

Abstract
The circadian clock synchronizes the activity level of an organism to the light-dark cycle of the environment. Energy intake, as well as energy metabolism, also has a diurnal rhythm. Although the role of the clock genes in the sleep-wake cycle is well characterized, their role in the generation of the metabolic rhythms is poorly understood. Here, we use mice deficient in the clock protein mPer2 to study how the circadian clock regulates two critical metabolic rhythms: glucocorticoid and food intake rhythms. Our findings indicate that mPer2−/− mice do not have a glucocorticoid rhythm even though the corticosterone response to hypoglycemia, ACTH, and restraint stress is intact. In addition, the diurnal feeding rhythm is absent in mPer2−/− mice. On high-fat diet, they eat as much during the light period as they do during the dark period and develop significant obesity. The diurnal rhythm of neuroendocrine peptide αMSH, a major effector of appetite control, is disrupted in the hypothalamus of mPer2−/− mice even though the diurnal rhythm of ACTH, the αMSH precursor, is intact. Peripheral injection of αMSH, which has been shown to enter the brain, restored the feeding rhythm and induced weight loss in mPer2−/− mice. These findings emphasize the requirement of mPer2 in appetite control during the inactive period and the potential role of peripherally administered αMSH in restoring night-day eating pattern in individuals with circadian eating disorders such as night-eating syndrome, which is also associated with obesity.
Clock protein mPer2 prevents obesity by suppressing appetite during the inactive period through production of hypothalamic anorexigen αMSH during the inactive period.
Circadian clocks generate circadian rhythms of physiology and behavior that are synchronized to the light-dark (LD) cycles of the environment (1). The circadian clocks, which are cell autonomous, are generated by autoregulated positive- and negative-feedback loops regulating the expression of the clock genes. In mammals the circadian rhythm is driven by the master clock in the hypothalamic suprachiasmatic nucleus (SCN) (2,3,4). The SCN neurons, which are sensitive to light, synchronize the peripheral clocks to temporally coordinate behavior and physiology to the environment. SCN lesions abolish glucocorticoid (5) and feeding rhythms (6). Studies using mutant mouse models bear out the central role of the clock genes in maintaining circadian rhythm. For example, disruptions of transcription factors involved in the activation limb of an autoregulatory feedback loop such as Bmal1 (7) or Clock (8), as well as those involved in the suppression limb such as mPer2 (9), have altered diurnal rhythms of behavior.
Because most of the food intake occurs during the active period of the LD cycle, i.e. daytime for man and nighttime for nocturnal animals such as mice, it is likely that circadian clocks play a role in regulating energy intake and metabolism. Indeed, metabolic studies using Clock-mutant mice indicate that this is the case (10). Mice consume almost 75% of their food during the dark period, but Clock-mutant mice consume almost as much food during the light period as the dark period. As a result, Clock-mutant mice consume significantly more food in a 24-h period and gain weight faster than wild-type mice. Moreover, both Bmal1−/− and Clock-mutant mice have suppressed diurnal variation in glucose and triglycerides (11). Suppression of gluconeogenesis is particularly noticeable in these mutant mice. Why a mutation of Clock results in increased food intake during the inactive period is not known. The mRNA levels of orexigenic and anorexigenic genes were both decreased in the hypothalamus of Clock-mutant mice (10).
Materials and Methods
Animals
All animals were maintained on a 12-h light (0600–1800 h), 12-h dark (1800–0600 h) cycle and were allowed free access to food and water. Unless indicated otherwise, mice were fed the standard facility diet 5021 (W.F. Fisher & Son, Inc. Somerville, NJ). mPer2−/− (B6.129S7-Per2tm1Brd) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The Tyrc-Brd allele was bred out by mating serially with C57BL/6J (The Jackson Laboratory). mPer2+/+ littermates from these matings were used as a wild-type control. For obesity and food intake studies, we used regular diet (12% calories by fat, Rodent NIH-31 auto 18-4, product no. 413110-75-56; Zeigler Brothers, Inc., Gardners, PA) or high-fat diet (HFD) D12492 (60% calories by fat; Research Diet, Inc., New Brunswick, NJ). The experiments involving leptin (data not shown) and αMSH injection (see Fig. 6) were performed with animals fed HFD. Animals’ care was in accordance with institutional guidelines, and all experiments were approved by the Animal Care and Use Committee of the National Heart Lung and Blood Institute, National Institutes of Health, and the University of Utah School of Medicine. Except for body weight studies, in which we switched to the indicated diet after weaning, all experiments were performed with 5- to 8-month-old female mice. Only mice used for activity measurements (Fig. 1A) had an activity wheel in the cage.
Figure 6.

