

Abstract
Although there is strong evidence that ligand activation of peroxisome proliferator-activated receptor (PPAR)-β/δ induces terminal differentiation and attenuates cell growth, some studies suggest that PPARβ/δ actually enhances cell proliferation. For example, it was suggested recently that retinoic acid (RA) is a ligand for PPARβ/δ and potentiates cell proliferation by activating PPARβ/δ. The present study examined the effect of ligand activation of PPARβ/δ on cell proliferation, cell cycle kinetics, and target gene expression in human HaCaT keratinocytes using two highly specific PPARβ/δ ligands [4-[[[2-[3-fluoro-4-(trifluoromethyl)phenyl]-4-methyl-5-thiazolyl]methyl]thio]-2-methylphenoxy acetic acid (GW0742) and 2-methyl-4-((4-methyl-2-(4-trifluoromethylphenyl)-1,3-thiazol-5-yl)-methylsulfanyl)phenoxy-acetic acid (GW501516)] and RA. Both PPARβ/δ ligands and RA inhibited cell proliferation of HaCaT keratinocytes. GW0742 and GW501516 increased expression of known PPARβ/δ target genes, whereas RA did not; RA increased the expression of known retinoic acid receptor/retinoid X receptor target genes, whereas GW0742 did not affect these genes. GW0742, GW501516, and RA did not modulate the expression of 3-phosphoinositide-dependent protein kinase or alter protein kinase B phosphorylation. GW0742 and RA increased annexin V staining as quantitatively determined by flow cytometry. The effects of GW0742 and RA were also examined in wild-type and PPARβ/δ-null primary mouse keratinocytes to determine the specific role of PPARβ/δ in modulating cell growth. Although inhibition of keratinocyte proliferation by GW0742 was PPARβ/δ-dependent, inhibition of cell proliferation by RA occurred in both genotypes. Results from these studies demonstrate that ligand activation of PPARβ/δ inhibits keratinocyte proliferation through PPARβ/δ-dependent mechanisms. In contrast, the observed inhibition of cell proliferation in mouse and human keratinocytes by RA is mediated by PPARβ/δ-independent mechanisms and is inconsistent with the notion that RA potentiates cell proliferation by activating PPARβ/δ.
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors and members of the nuclear hormone receptor superfamily. There are three PPAR isoforms, PPARα, PPARγ, and PPARβ (also referred to as PPARα and PPARβ/δ), and each regulates tissue-specific target genes involved in many biological processes (Lee et al., 2003; Peraza et al., 2006). For example, PPARα is the molecular target for the fibrate class of hypolipidemic drugs (Peters et al., 2005), and PPARγ is the molecular target of the thiazolidinedione class of insulin-sensitizing drugs (Willson et al., 2000). Although ligand activation of PPARβ/δ can increase serum high-density lipoprotein cholesterol, increase skeletal muscle fatty acid catabolism, and improve insulin sensitivity (Lee et al., 2006; Grimaldi, 2007), considerably less is known about the biological role of PPARβ/δ. In particular, the role of PPARβ/δ in tumorigenesis, apoptosis, and cell proliferation remains controversial. Given the pharmacological potential of PPARβ/δ agonists, which have been examined in clinical trials (Pelton, 2006), it is critical to determine the safety of this class of compounds in the appropriate model(s).
A number of independent laboratories have shown that ligand activation of PPARβ/δ can induce terminal differentiation of keratinocytes and epithelium (Burdick et al., 2006; Peters et al., 2008). Consistent with these findings, many laboratories have also demonstrated that PPARβ/δ inhibits cell growth in epithelium and other cell types, including keratinocytes, colonocytes, cardiomyocytes, lung fibroblasts, and cancer cell lines (Burdick et al., 2006; Peters et al., 2008). Despite a large body of literature demonstrating the induction of terminal differentiation and inhibition of cell growth that is mediated by PPARβ/δ, there are limited reports suggesting that ligand activation of PPARβ/δ can potentiate cell growth. For example, it was originally shown that PPARβ/δ can inhibit the expression of phosphatase and tensin homolog deleted on chromosome Ten (PTEN) and increase expression of 3-phosphoinositide-dependent-protein kinase 1 (PDPK1) and integrin-linked kinase (ILK) expression in keratinocytes during wound healing (Di-Poi et al., 2002). The combined effect of this PPARβ/δ-dependent regulation is increased phosphorylation of protein kinase B (Akt), leading to cell survival via inhibition of apoptosis that may be important during wound healing (Di-Poi et al., 2002). Subsequent work by others suggests that the antiapoptotic signaling mediated by PPARβ/δ during wound healing is also functional in colonic epithelium and human keratinocytes (Gupta et al., 2004; Wang et al., 2006; Schug et al., 2007). However, these changes in the PTEN/PDPK1/Akt pathway are not consistently observed in response to ligand activation of PPARβ/δ in mouse and human keratinocytes, colonic epithelium, or human cancer cell lines (Kim et al., 2006; Marin et al., 2006; Burdick et al., 2007; Hollingshead et al., 2007) and are in direct contrast to the large body of evidence showing that PPARβ/δ induces terminal differentiation and inhibits cell proliferation (Burdick et al., 2006; Peters et al., 2008).
There are a number of reasons that might explain the differences in the reported effects of PPARβ/δ ligands on cell proliferation and apoptosis, including differences in ligands and/or differences in experimental models. For example, GW501516 and GW0742 are two high-affinity ligands for PPARβ/δ (Berger et al., 1999; Sznaidman et al., 2003) that have a similar molecular structure but are structurally dissimilar with retinoic acid (RA), which was described recently as a PPARβ/δ ligand (Shaw et al., 2003). Structural differences between the ligands could explain why some investigators have reported that PPARβ/δ ligand potentiate cell growth, whereas others have reported that PPARβ/δ ligands inhibit cell proliferation. Differences in the approaches used to culture and treat cells and cell lines could also contribute to some of the variability in the literature. For example, studies examining the potential of lipophilic agonists to modulate apoptosis often culture cells in medium without serum or in medium containing a low percentage of charcoal-stripped serum to remove the influence of growth factors or other lipophilic compounds, because these are known to regulate apoptosis. This model system may not be optimal because it is unlikely that endogenous cells typically encounter conditions in the absence of normal serum and/or growth factors. Thus, there is potential for differences in ligands and experimental models to influence the effects of PPARβ/δ ligands on cell proliferation.
It was shown originally that ligand activation of PPARβ/δ induces terminal differentiation and inhibits cell proliferation of human keratinocytes (Burdick et al., 2007), which was consistent with findings from four independent laboratories showing similar effects in mouse keratinocytes (Tan et al., 2001; Westergaard et al., 2001; Schmuth et al., 2004; Kim et al., 2006). In contrast, others have suggested recently that all-trans retinoic acid (atRA) is a PPARβ/δ ligand and that retinoid-specific activation of PPARβ/δ promotes cell survival of human HaCaT keratinocytes by inducing the expression of PDPK1 and antiapoptotic signaling (Shaw et al., 2003; Schug et al., 2007). It was concluded from these studies that PPARβ/δ-specific activation by RA might explain the proproliferative and antiapoptotic effects of retinoic acid. However, this idea is inconsistent with the well established role for PPARβ/δ in promoting terminal differentiation. Thus, the present study critically evaluated the effect of two highly specific PPARβ/δ ligands (GW0742 and GW501516), atRA, and 9-cis retinoic acid (9-cis RA) on gene expression and modulation of cell proliferation in human and mouse keratinocytes.
Materials and Methods
Materials
GW0742 (Sznaidman et al., 2003) was synthesized by GlaxoSmithKline (Research Triangle Park, NC). GW501516 (Sznaidman et al., 2003) was synthesized as described previously (Girroir et al., 2008). atRA and 9-cis RA were purchased from Sigma-Aldrich (St. Louis, MO). GW0742 and GW501516 were dissolved in dimethyl sulfoxide (DMSO), and atRA and 9-cis RA were dissolved in ethanol (EtOH). Propidium iodine was purchased from Sigma-Aldrich. The FITC-Annexin V antibody was purchased from Invitrogen (Carlsbad, CA). The caspase 3/7 Glo reagent was purchased from Promega (Madison, WI).
Cell Culture
HaCaT human keratinocytes were kindly provided from Dr. Stuart Yuspa (National Cancer Institute, Bethesda, MD). These cells were maintained in Dulbecco’s minimal essential medium (DMEM) with 5% fetal bovine serum and 1% penicillin/streptomycin at 37°C and 5% CO2. Primary mouse keratinocytes from wild-type and PPARβ/δ-null mice were isolated from 2-day-old neonates as described previously (Kim et al., 2006). Keratinocytes were cultured in low calcium (0.05 mM) Eagle’s minimal essential medium with 8% chelexed fetal bovine serum at 37°C and 7% CO2 (Kim et al., 2006).
Cell Proliferation Analyses
HaCaT cells were plated on a 12-well plate at a density of 20,000 cells/well 24 h before cell counting at time 0. Cell proliferation was determined using a Z1 Coulter particle counter (Beckman Coulter, Hialeah, FL). Cells were then serum-starved or not for 24 h before ligand treatment. After this 24-h period, cells were maintained in DMEM with or without serum and treated with control (DMSO or EtOH), GW0742, GW501516, atRA, 9-cis RA, or combinations for 24, 48, or 72 h. The concentration of GW0742 and GW501516 used for all experiments ranged from 0.1 to 10.0 μM, because these concentrations have been shown to specifically activate PPARβ/δ (Kim et al., 2006). The concentration of atRA and 9-cis RA used for all experiments ranged from 0.1 to 1.0 μM. Cells were counted every 24 h. Triplicate samples for each treatment were used for each time point, and each replicate was counted three times. For the mouse primary keratinocyte proliferation assay, equivalent numbers (300,000) of cells from both genotypes were plated in 12-well plates. Two days after seeding, the day 0 plates were removed and counted. The remaining plates were switched to new low-calcium media until day 1. After day 1 counts, the remaining plates were treated with control (DMSO or EtOH), GW0742, atRA, or 9-cis RA for 24, 48, and 72 h in low-calcium media. The concentration of agonists examined was either 0.1 or 1.0 μM. Triplicate samples for each treatment were used for each time point, and replicates were counted three times.
