

Abstract
Vibrio cholerae causes a dehydrating diarrheal illness that can be rapidly fatal in the absence of specific treatment. The organism is an historic scourge and, like similar infectious diseases, may have influenced the evolution of the human genome. We report here the results of the first candidate gene association study of cholera. In a family-based study of 76 pedigrees from Dhaka, Bangladesh, we evaluated the association between cholera and five candidate genes -- the cystic fibrosis transmembrane receptor; lactoferrin; long palate, lung and nasal epithelium clone 1 (LPLUNC1); estrogen related receptor alpha; and calcium activated chloride channel 1. We found a significant association with a marker in the promoter region of LPLUNC1 (rs11906665), a member of a family of evolutionarily conserved innate immunity proteins. A previous microarray-based study of duodenal biopsies revealed significantly increased expression of LPLUNC1 in cholera patients compared to healthy control subjects. Our results suggest that variation in host innate immune responses may influence the outcome of exposure to V. cholerae in an endemic setting.
INTRODUCTION
Vibrio cholerae may have played an influential role in shaping the human genome. Historical documents describe a dehydrating diarrheal disease consistent with cholera as long ago as the 7th century, and seven pandemics with high rates of associated mortality have swept the globe in the last three centuries. Individuals with blood group O are at increased risk of severe cholera 1, and it has been hypothesized that V. cholerae infection may have selected for the low prevalence of the O blood group in the Ganges Delta region, a historic and current epicenter of cholera. 2
In a family-based study of cholera in Bangladesh, we have recently demonstrated that individuals who are first-degree relatives of an index patient with cholera have significantly greater odds of being infected with V. cholerae compared to unrelated or less closely related contacts, when living in the same household. This finding was independent of blood group phenotype in a multivariate analysis. 3
To begin to evaluate host genetic factors influencing susceptibility to cholera, we performed a family-based candidate gene association study in a cohort of individuals living in a cholera-endemic area of Bangladesh. We selected five candidate genes based on biologically plausible associations with V. cholerae infection. First, we included the cystic fibrosis transmembrane receptor (CFTR), since cholera has been speculated to be the selective pressure for the cystic fibrosis trait. 4,5 A recently-completed microarray-based study of duodenal biopsies of cholera patients from Bangladesh revealed increased expression of a number of innate immune effectors, compared to healthy control subjects. 6 We therefore included the two most highly upregulated genes identified in this study, lactoferrin (LTF) and long palate, lung and nasal epithelium clone 1 (LPLUNC1), as well as a nuclear steroid receptor that regulates lactoferrin production, estrogen related receptor alpha (ESRRA). Lastly, we included calcium activated chloride channel 1 (CLCA), which was detected in high levels in a proteomic evaluation of human cholera stools (unpublished data).
RESULTS
Characteristics of the study population
The study population is comprised of index cases with cholera and their family members, all of whom share household and cooking facilities. Index cases presented to the Dhaka hospital of the International Centre for Diarrhoeal Disease Research in Bangladesh (ICDDR,B) between January 2004 and November 2005. A diagnosis of cholera in an index case was made by the presence of acute watery diarrhea and a stool culture positive for V. cholerae. Family members of the index case were defined as affected with cholera if they had a positive rectal swab culture for V. cholerae or symptoms of diarrhea during 21 days of follow up. For this analysis, the index cases and affected relatives were classified as affected, and all others were classified as unaffected.
Two hundred seventy-seven individuals in 76 pedigrees were included in the study. In two-thirds of the pedigrees, the index case was a child. 73 pedigrees were nuclear families; 3 pedigrees additionally contained grandparents or grandchildren. Table 1 shows the structure of the families included in the association study, categorized by the number of affected children per family.
