significant to our understanding of the pathogenesis of cutaneous allodynia, as they suggest that processes within the trigeminal ganglion contribute to cutaneous allodynia. While there is undoubtedly a role for central sensitization in development of allodynia, the presence of allodynia across trigeminal dermatomes alone does not prove that central sensitization occurs, and, conversely, the existence of central sensitization does not preclude the contribution of intra-ganglionic mechanisms.
Several studies have shown reduction of pain-related behaviours by MEK inhibitors administered prior to or concurrent with injury. Our data show that U0126 reduced secondary allodynia when administered following the development of sensitization. Our results are similar to those of Seino and colleagues, who showed alleviation of pain by U0126 administered by intra-articular injection of the knee following development of inflammation in an arthritis model (59). The ability to modulate facial pain following development of allodynia has important implications for the treatment of migraine, because onset of allodynia signals the close of the temporal window during which triptan intervention is effective, ostensibly due to central sensitization (46, 60). Triptans are thought to function by acting on 5HT1B/D receptors in the trigeminal ganglion, and sumatriptan reduces CGRP release in cultured trigeminal ganglion neurons (61). An alternative argument for necessity of early triptan treatment is that triptans function by suppression of intra-ganglionic communication, and that allodynia signals closure of the window for preventing of cross-sensitization of ganglionic divisions. In that case, pharmacologic approaches targeting the periphery, such as ERK antagonism or reducing intraganglionic CGRP release, may be viable alternatives.
The results of this study suggest an intraganglionic component to facial allodynia. Intraganglionic sensitization may function in conjunction with central sensitization in the generalization of pain during the course of a trigeminal pain episode. Results suggest that oestrogen increases secondary sensitization by broadening the sensitized area via ERK activation in the trigeminal ganglion. These findings strongly suggest the possibility that modulation of signal transduction pathways in the periphery following the development of central sensitization may be a useful avenue in the management of trigeminal pain.
Figure 5.
Acknowledgments
This project was supported by NIH Grant P20 RR016475 from the INBRE Program of the National center for research Resources, R21 DE01582, P30 HDO2528, National Headache Foundation, KUMC Lied Foundation and Biomedical Resarch Training Programme. The authors thank Dr Kathy Roby for assistance with this project and Julia Berman for illustration work.
References
- 1.Manzoni GC, Granella F, Sandrini G, et al. Classification of chronic daily headache by International Headache Society criteria: limits and new proposals. Cephalalgia. 1995;15:37–43. doi: 10.1046/j.1468-2982.1995.1501037.x. [DOI] [PubMed] [Google Scholar]
- 2.Lyngberg AC, Rasmussen BK, Jorgensen T, Jensen R. Incidence of primary headache: a Danish epidemiologic follow-up study. Am J Epidemiol. 2005;161:1066–73. doi: 10.1093/aje/kwi139. [DOI] [PubMed] [Google Scholar]
- 3.Manzoni GC. Classification of primary headache and International Headache Society diagnostic criteria 1988: critical review. Ital J Neurol Sci. 1995;16:9–14. [PubMed] [Google Scholar]
- 4.Dworkin SF, Huggins KH, Wilson L, et al. A randomized clinical trial using research diagnostic criteria for temporomandibular disorders-axis II to target clinic cases for a tailored self-care TMD treatment program. J Orofac Pain. 2002;16:48–63. [PubMed] [Google Scholar]
- 5.Somerville BW. The role of estradiol withdrawal in the etiology of menstrual migraine. Neurology. 1972;22:355–65. doi: 10.1212/wnl.22.4.355. [DOI] [PubMed] [Google Scholar]
- 6.LeResche L, Mancl LA, Drangsholt MT, Saunders K, Korff MV. Relationship of pain and symptoms to pubertal development in adolescents. Pain. 2005;11.8:201–9. doi: 10.1016/j.pain.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 7.Lipton RB, Bigal ME. The epidemiology of migraine. Am J Med. 2005;118(Suppl 1):3S–10S. doi: 10.1016/j.amjmed.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 8.MacGregor EA, Hackshaw A. Prevalence of migraine on each day of the natural menstrual cycle. Neurology. 2004;63:351–3. doi: 10.1212/01.wnl.0000133134.68143.2e. [DOI] [PubMed] [Google Scholar]