Western Blot Analyses
HaCaT cells were cultured on 60-mm culture dishes. Cells were serum-starved for 24 h or not before ligand treatment. After this time, cells were maintained in DMEM with (5%) or without serum and treated with control (DMSO or EtOH), GW0742, GW501516, atRA, or 9-cis RA for 12 h. After 12 h of exposure, protein was isolated using a lysis buffer containing phosphatase and protease inhibitors (20 mM Tris, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1 mM β-glycerophosphate, 2 mM sodium pyrophosphate, and 1% Triton X). For analyzing the expression of retinoic acid receptors (RARs), HaCaT cells and primary keratinocytes from wild-type and PPARβ/δ-null mice were cultured on 100-mm culture dishes in triplicate. Soluble protein was isolated from confluent plates using MENG buffer (25 mM MOPS, 2 mM EDTA, 0.02% NaN3, and 10% glycerol, pH 7.5) containing 500 mM NaCl, 1% Nonidet P-40, and protease inhibitors.
Protein samples were isolated from control- and ligand-treated cells as described above. A total of 25 μg of protein per sample was resolved using SDS-polyacrylamide gels. The samples were transferred onto polyvinylidene fluoride membrane using an electroblotting method. The membranes were blocked with 5% dried milk in Tris-buffered saline/Tween 20 and incubated at 4°C overnight with primary antibodies. After incubation with biotinylated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA), immunoreactive proteins on the membrane were detected after incubation with 125I-labeled streptavidin (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). Hybridization signals for specific proteins were normalized to the hybridization signal of the housekeeping gene lactate dehydrogenase or β-actin. Independent triplicate samples were analyzed for each treatment group. The following antibodies were used: anti-Akt, anti-phospho-Akt, PARP (all from Cell Signaling Technology, Danvers, MA), and anti-lactate dehydrogenase (Rockland, Gilbertsville, PA); RARα, RARβ, RARγ, and RXRα were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The cleavage ratio of PARP was determined by the average ratio of normalized cleaved PARP to normalized uncleaved PARP.
Real-Time PCR
HaCaT cells were cultured on six-well plates. Cells were serum-starved for 24 h or not before ligand treatment. After this time, cells were maintained in DMEM with or without serum and treated with control (DMSO or EtOH); GW0742 or GW501516 (4, 8, or 24 h); or atRA or 9-cis RA (8 or 24 h). For isolation of mRNA from primary keratinocytes, a similar protocol was used. Keratinocytes were cultured to 90 to 95% confluence before treatment with control (DMSO or EtOH), GW0742, atRA, or 9-cis RA, for 8 or 24 h. Total RNA was isolated from cells using TRIzol reagent and the manufacturer’s recommended protocol. The mRNA encoding adipose differentiation-related protein (ADRP), angiopoietin-like protein 4 (Angptl4), PDPK1, transglutaminase 1, cytochrome P450 26A1 (CYP26A1), small proline-rich protein 1A (SPR1A), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was measured by quantitative real-time polymerase chain reaction analysis. cDNA was generated from 2.5 μg of total RNA using MultiScribe Reverse Transcriptase kit (Applied Biosystems, Foster City, CA). Real-time PCR primers for the above genes were designed using SciTools (Integrated DNA Technologies, Coralville, IA). The quantitative real-time PCR analysis was carried out using SYBR Green PCR master mix (Finnzymes, Espoo, Finland) in the iCycler and detected using the MyiQ Realtime PCR Detection System (Bio-Rad Laboratories, Hercules, CA). The following PCR reaction was used for all genes: 95°C for 10 s, 60°C for 30 s, and 72°C for 30 s, repeated for 45 cycles. Each PCR reaction included a no-template control reaction to control for contamination, and all real-time PCR reactions had greater than 85% efficiency. The relative mRNA value for each gene was normalized to the relative mRNA value for the housekeeping gene GAPDH. Statistical analysis of GAPDH expression for all treatment groups revealed no significant differences in expression, allowing for normalization to this gene product (data not shown).
Flow Cytometry
HaCaT cells were plated on a six-well plate at a density of 75,000 (with serum) or 250,000 (without serum) cells per well. Cells were then serum-starved or not 24 h before ligand treatment. After this 24-h period, cells were maintained in DMEM with or without serum and treated with control (DMSO or EtOH), GW0742, GW501516, atRA, 9-cis RA, or combinations for 24 and 48 h (without serum) or 48 and 72 h (with serum) with daily renewal of treatment. Independent triplicate samples for each treatment were used for each time point.
For Annexin V analysis of apoptosis, cells were trypsinized, washed in cold phosphate-buffered saline, and pelleted. The cells were then resuspended in 100 μl of annexin V buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2), and 5 μl of FITC-annexin V antibody was incubated at room temperature for 15 min. Ice-cold annexin V buffer (450 μl) was added to the cells with 2 μg of propidium iodine, and the cells were analyzed by flow cytometry. Approximately 10,000 cells/sample were analyzed using an EPICS-XL-MCL flow cytometer (Beckman Coulter Electronics) fitted with a single 15-mW argon ion laser providing excitation at 488 nm. Cells stained with FITC were monitored through a 525-nm bandpass filter. Early apoptosis was defined as the percentage of cells that were annexin V-positive and propidium iodide-negative, and late apoptosis/necrosis was defined as the percentage of cells that were annexin V-negative and propidium iodide-positive.
Caspase 3/7 Activity
HaCaT keratinocytes were cultured as described above, with and without culture medium serum for up to 72 h in the presence or absence of either GW0742 or retinoic acid. Caspase 3/7 activity was measured using the caspase 3/7 Glo reagent using the manufacturer’s recommended procedures. As a positive control, HaCaT keratinocytes were irradiated with 20,000 μJ/cm2 UV light using the CL-1000 Ultra Violet Cross-linker (UVP, Upland, CA) and examined 12-h after irradiation. Activity was normalized to protein content. Five independent samples per treatment group were examined.
Statistical Analysis
All analyses were made using either a one-way (Western blots) or two-way (proliferation, mRNA, and flow cytometry) analysis of variance (ANOVA) with Bonferroni’s multiple comparison test as mentioned in the figure legends. All results are reported as mean ± S.E.M.
Results
Activation of PPARβ/δ by Specific Ligands Inhibits Cell Proliferation of HaCaT Keratinocytes
To examine the effect of synthetic PPARβ/δ ligands on cell growth, Ha-CaT keratinocyte cell proliferation was quantified in the presence of either GW501516 or GW0742, with or without serum withdrawal. In the presence of culture medium with serum, inhibition of HaCaT cell proliferation was observed with 1.0 and 10 μM GW0742 (Fig. 1A). GW501516 did not influence cell growth of HaCaT cells in the presence of serum in the culture medium (Fig. 1B). Because growth factors and/or potential PPAR ligands present in serum could prevent the detection of significant changes in cell proliferation, these experiments were also performed in the absence of culture medium serum. When HaCaT cells were cultured in the absence of serum, both GW0742 and GW501516 inhibited cell growth (Fig. 1, C and D). These data do not distinguish between inhibition of cell cycle progression and cell death, but subsequent analysis examined these ideas.
Fig. 1.

Ligand activation of PPARβ/δ inhibits cell proliferation of HaCaT keratinocytes. HaCaT cells were treated with either GW0742 (A and C) or GW501516 (B and D) with the indicated concentration of ligand (arrow) in the presence (A and B) or absence (C and D) of culture medium with serum and cell number was quantified as described under Materials and Methods. Values represent the mean ± S.E.M. *, significantly different values (P < 0.05) from vehicle (DMSO) at the particular time point, as determined by ANOVA and Bonferroni’s multiple comparison test.