The effect of peripheral αMSH injection on feeding and activity rhythms on HFD. A, Total daily food intake of HFD was measured for mPer2+/+ (open symbols) and mPer2−/− (closed symbols) after injection with saline or αMSH (1 μg, ip) at 0600 h (n = 13–17, per genotype). Results are mean ± sem. Single-factor ANOVA for saline treated mPer2+/+ and mPer2−/− mice: P = 0.001, F = 13.27 (***). Single-factor ANOVA for saline and αMSH treatment for mPer2−/− mice: P = 0.0001, F = 19.18 (****). Two-way ANOVA (genotype × drug) was P = 0.008, F = 7.7. B, αMSH-induced change in food intake (mPer2−/− mice) during light and dark periods induced by αMSH injection relative to that by saline (n = 13–17, per genotype). Results are mean ± sem. ****, P < 0.0001, F = 10.8 between light and dark periods. C, The level of movement (in beam breaks) of mPer2−/− mice during the light period after saline or αMSH injection (n = 13–17, per genotype). D, Change in body weight during a 3-d course of saline or αMSH injection (n = 5–8, per genotype). Two-way ANOVA repeated measures: P = 0.028, F = 3.27 (*) for mPer2−/− mice (drug × time); no significance for mPer2+/+ mice (drug × time); P = 0.001, F = 7.44 (**) for αMSH treated (genotype × time); and no significance for saline treated (genotype × time). Two-way ANOVA (genotype × drug) was P = 0.0003, F = 16.6 (###) for d 2 and P = 0.01, F = 7.2 (##) for d 3.
Figure 1.

Diurnal rhythm of activity and corticosterone rhythm are attenuated in mPer2−/− mice. A, Locomotor activity during light period (0600–1800 h) and dark period (1800–0600 h) was monitored with wheel running activity (n = 4 per genotype). Five-month-old mPer2+/+ (open symbols) and mPer2−/− (closed symbols) mice were tested. Light and dark periods are indicated by white and black bars, respectively. B, Serum corticosterone levels in LD. Blood was collected from the tail vein with care to minimize the stress level. Results are mean ± sem. Statistical analysis was performed using GLM (see Materials and Methods). For corticosterone level, the overall F test is significant (F = 8.06; P < 0.0001), indicating strong evidence that the mean corticosterone levels for the 12 cells are different. Both time and genotype have significant effects on corticosterone level (F = 5.9, P < 0.0001 for time; and F = 34.17, P < 0.0001 for genotype). The significant interaction between time and genotype (F = 5; P = 0.0003) shows that the genotype effect depends also on time, i.e. differences between genotypes differ significantly across times. Comparison of corticosterone levels between genotypes using the F test performed at each specific time point further demonstrates that the significant differences occur during dark period: F = 19.98, P < 0.0001 (****) for 1800 h; F = 11.87, P = 0.0029 (**) for 2200 h; and F = 7.92, P = 0.013 (*) for 0200 h. Detailed information on the statistical analysis and the number of mice used is in supplemental Table 1.
Corticosterone measurement
Blood was collected from the tail vein, and serum corticosterone was analyzed by RIA following the manufacturer’s instructions (corticosterone kit from MP Biomedicals, LLC, Santa Ana, CA).
ACTH stimulation
Mice were injected (ip) with human ACTH from Sigma-Aldrich Corp. (St. Louis, MO) (10 μg/kg body weight in 0.1 ml PBS, and 0.5% BSA), at 0800 and 2000 h. Tail-vein blood was collected 60 min after ACTH injection, and serum corticosterone was measured by RIA as described previously.
Hypoglycemic stress
Mice were injected with recombinant regular human insulin (0.75 mIU/g) (Eli Lilly and Co., Indianapolis, IN) at 0800 and 2000 h. Tail vein blood was collected 50 min after insulin injection, and corticosterone was measured by RIA as described previously.
Restraint stress
A square piece of wire mesh was used to restrain the mice. After wrapping the mice with the wire mesh, the edges were then clamped with small clips to restrict the movement without interfering with respiration. Tail-vein blood was collected before, and 15, 30, and 55 min after 10 min restraint. Corticosterone levels were measured as described previously.
Locomotor activity and metabolic rate measurement
For wheel running, mice were placed into separate cages with a running wheel and allowed to acclimate for 1 wk before experimentation. Activity was measured with activity wheel counters (model 86061), an Animal Wheel Monitor Starter Interface (model 86056), and AWM software provided by Lafayette Instrument Co., Inc. (Lafayette, IN). Data were downloaded at 1-h intervals and expressed as total distance run (meters) in 1 h. Mice were also studied for a period of 72 h using the Comprehensive Laboratory Animal Monitoring System (Columbus Instruments, Columbus, OH) for measurements of O2 consumption, CO2 production, and heat production. Movement was measured by XY/Z laser beam interruption.
Measurements of leptin, αMSH, and ACTH
Mouse serum leptin was determined by using the Leptin (Mouse/Rat) EIA kit (catalog no. 022-LEP-E06; ALPCO Diagnostics, Salem, NH); hypothalamic neuropeptides were extracted according to the instructions (Phoenix Pharmaceuticals, Inc., Burlingame, CA). αMSH levels were measured using the RIA kit for acetylated αMSH (Phoenix Pharmaceuticals); ACTH levels were measured using a commercially available RIA kit for ACTH (Phoenix Pharmaceuticals).
TaqMan assay-based real-time PCR
Total RNA was extracted from the dissected pituitary gland and hypothalamus with the TRIzol reagent extraction kit (Invitrogen Corp., Carlsbad, CA), according to manufacturer’s instructions. RNA was subsequently reverse transcribed to cDNA with a high-capacity cDNA archive kit (part no. 4322171; Applied Biosystems, Foster City, CA). The mRNA levels were measured by real-time PCR using the TaqMan Gene Expression system using the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems).
Body composition
Body composition, including fat, fluid, and lean mass, were measured by nuclear magnetic resonance (NMR) spectroscopy using the Bruker Minispec (mq-7.5, 7.5 MHz) instrument (Bruker Optics, The Woodlands, TX).