Table 1. Structure of families included in the association study, categorized by the number of affected children in the family *.
| Number of affected children | Number of families | With 2 parents genotyped | With 1 parent genotyped | Number of healthy siblings |
|---|---|---|---|---|
| 0 | 12 | 10 | 2 | 16 |
| 1 | 37 | 30 | 5 | 16 |
| 2 | 21 | 14 | 4 | 21 |
| 3 | 4 | 3 | 1 | 1 |
| 4 | 2 | 1 | 0 | 4 |
| Total: | 76 | 58 | 12 | 58 |
Only the nuclear families from 3 multigenerational pedigrees were included
Table 2 shows the clinical features of the index cases as well as of the affected and unaffected relatives. The mean ages differed between unaffected relatives (26 ± 15 years), affected relatives (20 ± 14.5 years) and index cases (18.7 ± 14.5 years). The higher age among unaffected relatives likely reflects immunity acquired with age among individuals living in an endemic setting. Consistent with pre-existing immunity, there was a trend toward higher vibriocidal titers (representing antibodies directed at the whole organism) in unaffected individuals. As expected, the index cases and affected relatives were non-significantly enriched for blood group O, a previously identified risk factor for severe cholera.
Table 2. Clinical characteristics of cholera index cases and their affected and unaffected relatives *.
| Index Cases (n= 71) | Affected relatives (n= 95) | Unaffected relatives (n =107) | P value *** | |
|---|---|---|---|---|
| Age (years) ** | 18.7 ± 14.5 | 20.0 ± 14.5 | 26.0 ± 15.0 | 0.001 |
| Male gender | 35 (49%) | 43 (45%) | 53 (50%) | 0.805 |
| Vibriocidal Titer, Day 2 ** | 304 ± 732 | 516 ± 1163 | 785 ± 2725 | 0.239 |
| Blood Group O | 28 (39%) | 37 (39%) | 34 (32%) | 0.464 |
Table excludes 4 relatives with unknown affected status
Mean ± S.D.
Continuous variables were compared using one-way ANOVA and dichotomous variables were compared by two-sided Pearson Chi Square
Family-based association analysis of SNPs in five candidate genes with cholera
We evaluated SNPs in five candidate genes -- CLCA, CFTR, ESRRA, LTF and LPLUNC1 -- for an association with cholera in the 76 pedigrees. As outlined in Table 3, a total of 145 SNPs in the five genes passed quality control and were included in the analysis.
Table 3. Candidate genes evaluated.
| Gene | Chromosome | Number of SNPs that passed quality control |
|---|---|---|
| Calcium-activated chloride channel (CLCA) | 1p31 | 47 |
| Cystic fibrosis transmembrane receptor (CFTR) | 7q31 | 31 |
| Estrogen-related receptor alpha (ESRRA) | 11q13 | 9 |
| Lactoferrin (LTF) | 3q21 | 41 |
| Long palate, lung and nasal epithelium clone 1 (LPLUNC1) | 20q11 | 17 |
We performed transmission disequilibrium testing on SNPs in each of the five candidate genes, using Haploview version 4.1. Supplemental Table 1 contains the results of TDT testing for all SNPs. Table 4 shows the SNPs that were associated with cholera with a nominal p value <0.05. One variant in a coding region of ESRRA, 2 variants in different regions of the CFTR gene, and 9 variants in LPLUNC1, primarily in the promoter region, were associated with cholera. Following permutation testing, only rs11906665, a variant in the promoter region of LPLUNC1, remained significantly associated with cholera. In particular, transmission of the A allele was associated with cholera.