Activation of PPARβ/δ by Specific Ligands Increases Expression of ADRP and Angptl4 but Not PDPK1
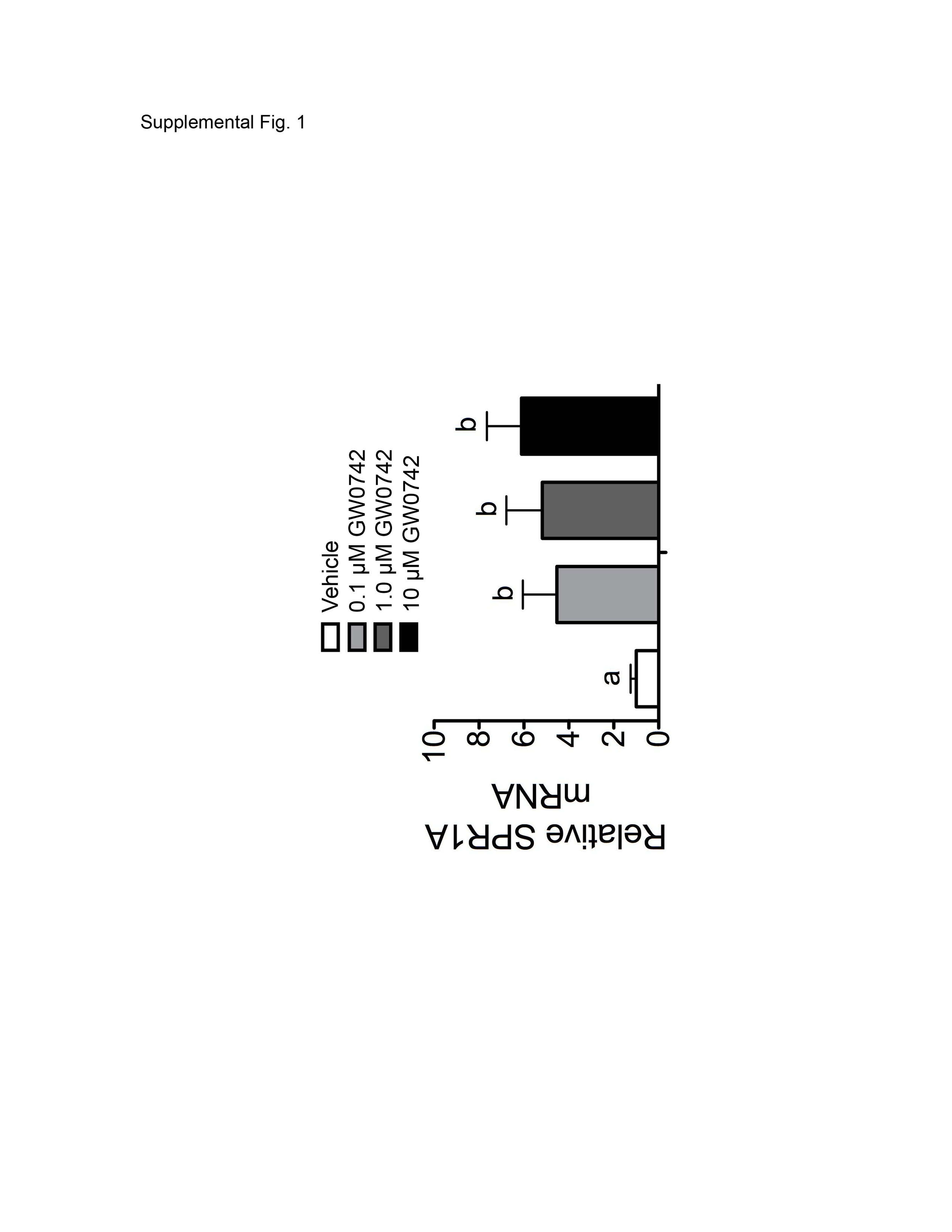
To verify that the inhibition of proliferation by GW0742 and GW501516 (Fig. 1) is associated with specific ligand activation of PPARβ/δ, expression of known and putative PPARβ/δ-dependent target genes was examined. The known PPARβ/δ-dependent target genes ADRP and Angptl4 were induced by GW0742 and GW501516 in a dose-dependent manner that was independent of culture medium serum (Fig. 2, A–D). Because increased expression of ADRP is a marker of keratinocyte differentiation (Westergaard et al., 2001; Schmuth et al., 2004; Kim et al., 2006; Burdick et al., 2007), expression of another mRNA marker of differentiation was also examined. Indeed, expression of SPR1A was increased by ligand activation of GW0742 (Supplemental Fig. 1), consistent with previous work demonstrating that ligand activation of PPARβ/δ induces terminal differentiation of keratinocytes (Tan et al., 2001; Westergaard et al., 2001; Schmuth et al., 2004; Kim et al., 2006; Burdick et al., 2007). In contrast, expression of the putative PPARβ/δ target gene PDPK1 was not altered by either PPARβ/δ ligand at either time point in the presence or absence of serum (Fig. 2E and F). PDPK1 was examined because others have suggested recently that activating PPARβ/δ in keratinocytes causes increased expression of this mRNA (Schug et al., 2007). These data demonstrate that HaCaT keratinocytes are responsive to PPARβ/δ ligands, as shown by the induction of two known PPARβ/δ-dependent target genes within 4 h of treatment. These data also suggest that PDPK1 is not a target of PPARβ/δ in human HaCaT keratinocytes, consistent with past studies (Kim et al., 2006; Marin et al., 2006; Burdick et al., 2007).
Fig. 2.

Modulation of gene expression by ligand activation of PPARβ/δ in HaCaT keratinocytes. HaCaT cells were treated for either 4 or 8 h with the indicated concentration of GW0742 (A, C, and E) or GW501516 (B, D, and F) in the presence or absence of serum. Quantitative real-time PCR was performed as described under Materials and Methods to examine the expression of mRNA encoding ADRP (A and B), Angptl4 (C and D), and PDPK1 (E and F) normalized to mRNA encoding GAPDH. Values are the average -fold change compared with control treatment and represent the mean ± S.E.M. Values with different letters are significantly different, P < 0.05, as determined by ANOVA and Bonferroni’s multiple comparison test.
Activation of PPARβ/δ by Specific Ligands Does Not Lead to Phosphorylation of Akt or Alter PARP Cleavage
Quantitative Western blotting was performed using protein from HaCaT cells treated with GW0742 and GW501516 for 12 h in the presence or absence of culture medium serum. This time point was examined because recent work by others suggested that ligand activation of PPARβ/δ leads to increased phosphorylation of Akt in HaCaT cells after 12 h (Schug et al., 2007). Likewise, because phosphorylated Akt is known to cause antiapoptotic activity, PARP cleavage was examined to determine whether ligand activation of PPARβ/δ would modulate this marker of apoptosis, with particular interest in potential changes that might occur after serum withdrawal when increased PARP cleavage should occur. No change in the expression of Akt protein and no evidence of altered Akt phosphorylation were observed in response to ligand activation of PPARβ/δ in the presence or absence of culture medium serum (Fig. 3, A–D). An increase in the average ratio of cleaved to uncleaved PARP was only observed in serum-deprived HaCaT keratinocytes compared with cells cultured in the presence of serum (Fig. 3, C and D, versus A and B). Neither PPARβ/δ ligand had any effect on PARP cleavage at any concentration in the presence of absence of culture medium serum.
Fig. 3.

Phosphorylation of Akt and PARP cleavage are not influenced by ligand activation of PPARβ/δ in HaCaT cells. HaCaT cells were treated for 12 h with either GW0742 (A and C) or GW501516 (B and D) with the indicated concentration of ligand in the presence (A and B) or absence (C and D) of serum as described under Materials and Methods to examine the quantitative expression of phosphorylated Akt and PARP cleavage. Values are the average -fold change compared with control treatment and represent the mean ± S.E.M. Values with different letters are significantly different, P < 0.05, as determined by ANOVA and Bonferroni’s multiple comparison test. The cleavage ratio is an indicator of apoptosis and is the average ratio of cleaved PARP to uncleaved PARP normalized values. E, Western blot analysis demonstrating specificity of the phospho-Akt antibody. Rat cerebrum lysate (+) was used as a positive control and representative samples from HaCaT cells treated with either GW0742, atRA, or 9-cis RA were used comparison. Phospho-Akt has a molecular mass of 60 kDa.