αMSH injection
The MSH analog [Ac-Cys4,d-Phe7,Cys10] αMSH-(4–13) was purchased from Bachem Americas, Inc. (Torrance, CA) (catalog no. H-9220). Mice were injected (ip) daily with αMSH (1 μg, in 0.1 ml PBS and 0.5% BSA) at the onset of light.
Statistical analysis
Data were analyzed by either single-factor ANOVA or two-way ANOVA depending on the number of variables. In studies involving multiple measurements from the same animal, two-way ANOVA repeated measures was used. To explore the difference in levels of corticosterone or αMSH between different genotypes and across time points, we performed statistical analysis using generalized linear models (GLMs):
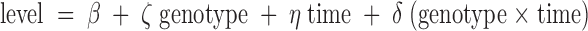 |
where ζ and η indicate, respectively, difference between genotypes and across times, and δ represents the interaction between genotypes and time. The SAS procedure PROC GLM (SAS 9.1; SAS Institute Inc., Cary, NC) was used to estimate the models. Both genotype and time are treated as class variables and, thus, forming 12 cells. The analysis results (P values and F values) are presented in supplemental Tables 1 and 2, which are published as supplemental data on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org, respectively, for corticosterone level and αMSH level. Significance was accepted at P < 0.05 and is indicated in all figure legends. Where there was no statistical significance, the P value was not indicated in the figures. Results are expressed as the mean ± sem.
Results
mPer2−/− mice have an abnormal activity rhythm
We characterized the locomotor activity of mPer2+/+ and mPer2−/− littermate mice (9) in a 12-h light, 12-dark cycle. In a 12-h light, 12-h dark cycle, light is turned on at 0600 h and off at 1800 h. Quantification of wheel running activity indicates that both mPer2−/− and mPer2+/+ mice have a robust LD pattern of activity, but mPer2−/− mice were less active during the dark (active) period. Representative 24-h wheel running is shown in Fig. 1A.
mPer2−/− mice do not have a corticosterone rhythm
Glucocorticoid is a hormone that boosts energy, blood pressure, muscle strength, and stress response. The activity of the hypothalamic-pituitary-adrenal (HPA) axis, which regulates the glucocorticoid production, has a very robust diurnal pattern (12,13). CRH released from the hypothalamus stimulates ACTH production in the pituitary gland. Secreted ACTH, in turn, stimulates the adrenal cortex to produce glucocorticoids such as corticosterone at the beginning of the active period. Although the SCN is required to generate the diurnal pattern of the HPA axis (14), the role of the clock genes in this process is not well characterized. To assess the circadian rhythm of the HPA axis, we measured the serum corticosterone levels in mPer2+/+ and mPer2−/− mice (Fig. 1B). As expected, mPer2+/+ mice had a robust diurnal pattern of corticosterone production, but mPer2−/− mice had no distinct diurnal pattern. Interaction between genotype and time was highly significant (P = 0.0003). Details of the statistical analysis are shown in supplemental Table 1.
mPer2−/− mice have intact glucocorticoid response to stress
The lack of corticosterone rhythm in mPer2−/− mice may be due to a defect in either corticosterone production or generation of HPA rhythm. To distinguish between these two possibilities, we stimulated the HPA axis with insulin-induced hypoglycemia and ACTH, using saline as a negative control. Insulin injection dramatically increased the corticosterone levels in both mPer2+/+ and mPer2−/− mice during both light and dark periods (Fig. 2A). ACTH injection also dramatically increased the corticosterone levels in both mPer2+/+ and mPer2−/− mice during both light and dark periods (Fig. 2B). To measure the time course of HPA response to restraint stress, we measured corticosterone levels before and varying time points after 10 min restraint stress in mPer2−/− mice (Fig. 2C). The time course of the corticosterone surge as well as its decline were identical between mPer2+/+ and mPer2−/− mice. These observations suggest that the HPA axis in mPer2−/− mice can respond to stress but cannot generate a rhythm.
Figure 2.

mPer2−/− mice have intact glucocorticoid response to stress. A, Serum corticosterone level 60 min after saline (light period, n = 7–12 per genotype; dark period n = 4 per genotype) or insulin (0.75 mIU/g) (light period, n = 7–9 per genotype; dark period, n = 5 per genotype) injection. Results are mean ± sem. Single-factor ANOVA between saline and insulin during the light period: P = 0.0001, F = 23 (*****) for mPer2+/+ mice; and P = 7e-05, F = 35 (*****) for mPer2−/− mice. Single-factor ANOVA between saline and insulin during the dark period: P = 0.0007, F = 34 (****) for mPer2+/+ mice; and P = 2e-05, F = 109 (*****) for mPer2−/− mice. B, Serum corticosterone level 60 min after saline (light period, n = 7–12 per genotype; dark period n = 4 per genotype) or ACTH (10 μg/kg) injection (light period, n = 7–12 per genotype; dark period, n = 5 per genotype). Results are mean ± sem. Single-factor ANOVA between saline and ACTH during the light period: P = 0.005, F = 10 (**) for mPer2+/+ mice; and P = 0.004, F = 14 (**) for mPer2−/− mice. Single-factor ANOVA between saline and ACTH during the dark period: P = 1e-05, F = 123 (****) for mPer2+/+ mice; and P = 2e-05, F = 97 (****) for mPer2−/− mice. C, Time course of corticosterone response. Serum corticosterone levels were measured before, and 15, 30, and 55 min after 10 min restraint stress (n = 5 per genotype). Results are mean ± sem. Single-factor ANOVA between basal and post-stress for mPer2+/+ mice: P = 4e-05, F = 68 (****) at 15 min; and P = 1e-08, F = 535 (*****) at 30 min. Single-factor ANOVA between basal and post-stress for mPer2−/− mice: P = 1e-10, F = 1654 (######) at 15 min; and P = 3e-06, F = 131 (#####) at 30 min.