- 9.LeResche L, Mancl L, Sherman JJ, Gandara B, Dworkin SF. Changes in temporomandibular pain and other symptoms across the menstrual cycle. Pain. 2003;106:253–61. doi: 10.1016/j.pain.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Glares AG, Urban D, Locke J. Headache and temporomandibular disorders: evidence for diagnostic and behavioural overlap. Cephalalgia. 2007;27:542–9. doi: 10.1111/j.1468-2982.2007.01325.x. [DOI] [PubMed] [Google Scholar]
- 11.Sarlani E, Greenspan JD. Evidence for generalized hyperalgesia in temporomandibular disorders patients. Pain. 2003;102:221–6. doi: 10.1016/S0304-3959(03)00095-2. [DOI] [PubMed] [Google Scholar]
- 12.Burstein R, Yarnitsky D, Goor-Aryeh I, Ransil BJ, Bajwa ZH. An association between migraine and cutaneous allodynia. Ann Neurol. 2000;47:614–24. [PubMed] [Google Scholar]
- 13.Fillingim RB, Edwards RR. The association of hormone replacement therapy with experimental pain responses in postmenopausal women. Pain. 2001;92:229–34. doi: 10.1016/s0304-3959(01)00256-1. [DOI] [PubMed] [Google Scholar]
- 14.MacGregor A. Oestrogen replacement and migraine aura. Headache. 1999;39:674–8. doi: 10.1046/j.1526-4610.1999.3909674.x. [DOI] [PubMed] [Google Scholar]
- 15.LeResche L, Saunders K, Von Korff MR, Barlow W, Dworkin SF. Use of exogenous hormones and risk of temporomandibular disorder pain. Pain. 1997;69:153–60. doi: 10.1016/s0304-3959(96)03230-7. [DOI] [PubMed] [Google Scholar]
- 16.Bereiter DA, Cioffi JL, Bereiter DF. Ooestrogen receptor-immunoreactive neurons in the trigeminal sensory system of male and cycling female rats. Arch Oral Biol. 2005;50:971–9. doi: 10.1016/j.archoralbio.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Bereiter DA, Barker DJ. Facial receptive fields of trigeminal neurons: increased size following oestrogen treatment in female rats. Neuroendocrinology. 1975;18:115–24. doi: 10.1159/000122389. [DOI] [PubMed] [Google Scholar]
- 18.Bereiter DA, Barker DJ. Hormone-induced enlargement of receptive fields in trigeminal mechanoreceptive neurons. I Time course, hormone, sex and modality specificity. Brain Res. 1980;184:395–410. doi: 10.1016/0006-8993(80)90808-2. [DOI] [PubMed] [Google Scholar]
- 19.Flake NM, Gold MS. Inflammation alters sodium currents and excitability of temporomandibular joint afferents. Neurosci Lett. 2005;384:294–9. doi: 10.1016/j.neulet.2005.04.091. [DOI] [PubMed] [Google Scholar]
- 20.Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–19. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- 21.Obata K, Yamanaka H, Dai Y, et al. Differential activation of MAPK in injured and uninjured DRG neurons following chronic constriction injury of the sciatic nerve in rats. Eur J Neurosci. 2004;20:2881–95. doi: 10.1111/j.1460-9568.2004.03754.x. [DOI] [PubMed] [Google Scholar]
- 22.Obata K, Yamanaka H, Dai Y, et al. Activation of extracellular signal-regulated protein kinase in the dorsal root ganglion following inflammation near the nerve cell body. Neuroscience. 2004;126:1011–21. doi: 10.1016/j.neuroscience.2004.04.036. [DOI] [PubMed] [Google Scholar]
- 23.Purves-Tyson TD, Keast JR. Rapid actions of estradiol on cyclic amp response-element binding protein phosphorylation in dorsal root ganglion neurons. Neuroscience. 2004;129:629–37. doi: 10.1016/j.neuroscience.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 24.Puri V, Puri S, Svojanovsky SR, et al. Effects of oestrogen on trigeminal ganglia in culture: implications for hormonal effects on migraine. Cephalalgia. 2006;26:33–42. doi: 10.1111/j.1468-2982.2005.00987.x. [DOI] [PubMed] [Google Scholar]
- 25.Burstein R, Cutrer MF, Yarnitsky D. The development of cutaneous allodynia during a migraine attack: clinical evidence for the sequential recruitment of spinal and supraspinal nociceptive neurons in migraine. Brain. 2000;123(Pt 8):1703–9. doi: 10.1093/brain/123.8.1703. [DOI] [PubMed] [Google Scholar]