Table 4. Results of transmission disequilibrium testing for SNPs with nominal p<0.05.
| Gene | SNP | Role | Overtransmitted allele (other allele) | Minor allele frequency genotyped (minor allele) | Number of fully genotyped trios | Transmission: non-transmissions to affected offspring | Chi Square | Nominal p value | Permutation p value * |
|---|---|---|---|---|---|---|---|---|---|
| ESRRA | rs2276014 | Coding | C (T) | 0.09 (T) | 52 | 19:6 | 6.76 | 0.0093 | 0.0575 |
| CFTR | rs1800130 | Coding | A (G) | 0.051 (G) | 53 | 8:1 | 5.444 | 0.0196 | 0.3451 |
| CFTR | rs713134 | Promoter | C (T) | 0.413 (C) | 52 | 36:19 | 5.255 | 0.0219 | 0.3703 |
| LPLUNC1 | rs11906665 | Promoter | A (G) | 0.076 (G) | 51 | 16:1 | 13.235 | 0.0003 | 0.0062 |
| LPLUNC1 | rs6057807 | Promoter | C (T) | 0.04 (T) | 56 | 12:2 | 7.143 | 0.0075 | 0.2106 |
| LPLUNC1 | rs8118559 | Promoter | C (T) | 0.03 (T) | 53 | 11:2 | 6.231 | 0.0126 | 0.3393 |
| LPLUNC1 | rs8118144 | Promoter | G (A) | 0.03 (A) | 57 | 12:3 | 5.4 | 0.0201 | 0.4677 |
| LPLUNC1 | rs17124508 | Promoter | T (G) | 0.03 (G) | 57 | 12:3 | 5.4 | 0.0201 | 0.4677 |
| LPLUNC1 | rs17307318 | Promoter | A (G) | 0.03 (G) | 57 | 12:3 | 5.4 | 0.0201 | 0.4677 |
| LPLUNC1 | rs6057810 | Promoter | T (C) | 0.03 (C) | 56 | 12:3 | 5.4 | 0.0201 | 0.4677 |
| LPLUNC1 | rs4398335 | Intron | G (A) | 0.03 (A) | 56 | 12:3 | 5.4 | 0.0201 | 0.4677 |
| LPLUNC1 | rs8115852 | Promoter | G (T) | 0.02 (T) | 55 | 10:2 | 5.333 | 0.0209 | 0.4809 |
Results of 100,000 cycles of permutation testing performed in Haploview v. 4.1
The FBAT statistical package extends the methodology of transmission disequilibrium testing to evaluate nuclear families that include both affected and unaffected offspring, and it also allows for missing parental data. Because of the varying affection status and family structure of these pedigrees, we performed focused association testing of the SNPs outlined in Table 4 using FBAT. Bi-allelic testing with an additive model was performed. The FBAT results, shown in Table 5, confirm the findings from TDT testing. The most highly associated SNP by FBAT was rs11906665 in LPLUNC1 (p = 0.0019).
Table 5. Results of FBAT testing for selected SNPs in candidate genes.
| Gene | SNP | Allele | Minor allele frequency (minor allele) | Number of informative families | S-E(S) | Var(S) | Z’ value | P value |
|---|---|---|---|---|---|---|---|---|
| ESRRA | rs2276014 | C | 0.079 (T) | 18 | 5.881 | 7.795 | 2.106 | 0.035 |
| CFTR | rs1800130 | A | 0.045 (G) | 7 | 5.5 | 4.25 | 2.668 | 0.0076 |
| CFTR | rs713134 | C | 0.441(C) | 38 | 11.286 | 16.641 | 2.767 | 0.0057 |
| LPLUNC1 | rs11906665 | A | 0.055 (G) | 16 | 7.167 | 5.306 | 3.111 | 0.0019 |
| LPLUNC1 | rs6057807 | C | 0.029 (T) | 8 | 5.5 | 3.75 | 2.84 | 0.0045 |
| LPLUNC1 | rs8118559 | C | 0.025 (T) | 5 | 5 | 3.5 | 2.673 | 0.0076 |
| LPLUNC1 | rs8118144 | G | 0.028 (A) | 8 | 5 | 4 | 2.5 | 0.012 |
| LPLUNC1 | rs17124508 | T | 0.03 (G) | 8 | 5 | 4 | 2.5 | 0.012 |
| LPLUNC1 | rs17307318 | A | 0.03 (G) | 8 | 5 | 4 | 2.5 | 0.012 |
| LPLUNC1 | rs6057810 | T | 0.03 (C) | 8 | 5 | 4 | 2.5 | 0.012 |
| LPLUNC1 | rs4398335 | G | 0.03 (A) | 8 | 5 | 4 | 2.5 | 0.012 |
| LPLUNC1 | rs8115852 | G | 0.02 (T) | 5 | 4 | 3 | 2.309 | 0.021 |
Haplotype-specific association analysis of LPLUNC
We performed a haplotype-based association analysis for SNPs in the candidate genes. Linkage disequilibrium (LD) between SNPs was inspected using Haploview, with haplotypes defined by solid spine of LD (D’ > 0.9). Common haplotypes with frequencies > 5% were observed in blocks for each candidate gene. After permutation testing was applied, no haplotype blocks, as defined by a D’ > 0.9, were significantly associated with cholera for any of the candidate genes (data not presented).