mPer2−/− mice have defective feeding rhythm
Like locomotor activity, feeding behavior has a diurnal rhythm. During the dark period, mPer2+/+ and mPer2−/− mice consumed 2.08 ± 0.09 and 1.62 ± 0.15 g regular diet, respectively (P = 0.016) (Fig. 3A). During the light period, mPer2+/+ and mPer2−/− mice consumed 0.86 ± 0.08 and 1.31 ± 0.12 g, respectively (P = 0.008). Thus, like Clock-mutant mice (10) and SCN-lesioned animals (6), mPer2−/− mice have defective feeding rhythm. Analysis by two-way ANOVA indicated that there was a statistically significant interaction between genotype and period (P = 0.0003). The light-period hyperphagia was even more dramatic when the mice were fed HFD (60% calories by fat) (Fig. 3B). During the light period, mPer2−/− and mPer2+/+ mice consumed 1.86 ± 0.07 and 1.09 ± 0.07 g, respectively (P < 0.0001). During the dark period, mPer2−/− and mPer2+/+ mice ate a similar amount of HFD (1.57 ± 0.07 g for mPer2−/− mice and 1.66 ± 0.08 g for mPer2+/+ mice). Analysis by two-way ANOVA indicated that there was a statistically significant interaction between genotype and period (P < 0.0001).
Figure 3.

mPer2−/− mice have attenuated feeding rhythm. A, The amount of regular diet (regular chow diet) consumed during light and dark periods (n = 5–6 per genotype). Results are mean ± sem. Single-factor ANOVA between mPer2+/+ and mPer2−/− mice was P = 0.008 and F = 10.1 (**) during light period, and P = 0.016 and F = 7.7 (*) during dark period. Two-way ANOVA (genotype × period): P = 0.0003, F = 17.4 for interaction; P = 0.95, F = 0.004 for genotype; and P < 0.0001, F = 50 for period. B, The amount of HFD consumed during light and dark periods (n = 13–17 per genotype). Results are mean ± sem. Single-factor ANOVA between mPer2+/+ and mPer2−/− mice during the light period: P = 3.3e-08 and F = 58 (*****). There was no difference in the dark period. Two-way ANOVA (genotype × period): P < 0.0001, F = 26.2 for interaction; P = 0.0004, F = 14.5 for genotype; and P = 0.0005, F = 13.8 for period. C, Twenty-four hour food intake of regular diet and HFD. Single-factor ANOVA between mPer2+/+ and mPer2−/− mice on HFD: P < 0.0001, F = 19.4 (***). Two-way ANOVA (genotype × diet): P < 0.001, F = 8.6 for interaction; P = 0.0002, F = 15.0 for genotype; and P = 0.04, F = 2.9 for food.
Despite the attenuated feeding rhythm, there was no difference in the total daily intake of regular diet between mPer2−/− and mPer2+/+ mice (2.9 ± 0.16 g/d for mPer2−/− mice and 2.9 ± 0.10 g/d for mPer2+/+ mice) (Fig. 3C). On HFD the total daily intake of mPer2−/− mice was significantly higher than that of mPer2+/+ mice (3.4 ± 0.11 vs. 2.8 ± 0.10 g; P < 0.0001). Thus, hyperphagia over a 24-h period was present with HFD but not regular diet. Analysis by two-way ANOVA (diet × genotype) indicates a high statistical significance for this interaction (P < 0.001).
To determine whether hyperphagia in mPer2−/− mice affected their body weight, we monitored their body weight on regular diet (Fig. 4A) and HFD (Fig. 4B). On regular diet, mPer2−/− mice weighed slightly more than mPer2+/+ mice from 2–4 months of age. On HFD the body weight difference was much more pronounced. At 4 months of age, mPer2−/− mice weighed almost 30% more than mPer2+/+ mice. Analysis by two-way ANOVA repeated measures (genotype × age) indicated significant interaction (P = 0.01). NMR spectroscopy revealed that HFD-fed mPer2−/− mice had significantly greater fat mass (P = 0.001) (Fig. 4C) but not lean or fluid mass compared with mPer2+/+ mice (Yang, S., and J. H. Chung, unpublished data).
Figure 4.

mPer2−/− mice are obese. A, Body weight of mPer2+/+ and mPer2−/− mice fed regular diet (n = 5–6 per genotype). Two-way (genotype × age) ANOVA repeated measures: P = 0.53, F = 0.76 for interaction; P < 0.0001, F = 101.3 for age; and P = 0.01, F = 9.8 for genotype. B, Body weight on HFD (n = 10–13 per genotype). Single-factor ANOVA between mPer2+/+ and mPer2−/− mice: P = 0.0004, F = 16.5 (***) at 2 months; P = 0.0005, F = 16 (***) at 3 months; and P = 0.0002, F = 18.8 (***) at 4 months. Two-way (genotype × age) ANOVA repeated measures: P = 0.01, F = 3.8 for interaction (#); P < 0.0001, F = 47.0 for age; and P < 0.0001, F = 40.0 for genotype. C, NMR spectroscopic quantification of the fat mass of mice on HFD (females) (n = 10–13 per genotype). Fat index (fat mass/body weight) was determined with live mice using a Bruker Minispec instrument as part of whole body composition analysis. Results are mean ± sem. Single-factor ANOVA between mPer2+/+ and mPer2−/− mice: P = 4.9e-06, F = 34 (*****) at 2 months; P = 8.5e-05, F = 22 (****) at 3 months; and P = 3.3e-05, F = 26 (****) at 4 months. Two-way (genotype × age) ANOVA repeated measures was: P = 0.001, F = 6.1 (##) for interaction; P < 0.0001, F = 83.3 for age; and P < 0.0001, F = 37.2 for genotype.