- 26.Lee J, Ro JY. Differential regulation of glutamate receptors in trigeminal ganglia following masseter inflammation. Neurosci Lett. 2007;421:91–5. doi: 10.1016/j.neulet.2007.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ro JY. Bite force measurement in awake rats: a behavioral model for persistent orofacial muscle pain and hyperalgesia. J Orofac Pain. 2005;19:159–67. [PubMed] [Google Scholar]
- 28.Oriowo MA, Landgren BM, Stenstrom B, Diczfalusy E. A comparison of the pharmacokinetic properties of three estradiol esters. Contraception. 1980;21:415–24. doi: 10.1016/s0010-7824(80)80018-7. [DOI] [PubMed] [Google Scholar]
- 29.Ambalavanar R, Moritani M, Moutanni A, et al. Deep tissue inflammation upregulates neuropeptides and evokes nociceptive behaviors which are modulated by a neuropeptide antagonist. Pain. 2006;120:53–68. doi: 10.1016/j.pain.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Ogawa A, Ren K, Tsuboi Y, et al. A new model of experimental parotitis in rats and its implication for trigeminal nociception. Exp Brain Res. 2003;152:307–16. doi: 10.1007/s00221-003-1538-x. [DOI] [PubMed] [Google Scholar]
- 31.Takeda M, Tanimoto T, Ikeda M, et al. Temporomandibular joint inflammation potentiates the excitability of trigeminal root ganglion neurons innervating the facial skin in rats. J Neurophysiol. 2005;93:2723–38. doi: 10.1152/jn.00631.2004. [DOI] [PubMed] [Google Scholar]
- 32.Kanarek RB, Mandillo S, Wiatr C. Chronic sucrose intake augments antinociception induced by injections of mu but not kappa opioid receptor agonists into the peri-aqueductal gray matter in male and female rats. Brain Res. 2001;920:97–105. doi: 10.1016/s0006-8993(01)03039-6. [DOI] [PubMed] [Google Scholar]
- 33.Anseloni VC, Weng HR, Terayama R, et al. Age-dependency of analgesia elicited by intraoral sucrose in acute and persistent pain models. Pain. 2002;97:93–103. doi: 10.1016/s0304-3959(02)00010-6. [DOI] [PubMed] [Google Scholar]
- 34.Terranova PF, Garza F. Relationship between the preovulatory luteinizing hormone (LH) surge and androstenedione synthesis of preantral follicles in the cyclic hamster: detection by in vitro responses to LH. Biol Reprod. 1983;29:630–6. doi: 10.1095/biolreprod29.3.630. [DOI] [PubMed] [Google Scholar]
- 35.Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis-part I. Headache. 2006;46:3–23. doi: 10.1111/j.1526-4610.2006.00309.x. [DOI] [PubMed] [Google Scholar]
- 36.Schneider JS, Denaro FJ, Olazabal UE, Leard HO. Stereotaxic atlas of the trigeminal ganglion in rat, cat, and monkey. Brain Res Bull. 1981;7:93–5. doi: 10.1016/0361-9230(81)90103-9. [DOI] [PubMed] [Google Scholar]
- 37.Whitehead JL, Ohara PT, Tauscher AN, LaVail JH. A procedure to deliver herpes simplex virus to the murine trigeminal ganglion. Brain Res Brain Res Protoc. 2003;12:60–6. doi: 10.1016/s1385-299x(03)00072-2. [DOI] [PubMed] [Google Scholar]
- 38.Duric V, McCarson KE. Neurokinin-1 (NK-1) receptor and brain-derived neurotrophic factor (BDNF) gene expression is, differentially modulated in the rat spinal dorsal horn and hippocampus during inflammatory pain. Mol Pain. 2007;3:32. doi: 10.1186/1744-8069-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–35. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ambalavanar R, Moutanni A, Dessem D. Inflammation of craniofacial muscle induces widespread mechanical allodynia. Neurosci Lett. 2006;399:249–54. doi: 10.1016/j.neulet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Ren K. An improved method for assessing mechanical allodynia in the rat. Physiol Behav. 1999;67:711–16. doi: 10.1016/s0031-9384(99)00136-5. [DOI] [PubMed] [Google Scholar]
- 42.Chesler EJ, Wilson SG, Lariviere WR, Rodriguez-Zas SL, Mogil JS. Influences of laboratory environment on behavior. Nat Neurosci. 2002;5:1101–2. doi: 10.1038/nn1102-1101. [DOI] [PubMed] [Google Scholar]
- 43.Leiser SC, Moxon KA. Relationship between physiological response type (RA and SA) and vibrissal receptive field of neurons within the rat trigeminal ganglion. J Neurophysiol. 2006;95:3129–45. doi: 10.1152/jn.00157.2005. [DOI] [PubMed] [Google Scholar]