Ligand Activation of PPARβ/δ Increases Annexin V Staining and Caspase 3/7 Activity
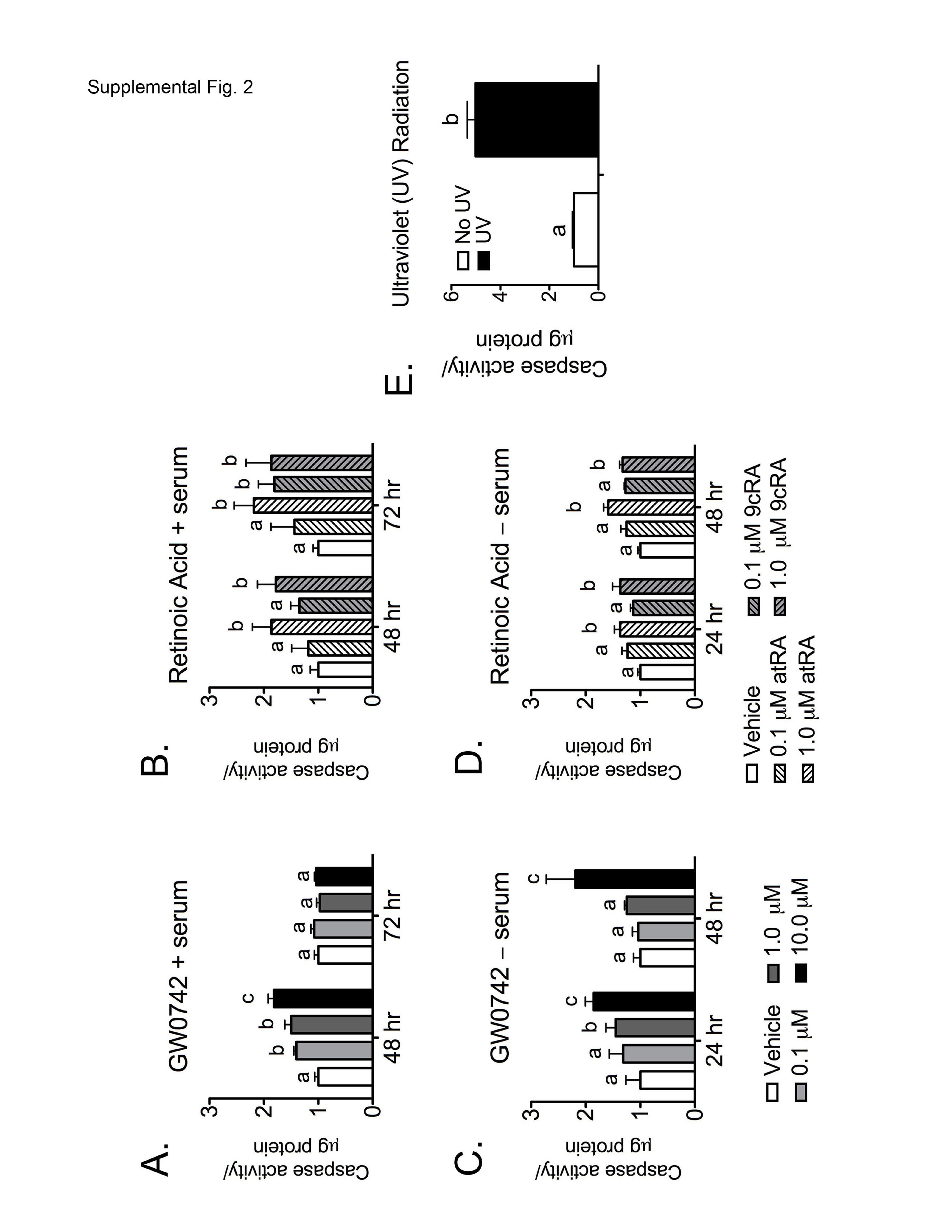
The observed decrease in cell proliferation (Fig. 1) could be due to the inhibition of cell cycle and/or modulation of apoptosis. Flow cytometric analysis using BrdU did not reveal significant differences in cell cycle progression (data not shown). Thus, flow cytometric analysis was performed to determine whether the observed decreases in cell proliferation by ligand activation of PPARβ/δ (Fig. 1) was due to modulation of apoptosis. The timing of this analysis corresponded to the time points just preceding and including the time point when a significant decrease in cell proliferation was observed [e.g., 48–72 h after ligand treatment in the presence of serum (Fig. 1A) and 24–48 h after ligand treatment in the absence of serum (Fig. 1C)]. A dose-dependent increase in the percentage of cells undergoing early apoptosis (annexin V-positive/propidium iodide-negative) was observed 48 h after ligand treatment in the presence of serum (Table 1), but these changes were not observed 72 h after ligand treatment (Table 1). In the absence of culture medium serum, an increase in cells undergoing early apoptosis was observed 72 h after ligand treatment in cells exposed to 10 μM GW0742 (Table 1). No changes in the percentage of cells undergoing late apoptosis (annexin V-negative/propidium iodide-positive) were observed for any treatment group. Consistent with the observed changes in annexin V staining, increased caspase 3/7 activity was also found after ligand activation of PPARβ/δ by GW0742 in HaCaT keratinocytes (Supplemental Fig. 2).
TABLE 1.
Flow cytometry analysis of annexin V/propidium iodide in HaCaT cells after ligand activation of PPARβ/δ
| GW0742 | 48 h |
72 h |
||
|---|---|---|---|---|
| Early Apoptosis | Late Apoptosis | Early Apoptosis | Late Apoptosis | |
| % | ||||
| + Serum | ||||
| Vehicle | 2.6 ± 0.1a | 0.4 ± 0.1a | 4.0 ± 0.5a | 0.6 ± 0.2a |
| 0.1 μM | 4.5 ± 0.4b | 0.3 ± 0.1a | 3.7 ± 0.2a | 0.5 ± 0.1a |
| 1.0 μM | 5.6 ± 0.2c | 0.4 ± 0.1a | 3.6 ± 0.5a | 0.9 ± 0.4a |
| 10.0 μM | 6.8 ± 0.3d | 0.5 ± 0.0a | 3.4 ± 0.4a | 0.5 ± 0.1a |
| − Serum | ||||
| Vehicle | 1.1 ± 0.2a | 0.9 ± 0.3a | 1.3 ± 0.1a | 0.2 ± 0.0a |
| 0.1 μM | 1.1 ± 0.2a | 0.6 ± 0.1a | 1.6 ± 0.3a | 0.2 ± 0.1a |
| 1.0 μM | 1.1 ± 0.1a | 0.5 ± 0.0a | 1.6 ± 0.2a | 0.2 ± 0.0a |
| 10.0 μM | 0.8 ± 0.2a | 0.4 ± 0.2a | 2.4 ± 0.5b | 0.6 ± 0.2a |
HaCaT cells were treated in triplicate for the indicated times with the indicated concentration of ligand in the presence (top) or absence (bottom) of culture medium serum. Values (mean ± S.E.M.) with different letters in each column are significantly different, P < 0.05, as determined by ANOVA and Bonferroni’s multiple comparison test. Early apoptosis was defined as the percentage of cells that were annexin V-positive and propidium iodide-negative, and late apoptosis/necrosis was defined as the percentage of cells that were annexin V-negative and propidium iodide-positive.
Retinoic Acid Inhibits HaCaT Cell Proliferation
To examine the effect of retinoic acid on cell growth, HaCaT keratinocyte proliferation was quantified in the presence of either atRA or 9-cis RA with or without culture medium serum withdrawal. atRA and 9-cis RA inhibited HaCaT cell proliferation in the presence of culture medium serum. Cells were more sensitive to 9-cis RA than atRA because inhibition of cell growth occurred at a lower concentration (0.1 μM) (Fig. 4, A and B). In the absence of culture medium serum, both 0.1 and 1.0 μM concentrations of atRA and 9-cis RA inhibited HaCaT cell proliferation with similar efficacy.
Fig. 4.

Effect of retinoic acid on HaCaT cell proliferation. HaCaT cells were treated with either atRA (A and C) or 9-cis RA (B and D) with the indicated concentration (arrow) in the presence (A and B) or absence (C and D) of culture medium serum, and cell number was quantified as described under Materials and Methods. Values represent the mean ± S.E.M. *, significantly different values (P < 0.05) from vehicle, as determined by ANOVA and Bonferroni’s multiple comparison test.
Retinoic Acid Regulates RAR-Dependent Target Genes but Does Not Regulate PPARβ/δ-Dependent Target Genes
Expression of PPARβ/δ-dependent target genes and RAR-dependent target genes was examined after exposure to retinoic acid. atRA and 9-cis RA did not increase expression of mRNA encoding the well characterized PPARβ/δ-dependent target Angptl4 after either 8 or 24 h of treatment (Fig. 5A). In contrast, a marked increase in the expression of Angptl4 mRNA was found in response to 0.2 μM GW0742 after 8 and 24 h of culture (Fig. 5A). Both atRA and 9-cis RA modulated expression of known RAR-dependent target genes; CYP26A1 was induced, and transglutaminase 1 was repressed (Fig. 5, B and C). These changes were not observed after exposure to the PPARβ/δ ligand GW0742. Neither atRA, 9-cis RA, nor GW0742 influenced expression of mRNA encoding PDPK1 (Fig. 5D).
Fig. 5.

Effect of retinoic acid on gene expression in HaCaT keratinocytes. HaCaT cells were treated for either 8 or 24 h with either GW0742 (0.2 μM), atRA, or 9-cis RA at the indicated concentration in the presence of serum. Quantitative real-time PCR was performed as described under Materials and Methods to examine the expression of mRNA encoding Angptl4 (A), CYP26A1 (B), TGM1 (C), and PDPK1 (D) normalized to mRNA encoding GAPDH. Values are the average -fold change compared with control treatment and represent the mean ± S.E.M. Values with different letters are significantly different, P < 0.05, as determined by ANOVA and Bonferroni’s multiple comparison test.
Retinoic Acid Does Not Lead to Phosphorylation of Akt or Alter PARP Cleavage
Quantitative Western blotting was performed on protein samples from HaCaT cells to determine whether retinoic acid can modulate phosphoryla-tion of Akt and/or PARP cleavage as markers of apoptotic signaling. No change in the expression or phosphorylation of Akt was found in response to either atRA or 9-cis RA in the presence of absence of culture medium serum (Fig. 6). Likewise, no change in PARP cleavage was observed in response to retinoic acid in the presence or absence of culture medium serum (Fig. 6). The only significant change in PARP cleavage was observed in serum-deprived cells compared with cells cultured in the presence of serum (Fig. 6).
Fig. 6.

Phosphorylation of Akt and PARP cleavage are not influenced by retinoic acid in HaCaT cells. HaCaT cells were treated for 12 h with either atRA or 9-cis RA at the indicated concentration in the presence (A) or absence (B) of culture medium serum as described under Materials and Methods to examine the quantitative expression of phosphorylated Akt and PARP cleavage. Values are the average -fold change compared with control treatment and represent the mean ± S.E.M. Values with different letters are significantly different, P < 0.05, as determined by ANOVA and Bonferroni’s multiple comparison test. The cleavage ratio is an indicator of apoptosis and is the average ratio of cleaved PARP to uncleaved PARP normalized values.
Retinoic Acid Increases Annexin V Staining and Caspase Activity
Flow cytometric analysis was performed to determine whether the observed decreases in cell proliferation by retinoic acid (Fig. 4) was due to modulation of apoptosis. The timing of this analysis corresponded to the time points just preceding and including the time point when a significant decrease in cell proliferation was observed [e.g., 48–72 h after retinoic acid treatment in the presence of serum (Fig. 4, A and B), and 24–48 h after retinoic acid treatment in the absence of serum (Fig. 4, C and D]. In the presence of culture medium serum, atRA and 9-cis RA significantly increased the percentage of cells undergoing early apoptosis 48 h after retinoic acid treatment (Table 2). The percentage of cells undergoing late apoptosis was also significantly increased by atRA and 9-cis RA 48 and 72 h after retinoic acid treatment (Table 2). In the absence of culture medium serum, no significant changes in the percentage of cells undergoing early or late apoptosis was observed at either time point. It is interesting that the percentage of cells undergoing apoptosis was higher in retinoic acid-treated cells (Table 2) compared with GW0742-treated cells (Table 1). Consistent with the observed changes in annexin V staining, increased caspase 3/7 activity was also found after exposure to retinoic acid in HaCaT keratinocytes (Supplemental Fig. 2).