Adiposity is determined not only by energy intake but also by energy expenditure. Using a metabolic chamber, we monitored energy expenditure of mPer2−/− mice for 72 h. Measurements of oxygen consumption (supplemental Fig. 1A), carbon dioxide elimination (supplemental Fig. 1B), and heat generation (supplemental Fig. 1C) were not lower in mPer2−/− mice. Although the differences were not statistically significant, mPer2−/− mice actually had a slightly higher thermogenesis rate, particularly during the light period. This was most likely caused by increased postprandial thermogenesis.
mPer2−/− mice are neither leptin deficient nor resistant
Although many physiological signals can control food intake and energy metabolism, leptin (15), a cytokine produced primarily by adipose tissue, is one of the most powerful regulators of the body weight set point. Deficiency in leptin-induced anorexia can lead to morbid obesity in animals and humans (16). The leptin receptor in the arcuate nucleus of the hypothalamus, when bound by leptin, suppresses appetite by inducing the firing of anorexigenic proopiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript neurons and activating POMC (17) and cocaine- and amphetamine-regulated transcript (18) expression. In addition, leptin inhibits the firing of orexigenic neuropeptide Y and agouti-related protein neurons and suppressing neuropeptide Y (19) and agouti-related protein (19) expression. Among the effectors of leptin signaling, POMC appears to have the most pronounced role in energy homeostasis. Deficiency of POMC in both humans (20) and mice (21) causes severe obesity secondary to hyperphagia. It is believed that the POMC-derived neuropeptide that mediates leptin signaling is the neuroendocrine peptide αMSH (22), which is derived from ACTH (23) after cleavage by proconvertase 2 (PC2) (24). αMSH produces the anorexigenic signal by binding to and activating melanocortin receptors (MCRs) 3 and 4. Consistent with this, deficiency of MCR3 (25) causes obesity in mice, and deficiency of MCR4 causes obesity in humans (26,27) and mice (27,28).
Because leptin production has a circadian pattern (29), we examined the possibility that defective leptin production caused hyperphagia in mPer2−/− mice. Measurements of serum leptin levels show that mPer2−/− mice have a robust diurnal pattern of leptin production, excluding a defect in leptin production as the source of their hyperphagia (Yang, S., and J. H. Chung, data not shown). The leptin levels in mPer2−/− mice tended to be higher, reflecting increased fat in these mice. To determine whether mPer2−/− mice are resistant to leptin, we performed a leptin-sensitivity test. In both mPer2+/+ and mPer2−/− mice, 3 d leptin injection significantly reduced the food intake and body weight (Yang, S., and J. H. Chung, data not shown), indicating that mPer2−/− mice are not leptin deficient or resistant.
Circadian clock and hypothalamic αMSH rhythm
To determine whether the expression of hypothalamic POMC mRNA was affected in mPer2−/− mice, we measured the POMC mRNA levels at different times of the day using real-time PCR. As shown in Fig. 5A, the expression pattern of POMC mRNA was similar between mPer2+/+ and mPer2−/− mice. To determine whether the hypothalamic production of POMC-derived neuropeptides ACTH and αMSH was altered in mPer2−/− mice, we extracted ACTH and αMSH from the hypothalamus at different times of the day and quantified their levels. In both mPer2+/+ and mPer2−/− mice, a prominent pulse of ACTH production occurred at 0600 h (Fig. 5B). A prominent pulse of αMSH production also occurred at 0600 h in mPer2+/+ mice, but it was significantly blunted in mPer2−/− mice (Fig. 5C). Interaction between genotype and time was significant (P = 0.0003). The details of the statistical analysis are shown in supplemental Table 2.
Figure 5.

mPer2−/− mice have an attenuated hypothalamic αMSH pulse. A, Expression levels of hypothalamic POMC mRNA at the indicated times were quantified with real-time PCR (n = 4–6 per genotype) using the 18S RNA level as the internal control [in arbitrary units (A.U.)]. There was no statistically significant difference between the two genotypes. Results are mean ± sem. Light and dark phases are indicated with a white and black bar, respectively. B, Levels of ACTH extracted from the hypothalamus at different times of day (n = 6–10 per time point for each genotype). There was no statistically significant difference between the two genotypes. C, Levels of αMSH extracted from the hypothalamus at different times of day. Statistical analysis was performed using GLM (see Materials and Methods). The overall F test is significant (F = 12.11; P < 0.0001), indicating strong evidence that the mean αMSH levels for the 12 cells are different. However, only time shows significant effects on αMSH level (F = 20.86; P < 0.0001). The interaction between time and genotype is also significant (F = 5.33; P = 0.0003), indicating that the genotype effect depends also on time, i.e. differences between genotypes differ significantly across times. Comparison of αMSH levels between genotypes using the F test performed at each specific time point further shows that αMSH levels differ significantly between genotypes at 0600 h (F = 8.6; P = 0.0093) (**). It should be pointed out that this does not contradict with the insignificance of the genotype effect (F = 2.26; P = 0.137) in the GLM model because the latter tests the overall genotype effects across all six time points. Detailed information on the statistical analysis and the number of mice used is in supplemental Table 2. Hypothalamic PC2 (D) and 7B2 (E) mRNA levels in mPer2+/+ and mPer2−/− mice measured at the indicated times (n = 9–11). There was no significant difference between the values for mPer2+/+ and mPer2−/− mice at either time points.