- 44.Karai L, Brown DC, Marines AJ, et al. Deletion of vanilloid receptor 1-expressing primary afferent neurons for pain control. J Clin Invest. 2004;113:1344–52. doi: 10.1172/JCI20449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flake NM, Bonebreak DB, Gold MS. Oestrogen and inflammation increase the excitability of rat temporomandibular joint afferent neurons. J Neurophysiol. 2005;93:1585–97. doi: 10.1152/jn.00269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burstein R, Jakubowski M. Analgesic triptan action in an animal model of intracranial pain: a race against the development of central sensitization. Ann Neurol. 2004;55:27–36. doi: 10.1002/ana.10785. [DOI] [PubMed] [Google Scholar]
- 47.Gazerani P, Andersen OK, Arendt-Nielsen L. A human experimental capsaicin model for trigeminal sensitization. Gender-specific differences. Pain. 2005;118:155–63. doi: 10.1016/j.pain.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 48.Karim F, Hu HJ, Adwanikar H, Kaplan DR, Gereau RWt. Impaired inflammatory pain and thermal hyperalgesia in mice expressing neuron-specific dominant negative mitogen activated protein Kinase Kinase (MEK) Mol Pain. 2006;2:2. doi: 10.1186/1744-8069-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doya H, Ohtori S, Takahashi K, et al. Extracellular signal-regulated kinase mitogen-activated protein kinase activation in the dorsal root ganglion (DRG) and spinal cord after DRG injury in rats. Spine. 2005;30:2252–6. doi: 10.1097/01.brs.0000182091.53834.08. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki I, Harada T, Asano M, et al. Phosphorylation of ERK in trigeminal spinal nucleus neurons following passive jaw movement in rats with chronic temporomandibular joint inflammation. J Orofac Pain. 2007;21:225–31. [PubMed] [Google Scholar]
- 51.Shimizu K, Asano M, Kitagawa J, et al. Phosphorylation of extracellular signal-regulated kinase in medullary and upper cervical cord neurons following noxious tooth pulp stimulation. Brain Res. 2006;1072:99–109. doi: 10.1016/j.brainres.2005.12.040. [DOI] [PubMed] [Google Scholar]
- 52.Waeber C, Moskowitz MA. Migraine as an inflammatory disorder. Neurology. 2005;64:S9–15. doi: 10.1212/wnl.64.10_suppl_2.s9. [DOI] [PubMed] [Google Scholar]
- 53.Ulrich-Lai YM, Flores CM, Harding-Rose CA, Goodis HE, Hargreaves KM. Capsaicin-evoked release of immunoreactive calcitonin gene-related peptide from rat trigeminal ganglion: evidence for intraganglionic neurotransmission. Pain. 2001;91:219–26. doi: 10.1016/S0304-3959(00)00439-5. [DOI] [PubMed] [Google Scholar]
- 54.Matsuka Y, Neubert JK, Maidment NT, Spigelman I. Concurrent release of ATP and substance P within guinea pig trigeminal ganglia in vivo. Brain Res. 2001;915:248–55. doi: 10.1016/s0006-8993(01)02888-8. [DOI] [PubMed] [Google Scholar]
- 55.Takeda M, Tanimoto T, Nasu M, et al. Activation of NK1 receptor of trigeminal root ganglion via substance P paracrine mechanism contributes to the mechanical allodynia in the temporomandibular joint inflammation in rats. Pain. 2005;116:375–85. doi: 10.1016/j.pain.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 56.Thalakoti S, Patil VV, Damodaram S, et al. Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache. 2007;47:1008–23. doi: 10.1111/j.1526-4610.2007.00854.x. discussion 1024–05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Obata K, Yamanaka H, Dai Y, et al. Differential activation of extracellular signal-regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. J Neurosci. 2003;23:4117–26. doi: 10.1523/JNEUROSCI.23-10-04117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burstein R, Yamamura H, Malick A, Strassman AM. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79:964–82. doi: 10.1152/jn.1998.79.2.964. [DOI] [PubMed] [Google Scholar]
- 59.Seino D, Tokunaga A, Tachibana T, et al. The role of ERK signaling and the P2X receptor on mechanical pain evoked by movement of inflamed knee joint. Pain. 2006;123:193–203. doi: 10.1016/j.pain.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 60.Burstein R, Collins B, Jakubowski M. Defeating migraine pain with triptans: a race against the development of cutaneous allodynia. Ann Neurol. 2004;55:19–26. doi: 10.1002/ana.10786. [DOI] [PubMed] [Google Scholar]
- 61.Bellamy J, Bowen EJ, Russo AF, Durham PL. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci. 2006;23:2057–66. doi: 10.1111/j.1460-9568.2006.04742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]