TABLE 2.
Flow cytometry analysis of annexin V/propidium iodide in HaCaT cells after atRA or 9-cis RA
| Treatment | 48 h |
72 h |
||
|---|---|---|---|---|
| Early Apoptosis | Late Apoptosis | Early Apoptosis | Late Apoptosis | |
| % | ||||
| + Serum | ||||
| Vehicle | 6.1 ± 0.5a | 1.7 ± 0.3a | 4.7 ± 0.3a | 1.8 ± 0.7a |
| 0.1 μM atRA | 8.5 ± 0.8a | 2.4 ± 0.6a | 4.4 ± 0.3a | 1.9 ± 0.5a |
| 1.0 μM atRA | 11.6 ± 0.7b | 5.6 ± 0.9b | 4.1 ± 0.3a | 4.1 ± 0.1b |
| 0.1 μM 9-cis RA | 10.7 ± 1.0b | 3.8 ± 1.1a | 5.2 ± 0.4a | 2.6 ± 0.7a |
| 1.0 μM 9-cis RA | 9.2 ± 1.0b | 4.9 ± 1.4b | 4.4 ± 0.1a | 6.7 ± 1.0c |
| − Serum | ||||
| Vehicle | 0.6 ± 0.1a | 1.0 ± 0.1a | 1.0 ± 0.0a | 0.4 ± 0.1a |
| 0.1 μM atRA | 0.5 ± 0.1a | 0.6 ± 0.0a | 0.8 ± 0.1a | 0.2 ± 0.0a |
| 1.0 μM atRA | 0.4 ± 0.1a | 1.0 ± 0.1a | 1.0 ± 0.1a | 0.6 ± 0.4a |
| 0.1 μM 9-cis RA | 0.4 ± 0.1a | 1.0 ± 0.2a | 0.9 ± 0.1a | 0.2 ± 0.0a |
| 1.0 μM 9-cis RA | 0.4 ± 0.1a | 1.1 ± 0.3a | 1.1 ± 0.1a | 0.4 ± 0.1a |
HaCaT cells were treated in triplicate for the indicated times with the indicated concentration of RA in the presence (top) or absence (bottom) of culture medium serum. Values (mean ± S.E.M.) with different letters in each column are significantly different, P < 0.05, as determined by ANOVA and Bonferroni’s multiple comparison test.
GW0742 and Retinoic Acid Decrease Mouse Primary Keratinocyte Cell Proliferation
Primary mouse keratin-ocytes from wild-type and PPARβ/δ-null mice were used to assess the specific role of PPARβ/δ in modulating cell growth. Keratinocytes from PPARβ/δ-null mice proliferated much faster compared with wild-type keratinocytes, consistent with previous studies (Kim et al., 2006). Inhibition of cell proliferation was observed in wild-type mouse keratinocytes after exposure to GW0742, and this effect was not found in similarly treated PPARβ/δ-null keratinocytes (Fig. 7A). In contrast, atRA and 9-cis RA inhibited cell proliferation in both wild-type and PPARβ/δ-null keratinocytes (Fig. 7, B and C).
Fig. 7.

Role of PPARβ/δ in the modulation of cell growth of mouse primary keratinocytes by GW0742 or retinoic acid. Primary keratinocytes from wild-type +/+) and PPARβ/δ-null (−/−) mice were treated with the indicated concentration of either GW0742 (A), atRA (B), or 9-cis RA (C; arrow), and cell number was quantified as described under Materials and Methods. Values represent the mean ± S.E.M. *, significantly different values (P < 0.05) from vehicle, as determined by ANOVA and Bonferroni’s multiple comparison test.
Retinoic Acid Increases the Expression of an RAR-Dependent Target Gene but Does Not Increase the Expression of a PPARβ/δ Target Gene in Mouse Primary Keratinocytes
Primary keratinocytes from wild-type and PPARβ/δ-null mice were used to examine changes in gene expression of RAR- and PPARβ/δ-dependent target genes. At a concentration that specifically activates PPARβ/δ (0.2 μM), GW0742 increased the expression of Angptl4 mRNA in wild-type keratinocytes at both time points examined, and this increase was not observed in similarly treated PPARβ/δ-null keratinocytes (Fig. 8, A and B). atRA did not increase Angptl4 mRNA expression (Fig. 8, A and B), but 9-cis RA did cause an increase after 24 h of treatment (Fig. 8, A and B). This is of interest because the increase in Angptl4 mRNA expression did not occur in PPARβ/δ-null keratinocytes. This is consistent with previous work showing that 9-cis RA can activate PPAR/RXR heterodimers and increase the expression of PPAR target genes (Mukherjee et al., 1997). atRA and 9-cis RA both increased expression of the RAR-dependent target gene CYP26A1 in wild-type and PPARβ/δ-null keratinocytes, whereas GW0742 had no effect on CYP26A1 mRNA in either genotype (Fig. 8, C and D).
Fig. 8.

Role of PPARβ/δ in the modulation of gene expression by GW0742 or retinoic acid in mouse primary keratinocytes. Primary keratinocytes from wild-type (+/+) and PPARβ/δ-null (−/−) mice were treated for 8 (left) or 24 (right) h with either GW0742, atRA, or 9-cis RA at the indicated concentration. Quantitative real-time PCR was performed as described under Materials and Methods to examine the expression of mRNA encoding Angptl4 (A and B) or CYP26A1 (C and D) normalized to mRNA encoding GAPDH. Values are the average -fold change compared with control treatment and represent the mean ± S.E.M. Values with different letters are significantly different, P < 0.05, as determined by ANOVA and Bonferroni’s multiple comparison test.
HaCaT and Mouse Primary Keratinocytes Differentially Express RAR Isoforms
To confirm that retinoic acid receptors (RARα, RARβ, RARγ, and RXRα) are expressed in HaCaT keratinocytes and primary keratinocytes, quantitative Western blotting was performed on soluble cellular ly-sates from HaCaT cells and wild-type and PPARβ/δ-null primary keratinocytes. Expression of all three RAR isoforms was detected in HaCaT keratinocytes; however, RARγ was only expressed in primary keratinocytes (Fig. 9). The expression of RXRα, the heterodimerization partner of PPARβ/δ and RARs, was highly expressed in both HaCaT and primary mouse keratinocytes.
Fig. 9.
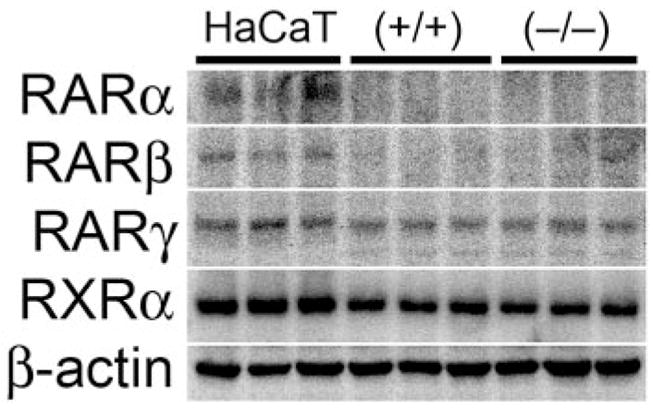
Expression of retinoid receptors in mouse keratinocytes and HaCaT keratinocytes. HaCaT and primary keratinocytes from wild-type (+/+) and PPARβ/δ-null (−/−) mice were cultured as described under Materials and Methods to examine the quantitative expression of RARα, RARβ, RARγ, and RXRα.
Cotreatment of GW0742 and Retinoic Acid Decreases HaCaT Cell Proliferation and Independently Regulate Receptor-Specific Target Genes
If retinoids and PPARβ/δ ligands were functioning to promote antiapoptotic signaling, then combining the two ligands might allow us to observe this effect that we were not observing with only the ligand. Toward this goal, both cell proliferation and markers of gene expression were examining using combinations of ligands using concentrations that are known to specifically activate the respective receptor. As noted above, others have suggested that ligand activation of PPARβ/δ will lead to increased expression of PDPK1 and subsequent antiapoptotic activity (Schug et al., 2007). To begin to examine whether inhibition of cell proliferation by retinoic acid and GW0742 could lead to additive or synergistic effects, HaCaT cell proliferation was examined after cotreatment with retinoic acid and GW0742. Similar to the results described above, atRA and 9-cis RA significantly inhibited Ha-CaT cell proliferation (Fig. 10, A–C). GW0742 did not inhibit cell proliferation, but this concentration (0.2 μM) was used because it specifically activates PPARβ/δ without inhibiting cell growth (Figs. 1A and 5A). Cotreatment of atRA or 9-cis RA with 0.2 μM GW0742 did not lead to enhanced inhibition of cell proliferation compared with inhibition observed with atRA or 9-cis RA alone in the presence or absence of culture medium serum (Fig. 10). However, combining atRA with 9-cis RA caused a significantly greater inhibition of cell proliferation in the absence of culture medium serum compared with inhibition observed with atRA or 9-cis RA alone (Fig. 10).
Fig. 10.