Two potential targets of mPer2 regulation are PC2, which converts ACTH to αMSH, and 7B2 (30), the helper protein for PC2. However, measurements of PC2 (Fig. 5D) and 7B2 (Fig. 5E) mRNA in the hypothalamus indicate that their expression was not significantly affected in mPer2−/− mice.
αMSH pulse and light-period hyperphagia
These results prompted us to investigate whether the deficiency of the αMSH pulse at the beginning of the light period caused the light-period hyperphagia in mPer2−/− mice. It is well established that peripherally administered αMSH crosses the blood-brain barrier by a nonsaturable process (31). The rate of entry of αMSH into cerebrospinal fluid is similar to that of peptides such as insulin (32). Previously, Yaswen et al. (21) took advantage of the blood-brain barrier permeability of αMSH to reverse hyperphagia and obesity in POMC-deficient mice by administering αMSH peripherally. Using the same dose of αMSH administered by Yaswen et al. (21), we tested whether peripherally injecting αMSH at 0600 h to mimic the αMSH pulse could reverse the light-period hyperphagia in mPer2−/− mice. Compared with saline injections, αMSH injections reduced the total HFD intake by 0.5–0.6 g/d in mPer2−/− mice (P = 0.0001) (Fig. 6A). In contrast, αMSH injections had no effect on the total daily food intake of mPer2+/+ mice. Analysis by two-way ANOVA indicated that the interaction between genotype and drug was highly significant (P = 0.008). The normalization of food intake in mPer2−/− mice was caused predominantly by a reduction in light-period feeding (P < 0.0001) (Fig. 6B). The number of beam breaks during light period did not change with αMSH injection in mPer2−/− mice (Fig. 6C), indicating that αMSH did not suppress the light-period activity level. The number of beam breaks during dark period also did not change with αMSH injection (Yang, S., and J. H. Chung, data not shown). Consistent with these findings, mPer2−/− mice treated with αMSH, but not saline, lost weight (Fig. 6D). mPer2+/+ mice did not lose weight with either saline or αMSH drug. These results support our hypothesis that mPer2 suppresses feeding during the inactive period by regulating the αMSH rhythm in the hypothalamus.
Discussion
mPer2-deficient mice have been a valuable tool in understanding the physiological functions of the core clock protein mPer2. Not surprisingly, mPer2-mutant mice have abnormal circadian rhythm of behavior as evidenced by the loss of rhythm in constant darkness (9). Interestingly, mPer2-mutant mice also have an abnormal response to DNA damage and are more prone to develop tumors after ionizing radiation (33). Moreover, mPer2-mutant mice are hypersensitive to cocaine and consume more alcohol (34,35).
In this study we used mPer2−/− mice to study the role of mPer2 in the regulation of the glucocorticoid rhythm. We find that mPer2−/− mice lack a recognizable corticosterone rhythm even though they can produce corticosterone normally after different forms of stress. mPer2 may be important for generating the ACTH rhythm and/or the adrenal cortical rhythm. Unlike in humans, the ACTH rhythm is not robust and is difficult to discern (36). Therefore, we were not able to address the central role of mPer2 in the production of corticosterone rhythm. However, a report published while this work was in progress demonstrated that the circadian rhythm of corticosterone production is regulated by an additional gating mechanism residing in the adrenal cortical clock that is mPer2 dependent (37).
In addition to an abnormal corticosterone rhythm, mPer2−/− mice have an attenuated feeding rhythm. Like Clock-mutated mice (10), mPer2−/− mice consume significantly more food during the light period. We propose that insufficient production of hypothalamic αMSH at the beginning of the light period caused the light-period hyperphagia. This is supported by our finding that the light-period food intake decreased with αMSH supplementation at the beginning of the light period and by a previous report that injection of SHUR9119, a MCR antagonist, at the beginning of the light period increased the light-period food intake (38).
Because the hypothalamic production of ACTH, the precursor of αMSH, is intact in mPer2−/− mice, we postulated that processing of ACTH to αMSH was deregulated in mPer2−/− mice. However, the expression of PC2 and 7B2, which mediate ACTH processing, appears normal in mPer2−/− mice. We speculate that mPer2 affects expression of other gene(s) involved in modifying the function of PC2 and/or a downstream step(s) such as disposal of αMSH in the hypothalamus.
It is interesting that SCN lesion abolishes the feeding rhythm but does not induce overeating over the 24-h period (6). This observation, together with our result that mPer2−/− mice on regular diet have a defective feeding rhythm but do not overeat, indicates that loss of feeding rhythm alone does not cause overeating. We postulate that mPer2−/− fed HFD overeat as a result of a combined effect of calorie-rich food and defective feeding rhythm.
Normal individuals consume only 10% of their total daily intake after the evening meal (39). One trivial reason for the decreased intake after the evening meal is that sleep occupies most of that time. However, αMSH injection suppressed the light-period appetite without affecting the light-period activity level in mPer2−/− mice. We propose that the anorexia induced by the αMSH pulse at the beginning of the inactive period adds a layer of control that further limits feeding to the active period.
The feeding abnormality of mPer2−/− mice resembles that of the night-eating syndrome (NES) (40,41,42), which has combined features of a circadian rhythm disorder and an eating disorder. Unlike normal individuals, who eat more than 9-fold in the first 8 h of the day (0600–1400 h) compared with the last 8 h of the day (2200–0600 h), individuals with NES eat similar amounts during these two periods (39). They are also noted for consuming excessive amounts of calorie-rich foods (41). Although the prevalence of NES in the general population is only 1.5%, the prevalence of NES among the patients undergoing gastric surgery for obesity is almost 30% (43). Administration of αMSH in the evening may decrease night eating for individuals with NES.
It is becoming clear that sleep is much more than a time of rest. Inadequate sleep leads to increased appetite and obesity (44), poor memory consolidation (45), and increased mortality (46). Thus, by inducing anorexia during the sleep period, we propose that the circadian clock enhances the quality of sleep and, ultimately, the vitality of an organism.
Supplementary Material
Acknowledgments
We thank Dr. Oksana Gavrilova (National Institute of Diabetes and Digestive and Kidney Diseases), Yiying Tsai, Dalton Saunders, Amy Mhatre, Veronica Mills, and Prabha Singh for their technical assistance, and Dr. Peng Loh (National Institute of Child Health and Human Development) for scientific advice.
Footnotes
This work was supported by the Intramural Research Program, National Heart Lung and Blood Institute, National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
First Published Online January 29, 2009
Abbreviations: GLM, Generalized linear model; HFD, high-fat diet; HPA, hypothalamic-pituitary-adrenal; LD, light-dark; MCR, melanocortin receptor; NES, night-eating syndrome; NMR, nuclear magnetic resonance; PC2, proconvertase 2; POMC, proopiomelanocortin; SCN, suprachiasmatic nucleus.
References
- Reppert SM, Weaver DR 2002 Coordination of circadian timing in mammals. Nature 418:935–941 [DOI] [PubMed] [Google Scholar]
- Ibuka N, Inouye SI, Kawamura H 1977 Analysis of sleep-wakefulness rhythms in male rats after suprachiasmatic nucleus lesions and ocular enucleation. Brain Res 122:33–47 [DOI] [PubMed] [Google Scholar]
- Ibuka N, Nihonmatsu I, Sekiguchi S 1980 Sleep-wakefulness rhythms in mice after suprachiasmatic nucleus lesions. Waking Sleeping 4:167–173 [PubMed] [Google Scholar]
- Welsh D, Richardson GS, Dement WC 1988 Effect of running wheel availability on circadian patterns of sleep and wakefulness in mice. Physiol Behav 43:771–777 [DOI] [PubMed] [Google Scholar]
- Moore RY, Eichler VB 1972 Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res 42:201–206 [DOI] [PubMed] [Google Scholar]
- Nagai K, Nishio T, Nakagawa H, Nakamura S, Fukuda Y 1978 Effect of bilateral lesions of the suprachiasmatic nuclei on the circadian rhythm of food-intake. Brain Res 142:384–389 [DOI] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA 2000 Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103:1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS 1994 Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 264:719–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Larkin DW, Albrecht U, Sun ZS, Sage M, Eichele G, Lee CC, Bradley A 1999 The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature 400:169–173 [DOI] [PubMed] [Google Scholar]
- Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J 2005 Obesity and metabolic syndrome in circadian clock mutant mice. Science 308:1043–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, Fitzgerald GA 2004 BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol 2:e377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman MF, Engeland WC, Rose JC, Wilkinson CW, Shinsako J, Siedenburg F 1978 Nyctohemeral rhythm in adrenal responsiveness to ACTH. Am J Physiol 235:R210–R218 [DOI] [PubMed] [Google Scholar]
- Krieger DT 1975 Circadian pituitary adrenal rhythms. Adv Exp Med Biol 54:169–189 [DOI] [PubMed] [Google Scholar]
- Abe K, Kroning J, Greer MA, Critchlow V 1979 Effects of destruction of the suprachiasmatic nuclei on the circadian rhythms in plasma corticosterone, body temperature, feeding and plasma thyrotropin. Neuroendocrinology 29:119–131 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM 1994 Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432 [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Keogh JM, Kamath S, Jones S, Gibson WT, Trussell R, Jebb SA, Lip GY, O'Rahilly S 2001 Partial leptin deficiency and human adiposity. Nature 414:34–35 [DOI] [PubMed] [Google Scholar]
- Boston BA, Blaydon KM, Varnerin J, Cone RD 1997 Independent and additive effects of central POMC and leptin pathways on murine obesity. Science 278:1641–1644 [DOI] [PubMed] [Google Scholar]
- Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, Clausen JT, Jensen PB, Madsen OD, Vrang N, Larsen PJ, Hastrup S 1998 Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature 393:72–76 [DOI] [PubMed] [Google Scholar]