Effect of combining GW0742 with retinoic acid on cell proliferation of HaCaT keratinocytes. HaCaT cells were treated with either GW0742 (0.2 μM), atRA (1.0 μM), or 9-cis RA (1.0 μM) (A and C) or combinations of GW0742, atRA, and 9-cis RA (arrow) (B and D) in the presence (A and B) or absence (C and D) of culture medium serum, and cell number was quantified as described under Materials and Methods. Values represent the mean ± S.E.M. *, significantly different values (P < 0.05) from vehicle (DMSO) at the particular time point, as determined by ANOVA and Bonferroni’s multiple comparison test.
Ligand activation of PPARβ/δ caused an increase in the expression of mRNA encoding Angptl4 in HaCaT cells, whereas atRA and 9-cis RA had no effect on this PPARβ/δ target gene (Fig. 11, A and B). Combining atRA or 9-cis RA with GW0742 did not consistently alter the induction of Angptl4, but a modest enhancement was observed after 8 h of treatment with atRA and GW0742 (Fig. 11A). Increased expression of CYP26A1 mRNA was observed in atRA- and 9-cis RA-treated HaCaT cells, but this effect was not consistently altered by cotreatment with GW0742 (Fig. 11, C and D). Expression of mRNA encoding PDPK1 was not altered by atRA, 9-cis RA, or GW0742 (Fig. 11, E and F). No consistent changes in PDPK1 mRNA were observed after cotreatment with either atRA or 9-cis RA with GW0742, but a decrease in PDPK1 mRNA was found after cotreatment of GW0742 and 9-cis RA or cotreatment of atRA and 9-cis RA (Fig. 11, E and F).
Fig. 11.

Effect of combining GW0742 with retinoic acid on gene expression in HaCaT keratinocytes. HaCaT cells were treated for either 8 (left) or 24 (right) h with either GW0742 (0.2 μM), atRA, or 9-cis RA at the indicated concentration in the presence of serum. Quantitative real-time PCR was performed as described under Materials and Methods to examine the expression of mRNA encoding Angptl4 (A and B), CYP26A1 (C and D), and PDPK1 (E and F) normalized to mRNA encoding GAPDH. Values are the average -fold change compared with control treatment and represent the mean ± S.E.M. Values with different letters are significantly different, P < 0.05, as determined by ANOVA and Bonferroni’s multiple comparison test.
Discussion
Results from the present study clearly indicate that ligand activation of PPARβ/δ inhibits cell proliferation in human HaCaT keratinocytes (Fig. 12). This observation is consistent with previous work showing PPARβ/δ-dependent inhibition of cell proliferation in keratinocytes (Peters et al., 2000; Michalik et al., 2001; Westergaard et al., 2001; Kim et al., 2004, 2005, 2006; Burdick et al., 2007; Man et al., 2008) and many other cell types (Burdick et al., 2006; Peters et al., 2008). Because the observed inhibition of cell proliferation by ligand activation of PPARβ/δ is not found in mouse keratinocytes that do not express PPARβ/δ, this demonstrates that this effect requires a functional receptor. The specific mechanism(s) that lead to inhibition of cell proliferation in human HaCaT keratinocytes cannot be determined from the present studies. However, because inhibition of cell proliferation is typically associated with terminal differentiation, it is important to note that increased expression of known differentiation markers (e.g., ADRP and SPR1A) was observed in the present study and that PPARβ/δ has been linked with modulation of terminal differentiation in keratinocytes (Matsuura et al., 1999; Tan et al., 2001; Schmuth et al., 2004; Kim et al., 2006; Burdick et al., 2007; Man et al., 2008) and other cell types, including intestinal epithelium (Burdick et al., 2006; Peters et al., 2008). It is interesting that the induction of terminal differentiation of keratinocytes is associated with increased activity of proapoptotic-like signaling (Weil et al., 1999). Thus, the increase in annexin V-positive cells and caspase 3/7 activity found in response to ligand activation of PPARβ/δ is also consistent with the idea that PPARβ/δ mediates terminal differentiation and might explain in part the decreased cell proliferation observed after activation of PPARβ/δ in HaCaT keratinocytes. Given that HaCaT keratinocytes are relatively resistant to the induction of apoptosis (Henseleit et al., 1996), the observed increase in apoptosis with ligand activation of PPARβ/δ illustrates a unique function of PPARβ/δ in this cell type.
Fig. 12.

PPARβ/δ- and RAR/RXR-dependent modulation of keratinocyte cell growth. In response to ligand activation, PPARβ/δ heterodimerizes with RXR, leading to up-regulation of target genes that cause terminal differentiation and apoptotic signaling culminating in the inhibition of cell growth. 9-cis RA may also interact with this signaling by enhancing this effect. atRA or 9-cis RA activate RAR and RXR, respectively, and lead to either heterodimerization or homodimerization with RXR, respectively, and up-regulation of target genes that cause an increase in apoptotic signaling and inhibition of cell growth.
Previous studies by others suggested that ligand activation of PPARβ/δ in keratinocytes promotes cell survival by modulating PTEN/PDPK1/ILK/Akt activity, leading to antiapoptotic signaling (Di-Poi et al., 2002). However, this signaling seems to be context-specific because changes in these signaling proteins may occur in keratinocytes during wound healing but are clearly not found in normal mouse or human keratinocytes, based on results reported from the present study and from previous work (Kim et al., 2006; Burdick et al., 2007). This is also supported by the lack of changes in the PTEN/PDPK1/ILK/Akt expression and/or activity after ligand activation of PPARβ/δ in colon and human colon cancer cell lines (Marin et al., 2006; Hollingshead et al., 2007). Together, earlier work and results from the present study strongly support the idea that ligand activation of PPARβ/δ inhibits cell proliferation by inducing terminal differentiation and apoptotic signaling. Furthermore, these findings do not support the hypothesis that PPARβ/δ promotes cell survival of keratinocytes by modulating PTEN/PDPK1/ILK/Akt activity leading to antiapoptotic signaling, as suggested by others (Di-Poi et al., 2002)
Because recent evidence suggests that retinoic acid is a ligand for PPARβ/δ (Shaw et al., 2003), the effect of retinoic acid on HaCaT cell proliferation was also examined. Results from the present study provide convincing evidence that atRA and 9-cis RA inhibit cell proliferation of both human HaCaT keratinocytes and mouse primary keratinocytes, and that this effect is associated with an increase in apoptosis. It is interesting that the relative percentage of cells undergoing apoptosis in response to atRA and 9-cis RA is significantly greater than the percentage of cells undergoing apoptosis in response to a potent PPARβ/δ ligand. These observations are consistent with the inhibition of cell proliferation found in HaCaT keratinocytes, other human keratinocyte cell lines, and various human cancers after administration of retinoic acid (Chen et al., 2000; Hansen et al., 2000; Kanekura et al., 2000; Klaassen et al., 2001; Memezawa et al., 2007). Retinoic acid also inhibits cell proliferation in mouse primary keratinocytes, which is consistent with previous studies (Tong et al., 1988) and with the inhibition of skin cancer by retinoids observed in several mouse models (Verma et al., 1980; Verma, 1987, 1988; Chen et al., 1994a,b; Tennenbaum et al., 1998; Xu et al., 2006). It is also worth noting that loss of RAR isoforms has been shown to enhance tumorigenesis (Darwiche et al., 1995, 1996; Chen et al., 2004). Together, results from the present studies demonstrate that retinoic acid inhibits cell proliferation in mouse primary keratinocytes and human HaCaT keratinocytes (Fig. 12).
In contrast with results from the present study and other published reports, it was suggested recently that retinoic acid acts as a PPARβ/δ ligand and promotes cell survival and increased cell growth of HaCaT keratinocytes (Schug et al., 2007). This was an attractive hypothesis to potentially explain the known differential effects of retinoic acid reported in the literature showing that retinoic acid inhibits cell proliferation in some models but increases cell proliferation in other models. However, the former analysis was limited in scope and only examined the expression of an mRNA encoding a putative PPARβ/δ target gene (e.g., PDPK1) and did not critically evaluate cell proliferation and apoptosis in HaCaT keratinocytes as performed in the present analysis. Given the significant weight of evidence from multiple laboratories demonstrating PPARβ/δ-dependent inhibition of cell proliferation in keratinocytes (Westergaard et al., 2001; Kim et al., 2004, 2005, 2006; Martinasso et al., 2006; Burdick et al., 2007; Man et al., 2008) and many other cell types (Burdick et al., 2006; Peters et al., 2008), it is surprising that the studies by Schug et al. (2007) did not address this inconsistency in their work. Indeed, the present study in which cell proliferation was examined with quantitative measures under several culture conditions revealed multiple inconsistencies with the hypothesis that ligand activation of PPARβ/δ by retinoic acid promotes cell survival. For example, increased expression of PDPK1 is not found in HaCaT keratinocytes cultured in the presence of retinoic acid, despite demonstration of increased expression of known RA-responsive genes (e.g., CYP26A1). Both atRA and 9-cis RA also failed to increase the expression of known PPARβ/δ target genes in HaCaT keratinocytes, whereas PPARβ/δ ligands activated the expression of ADRP and Angptl4. More importantly, retinoic acid did not alter phosphorylation of Akt, inhibit serum withdrawal-induced cleavage of PARP, or reduce annexin V-positive cells, and it is noteworthy that there was no increase in cell proliferation. These observations demonstrate that retinoic acid does not potentiate cell proliferation of HaCaT keratinocytes. Because combining ligand activation did not counteract the growth-inhibitory effects of retinoic acid in HaCaT keratinocytes, this provides more indirect support that retinoic acid does not function differentially through both PPARβ/δ and RAR/RXRs. In addition, atRA and 9-cis RA inhibited cell proliferation in both wild-type and PPARβ/δ-null mouse primary keratinocytes. This demonstrates that retinoic acid inhibits cell proliferation and that the mechanisms underlying this inhibition do not require PPARβ/δ. These results are not surprising, given recent studies demonstrating that atRA does not bind to or activate PPARβ/δ, and that atRA does not cause PPARβ/δ to interact with a coactivator peptide in a time-resolved fluorescence resonance energy transfer assay (Rieck et al., 2008). Together, these findings are in contrast to studies reported previously by others (Schug et al., 2007). These results demonstrate that retinoic acid modulates HaCaT keratinocyte cell proliferation by increasing apoptosis thereby inhibiting growth but provide no evidence that retinoic acid potentiates cell proliferation by activating PPARβ/δ as suggested previously (Schug et al., 2007).
In summary, the present findings provide additional observations to the increasing body of evidence demonstrating that ligand activation of PPARβ/δ inhibits cell proliferation. This conclusion is based on comprehensive analysis using two high-affinity ligands and quantitative measures of cell proliferation, differentiation, and apoptosis. It is of interest to note that inhibition of keratinocyte proliferation by PPARα and PPARγ agonists have also been observed (Hanley et al., 1998; Demerjian et al., 2006), suggesting that there may be redundancy in the target genes modulated by PPARs in keratinocytes that mediate this effect. Results from the present study also clearly demonstrate that retinoic acid inhibits proliferation of mouse and human keratinocytes but does not activate PPARβ/δ. These findings also strongly suggest that the mechanisms underlying the differential effects of retinoids on cell proliferation are not mediated by PPARβ/δ. Further studies will be necessary to determine how retinoids can increase cell growth in some models and inhibit cell growth in others.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We gratefully acknowledge Drs. Andrew N. Billin and Timothy M. Willson for providing GW0742 and Dr. Pengfei He and the Penn State University Flow Cytometry Core Facility (Elaine Kunze, Susan Magargee, and Nicole Bem) for their technical support with flow cytometry and data analysis.
This work was supported by a National Science Foundation Graduate Research Fellowship and by National Institutes of Health grants CA124533 (to J.M.P.) and CA90214 (to A.C.R.).
ABBREVIATIONS
- PPAR
peroxisome proliferator-activated receptor
- 9-cis RA
9-cis retinoic acid
- ADRP
adipocyte differentiation-related protein
- ANOVA
analysis of variance
- Angptl4
angiopoietin-like protein 4
- Akt
protein kinase B
- atRA
all-trans retinoic acid
- DMSO
dimethyl sulfoxide
- DMEM
Dulbecco’s minimal essential medium
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- ILK
integrin-linked kinase
- PDPK1
3-phosphoinositide-dependent protein kinase 1
- PCR
polymerase chain reaction
- PTEN
phosphatase and tensin homolog deleted on chromosome ten
- MOPS
3-(N-morpholino)propanesulfonic acid
- PARP
poly(ADP-ribose) polymerase
- PI
propidium iodine
- RA
retinoic acid
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- GW0742
4-[[[2-[3-fluoro-4-(trifluoromethyl)phenyl]-4-methyl-5-thiazolyl]methyl]thio]-2-methylphenoxy acetic acid
- GW501516
2-methyl-4-((4-methyl-2-(4-trifluoromethylphenyl)-1,3-thiazol-5-yl)-methylsulfanyl)phenoxy-acetic acid
- FITC
fluorescein isothiocyanate
- SPR1A
small proline-rich protein 1A
Footnotes
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
References
- Berger J, Leibowitz MD, Doebber TW, Elbrecht A, Zhang B, Zhou G, Biswas C, Cullinan CA, Hayes NS, Li Y, Tanen M, Ventre J, Wu MS, Berger GD, Mosley R, Marquis R, Santini C, Sahoo SP, Tolman RL, Smith RG, Moller DE. Novel peroxisome proliferator-activated receptor (PPAR) γ and PPARδ ligands produce distinct biological effects. J Biol Chem. 1999;274:6718–6725. doi: 10.1074/jbc.274.10.6718. [DOI] [PubMed] [Google Scholar]
- Burdick AD, Bility MT, Girroir EE, Billin AN, Willson TM, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) inhibits cell growth of human N/TERT-1 keratinocytes. Cell Signal. 2007;19:1163–1171. doi: 10.1016/j.cellsig.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick AD, Kim DJ, Peraza MA, Gonzalez FJ, Peters JM. The role of peroxisome proliferator-activated receptor-β/δ in epithelial growth and differentiation. Cell Signal. 2006;18:9–20. doi: 10.1016/j.cellsig.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Chen CF, Goyette P, Lohnes D. RARγ acts as a tumor suppressor in mouse keratinocytes. Oncogene. 2004;23:5350–5359. doi: 10.1038/sj.onc.1207682. [DOI] [PubMed] [Google Scholar]
- Chen LC, Kirchhoff S, De Luca LM. Effect of excess dietary retinoic acid on skin papilloma and carcinoma formation induced by a complete carcinogenesis protocol in female Sencar mice. Cancer Lett. 1994a;78:63–67. doi: 10.1016/0304-3835(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Chen LC, Sly L, De Luca LM. High dietary retinoic acid prevents malignant conversion of skin papillomas induced by a two-stage carcinogenesis protocol in female SENCAR mice. Carcinogenesis. 1994b;15:2383–2386. doi: 10.1093/carcin/15.10.2383. [DOI] [PubMed] [Google Scholar]
- Chen WC, Sass JO, Seltmann H, Nau H, Orfanos CE, Zouboulis CC. Biological effects and metabolism of 9-cis-retinoic acid and its metabolite 9,13-di-cis-retinoic acid in HaCaT keratinocytes in vitro: comparison with all-trans-retinoic acid. Arch Dermatol Res. 2000;292:612–620. doi: 10.1007/s004030000189. [DOI] [PubMed] [Google Scholar]
- Darwiche N, Celli G, Tennenbaum T, Glick AB, Yuspa SH, De Luca LM. Mouse skin tumor progression results in differential expression of retinoic acid and retinoid X receptors. Cancer Res. 1995;55:2774–2782. [PubMed] [Google Scholar]
- Darwiche N, Scita G, Jones C, Rutberg S, Greenwald E, Tennenbaum T, Collins SJ, De Luca LM, Yuspa SH. Loss of retinoic acid receptors in mouse skin and skin tumors is associated with activation of the ras(Ha) oncogene and high risk for premalignant progression. Cancer Res. 1996;56:4942–4949. [PubMed] [Google Scholar]
- Demerjian M, Man MQ, Choi EH, Brown BE, Crumrine D, Chang S, Mauro T, Elias PM, Feingold KR. Topical treatment with thiazolidinediones, activators of peroxisome proliferator-activated receptor-γ, normalizes epidermal homeostasis in a murine hyperproliferative disease model. Exp Dermatol. 2006;15:154–160. doi: 10.1111/j.1600-0625.2006.00402.x. [DOI] [PubMed] [Google Scholar]
- Di-Poï N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARβ in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell. 2002;10:721–733. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- Girroir EE, Hollingshead HE, Billin AN, Willson TM, Robertson GP, Sharma AK, Amin S, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-β/δ (PPAR β/δ) ligands inhibit growth of UACC903 and MCF7 human cancer cell lines. Toxicology. 2008;243:236–243. doi: 10.1016/j.tox.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi PA. Regulatory functions of PPARβ in metabolism: implications for the treatment of metabolic syndrome. Biochim Biophys Acta. 2007;1771:983–990. doi: 10.1016/j.bbalip.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-δ accelerates intestinal adenoma growth. Nat Med. 2004;10:245–247. doi: 10.1038/nm993. [DOI] [PubMed] [Google Scholar]
- Hanley K, Jiang Y, He SS, Friedman M, Elias PM, Bikle DD, Williams ML, Feingold KR. Keratinocyte differentiation is stimulated by activators of the nuclear hormone receptor PPARα. J Invest Dermatol. 1998;110:368–375. doi: 10.1046/j.1523-1747.1998.00139.x. [DOI] [PubMed] [Google Scholar]
- Hansen LA, Sigman CC, Andreola F, Ross SA, Kelloff GJ, De Luca LM. Retinoids in chemoprevention and differentiation therapy. Carcinogenesis. 2000;21:1271–1279. [PubMed] [Google Scholar]
- Henseleit U, Rosenbach T, Kolde G. Induction of apoptosis in human HaCaT keratinocytes. Arch Dermatol Res. 1996;288:676–683. doi: 10.1007/BF02505277. [DOI] [PubMed] [Google Scholar]
- Hollingshead HE, Killins RL, Borland MG, Girroir EE, Billin AN, Willson TM, Sharma AK, Amin S, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) ligands do not potentiate growth of human cancer cell lines. Carcinogenesis. 2007;28:2641–2649. doi: 10.1093/carcin/bgm183. [DOI] [PubMed] [Google Scholar]
- Kanekura T, Higashi Y, Kanzaki T. Inhibitory effects of 9-cis-retinoic acid and pyrrolidinedithiocarbamate on cyclooxygenase (COX)-2 expression and cell growth in human skin squamous carcinoma cells. Cancer Lett. 2000;161:177–183. doi: 10.1016/s0304-3835(00)00604-2. [DOI] [PubMed] [Google Scholar]
- Kim DJ, Akiyama TE, Harman FS, Burns AM, Shan W, Ward JM, Kennett MJ, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor β (δ) -dependent regulation of ubiquitin C expression contributes to attenuation of skin carcinogenesis. J Biol Chem. 2004;279:23719–23727. doi: 10.1074/jbc.M312063200. [DOI] [PubMed] [Google Scholar]