- Korner J, Savontaus E, Chua Jr SC, Leibel RL, Wardlaw SL 2001 Leptin regulation of Agrp and Npy mRNA in the rat hypothalamus. J Neuroendocrinol 13:959–966 [DOI] [PubMed] [Google Scholar]
- Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A 1998 Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 19:155–157 [DOI] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U 1999 Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 5:1066–1070 [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD 1999 Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet 21:119–122 [DOI] [PubMed] [Google Scholar]
- Smith AI, Funder JW 1988 Proopiomelanocortin processing in the pituitary, central nervous system, and peripheral tissues. Endocr Rev 9:159–179 [DOI] [PubMed] [Google Scholar]
- Benjannet S, Rondeau N, Day R, Chreéien M, Seidah NG 1991 PC1 and PC2 are proprotein convertases capable of cleaving proopiomelanocortin at distinct pairs of basic residues. Proc Natl Acad Sci USA 88:3564–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH 2000 Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet 26:97–102 [DOI] [PubMed] [Google Scholar]
- Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O'Rahilly S 1998 A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet 20:111–112 [DOI] [PubMed] [Google Scholar]
- Vaisse C, Clement K, Guy-Grand B, Froguel P 1998 A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet 20:113–114 [DOI] [PubMed] [Google Scholar]
- Butler AA, Marks DL, Fan W, Kuhn CM, Bartolome M, Cone RD 2001 Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat Neurosci 4:605–611 [DOI] [PubMed] [Google Scholar]
- Sinha MK, Ohannesian JP, Heiman ML, Kriauciunas A, Stephens TW, Magosin S, Marco C, Caro JF 1996 Nocturnal rise of leptin in lean, obese, and non-insulin-dependent diabetes mellitus subjects. J Clin Invest 97:1344–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbikay M, Seidah NG, Chretien M 2001 Neuroendocrine secretory protein 7B2: structure, expression and functions. Biochem J 357(Pt 2):329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ 1995 Permeability of the blood-brain barrier to melanocortins. Peptides 16:1157–1161 [DOI] [PubMed] [Google Scholar]
- Wilson JF, Anderson S, Snook G, Llewellyn KD 1984 Quantification of the permeability of the blood-CSF barrier to α-MSH in the rat. Peptides 5:681–685 [DOI] [PubMed] [Google Scholar]
- Fu L, Pelicano H, Liu J, Huang P, Lee C 2002 The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111:41–50 [DOI] [PubMed] [Google Scholar]
- Abarca C, Albrecht U, Spanagel R 2002 Cocaine sensitization and reward are under the influence of circadian genes and rhythm. Proc Natl Acad Sci USA 99:9026–9030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R, Pendyala G, Abarca C, Zghoul T, Sanchis-Segura C, Magnone MC, Lascorz J, Depner M, Holzberg D, Soyka M, Schreiber S, Matsuda F, Lathrop M, Schumann G, Albrecht U 2005 The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nat Med 11:35–42 [DOI] [PubMed] [Google Scholar]
- Kwak SP, Morano MI, Young EA, Watson SJ, Akil H 1993 Diurnal CRH mRNA rhythm in the hypothalamus: decreased expression in the evening is not dependent on endogenous glucocorticoids. Neuroendocrinology 57:96–105 [DOI] [PubMed] [Google Scholar]
- Oster H, Damerow S, Kiessling S, Jakubcakova V, Abraham D, Tian J, Hoffmann MW, Eichele G 2006 The circadian rhythm of glucocorticoids is regulated by a gating mechanism residing in the adrenal cortical clock. Cell Metab 4:163–173 [DOI] [PubMed] [Google Scholar]
- Sutton GM, Josephine Babin M, Gu X, Hruby VJ, Butler AA 2008 A derivative of the melanocortin receptor antagonist SHU9119 (PG932) increases food intake when administered peripherally. Peptides 29:104–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reardon JP, Ringel BL, Dinges DF, Allison KC, Rogers NL, Martino NS, Stunkard AJ 2004 Circadian eating and sleeping patterns in the night eating syndrome. Obes Res 12:1789–1796 [DOI] [PubMed] [Google Scholar]
- Stunkard AJ, Grace WJ, Wolff HG 1955 The night-eating syndrome; a pattern of food intake among certain obese patients. Am J Med 19:78–86 [DOI] [PubMed] [Google Scholar]
- Birketvedt GS, Florholmen J, Sundsfjord J, Osterud B, Dinges D, Bilker W, Stunkard A 1999 Behavioral and neuroendocrine characteristics of the night-eating syndrome. JAMA 282:657–663 [DOI] [PubMed] [Google Scholar]
- Gluck ME, Geliebter A, Satov T 2001 Night eating syndrome is associated with depression, low self-esteem, reduced daytime hunger, and less weight loss in obese outpatients. Obes Res 9:264–267 [DOI] [PubMed] [Google Scholar]
- Rand CS, Macgregor AM, Stunkard AJ 1997 The night eating syndrome in the general population and among postoperative obesity surgery patients. Int J Eat Disord 22:65–69 [DOI] [PubMed] [Google Scholar]
- Taheri S, Lin L, Austin D, Young T, Mignot E 2004 Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med 1:e62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickgold R, Walker MP 2007 Sleep-dependent memory consolidation and reconsolidation. Sleep Med 8:331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrie JE, Shipley MJ, Cappuccio FP, Brunner E, Miller MA, Kumari M, Marmot MG 2007 A prospective study of change in sleep duration: associations with mortality in the Whitehall II cohort. Sleep 30:1659–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.