- Kim DJ, Bility MT, Billin AN, Willson TM, Gonzalez FJ, Peters JM. PPARβ/δ selectively induces differentiation and inhibits cell proliferation. Cell Death Differ. 2006;13:53–60. doi: 10.1038/sj.cdd.4401713. [DOI] [PubMed] [Google Scholar]
- Kim DJ, Murray IA, Burns AM, Gonzalez FJ, Perdew GH, Peters JM. Peroxisome Proliferator-activated Receptor-β/δ Inhibits Epidermal Cell Proliferation by Down-regulation of Kinase Activity. J Biol Chem. 2005;280:9519 –9527. doi: 10.1074/jbc.M413808200. [DOI] [PubMed] [Google Scholar]
- Klaassen I, Brakenhoff RH, Smeets SJ, Snow GB, Braakhuis BJ. Metabolism and growth inhibition of four retinoids in head and neck squamous normal and malignant cells. Br J Cancer. 2001;85:630–635. doi: 10.1054/bjoc.2001.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Olson P, Evans RM. Lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144:2201–2207. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- Lee CH, Olson P, Hevener A, Mehl I, Chong LW, Olefsky JM, Gonzalez FJ, Ham J, Kang H, Peters JM, et al. PPARδ regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci U S A. 2006;103:3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man MQ, Barish GD, Schmuth M, Crumrine D, Barak Y, Chang S, Jiang Y, Evans RM, Elias PM, Feingold KR. Deficiency of PPARβ/δ in the epidermis results in defective cutaneous permeability barrier homeostasis and increased inflammation. J Invest Dermatol. 2008;128:370–377. doi: 10.1038/sj.jid.5701026. [DOI] [PubMed] [Google Scholar]
- Marin HE, Peraza MA, Billin AN, Willson TM, Ward JM, Kennett MJ, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor β inhibits colon carcinogenesis. Cancer Res. 2006;66:4394–4401. doi: 10.1158/0008-5472.CAN-05-4277. [DOI] [PubMed] [Google Scholar]
- Martinasso G, Maggiora M, Trombetta A, Canuto RA, Muzio G. Effects of di(2-ethylhexyl) phthalate, a widely used peroxisome proliferator and plasticizer, on cell growth in the human keratinocyte cell line NCTC 2544. J Toxicol Environ Health A. 2006;69:353–365. doi: 10.1080/15287390500227522. [DOI] [PubMed] [Google Scholar]
- Matsuura H, Adachi H, Smart RC, Xu X, Arata J, Jetten AM. Correlation between expression of peroxisome proliferator-activated receptor β and squamous differentiation in epidermal and tracheobronchial epithelial cells. Mol Cell Endocrinol. 1999;147:85–92. doi: 10.1016/s0303-7207(98)00214-7. [DOI] [PubMed] [Google Scholar]
- Memezawa A, Takada I, Takeyama K, Igarashi M, Ito S, Aiba S, Kato S, Kouzmenko AP. Id2 gene-targeted crosstalk between Wnt and retinoid signaling regulates proliferation in human keratinocytes. Oncogene. 2007;26:5038–5045. doi: 10.1038/sj.onc.1210320. [DOI] [PubMed] [Google Scholar]
- Michalik L, Desvergne B, Tan NS, Basu-Modak S, Escher P, Rieusset J, Peters JM, Kaya G, Gonzalez FJ, Zakany J, et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR) α and PPARβ mutant mice. J Cell Biol. 2001;154:799–814. doi: 10.1083/jcb.200011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee R, Jow L, Croston GE, Paterniti JR., Jr Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARγ2 versus PPARγ1 and activation with retinoid X receptor agonists and antagonists. J Biol Chem. 1997;272:8071–8076. doi: 10.1074/jbc.272.12.8071. [DOI] [PubMed] [Google Scholar]
- Pelton P. GW-501516 GlaxoSmithKline/Ligand. Curr Opin Investig Drugs. 2006;7:360–370. [PubMed] [Google Scholar]
- Peraza MA, Burdick AD, Marin HE, Gonzalez FJ, Peters JM. The toxicology of ligands for peroxisome proliferator-activated receptors (PPAR) Toxicol Sci. 2006;90:269–295. doi: 10.1093/toxsci/kfj062. [DOI] [PubMed] [Google Scholar]
- Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? J Mol Med. 2005;83:774–785. doi: 10.1007/s00109-005-0678-9. [DOI] [PubMed] [Google Scholar]
- Peters JM, Hollingshead HE, Gonzalez FJ. Role of peroxisome-proliferator-activated receptor β/δ (PPARβ/δ) in gastrointestinal tract function and disease. Clin Sci (Lond) 2008;115:107–127. doi: 10.1042/CS20080022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, Reitman ML, Hudson LD, Gonzalez FJ. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β (δ) Mol Cell Biol. 2000;20:5119–5128. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieck M, Meissner W, Ries S, Müller-Brüsselbach S, Müller R. Ligand-mediated regulation of peroxisome proliferator-activated receptor (PPAR) β/δ: A comparative analysis of PPAR-selective agonists and all-trans retinoic acid. Mol Pharmacol. 2008;74:1269–1277. doi: 10.1124/mol.108.050625. [DOI] [PubMed] [Google Scholar]
- Schmuth M, Haqq CM, Cairns WJ, Holder JC, Dorsam S, Chang S, Lau P, Fowler AJ, Chuang G, Moser AH, et al. Peroxisome proliferator-activated receptor (PPAR)-β/δ stimulates differentiation and lipid accumulation in keratinocytes. J Invest Dermatol. 2004;122:971–983. doi: 10.1111/j.0022-202X.2004.22412.x. [DOI] [PubMed] [Google Scholar]
- Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129:723–733. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw N, Elholm M, Noy N. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor β/δ. J Biol Chem. 2003;278:41589–41592. doi: 10.1074/jbc.C300368200. [DOI] [PubMed] [Google Scholar]
- Sznaidman ML, Haffner CD, Maloney PR, Fivush A, Chao E, Goreham D, Sierra ML, LeGrumelec C, Xu HE, Montana VG, et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor δ (PPARδ)–synthesis and biological activity. Bioorg Med Chem Lett. 2003;13:1517–1521. doi: 10.1016/s0960-894x(03)00207-5. [DOI] [PubMed] [Google Scholar]
- Tan NS, Michalik L, Noy N, Yasmin R, Pacot C, Heim M, Flühmann B, Desvergne B, Wahli W. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes Dev. 2001;15:3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennenbaum T, Lowry D, Darwiche N, Morgan DL, Gartsbein M, Hansen L, De Luca LM, Hennings H, Yuspa SH. Topical retinoic acid reduces skin papilloma formation but resistant papillomas are at high risk for malignant conversion. Cancer Res. 1998;58:1435–1443. [PubMed] [Google Scholar]
- Tong PS, Mayes DM, Wheeler LA. Differential effects of retinoids on DNA synthesis in calcium-regulated murine epidermal keratinocyte cultures. J Invest Dermatol. 1988;90:861–868. doi: 10.1111/1523-1747.ep12462107. [DOI] [PubMed] [Google Scholar]
- Verma AK. Inhibition of both stage I and stage II mouse skin tumour promotion by retinoic acid and the dependence of inhibition of tumor promotion on the duration of retinoic acid treatment. Cancer Res. 1987;47:5097–5101. [PubMed] [Google Scholar]
- Verma AK. Inhibition of tumor promoter 12-O-tetradecanoylphorbol-13-acetate-induced synthesis of epidermal ornithine decarboxylase messenger RNA and diacylglycerol-promoted mouse skin tumor formation by retinoic acid. Cancer Res. 1988;48:2168–2173. [PubMed] [Google Scholar]
- Verma AK, Slaga TJ, Wertz PW, Mueller GC, Boutwell RK. Inhibition of skin tumor promotion by retinoic acid and its metabolite 5,6-epoxyretinoic acid. Cancer Res. 1980;40:2367–2371. [PubMed] [Google Scholar]
- Wang D, Wang H, Guo Y, Ning W, Katkuri S, Wahli W, Desvergne B, Dey SK, DuBois RN. From the cover: crosstalk between peroxisome proliferator-activated receptor δ and VEGF stimulates cancer progression. Proc Natl Acad Sci U S A. 2006;103:19069–19074. doi: 10.1073/pnas.0607948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil M, Raff MC, Braga VM. Caspase activation in the terminal differentiation of human epidermal keratinocytes. Curr Biol. 1999;9:361–364. doi: 10.1016/s0960-9822(99)80162-6. [DOI] [PubMed] [Google Scholar]
- Westergaard M, Henningsen J, Svendsen ML, Johansen C, Jensen UB, Schrøder HD, Kratchmarova I, Berge RK, Iversen L, Bolund L, et al. Modulation of keratinocyte gene expression and differentiation by PPAR-selective ligands and tetradecylthioacetic acid. J Invest Dermatol. 2001;116:702–712. doi: 10.1046/j.1523-1747.2001.01329.x. [DOI] [PubMed] [Google Scholar]
- Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- Xu H, Cheepala S, McCauley E, Coombes K, Xiao L, Fischer SM, Clifford JL. Chemoprevention of skin carcinogenesis by phenylretinamides: retinoid receptor-independent tumor suppression. Clin Cancer Res. 2006;12:969–979. doi: 10.1158/1078-0432.CCR-05-1648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.