Because we observed the strongest association with a variant in LPLUNC1 (rs11906665), we additionally evaluated a 3 kb block comprised of rs17124508, rs11906665, rs1884884, rs17307318 and rs8115852, defined by a D’ of 1.0. Figure 1 shows this block, which is located in the 5′ promoter region of LPLUNC1. Table 1 shows the haplotype frequencies and the results of transmission disequilibrium testing for this block. Two haplotypes that differed only by the alleles of rs11906665 were significantly associated with cholera after permutation testing (p = 0.001 and p = 0.012).
Figure 1. LPLUNC1 haplotype block.
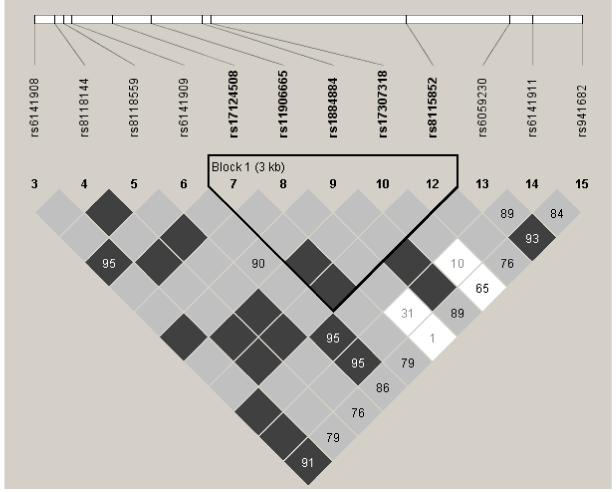
The location of each tested SNP along the chromosome is indicated at the top. The number in each diamond indicates the magnitude of LD in percent between respective pairs of SNPs. Diamonds without values represent perfect LD (D’ = 1.0). Dark grey diamonds represent strong evidence of LD; light grey diamonds are uninformative; and white diamonds represent strong evidence of recombination.
DISCUSSION
Substantial observational evidence has suggested that human genetic factors may influence susceptibility to V. cholerae infection. To date, no specific genetic association studies have been performed in cholera patients, although individuals with blood group O have been observed to be at higher risk of severe disease than those of other blood groups. 1 In this study, we used a family-based cohort in an endemic area to investigate the association between cholera and SNPs in five candidate genes.We found a variant in the promoter region of LPLUNC1 to be significantly associated with cholera. This association was robust and was identified using two family-based tests, the TDT and FBAT.
LPLUNC1 is a member of a family of evolutionarily conserved innate immunity proteins that have been best studied in the palate, lung and nasal epithelium. The short palate, lung and nasal epithelium clone 1 protein (SPLUNC1), a close relative of LPLUNC1, has been shown to inhibit growth of Pseudomonas aeruginosa in vitro and to bind to lipopolysaccharide of the organism. 7 A recent whole-genome microarray screen of gene expression in the duodenal mucosa of patients with cholera revealed LPLUNC1 to be greater than 7-fold more highly expressed during the acute phase of infection than during the convalescent phase 6; these findings were further confirmed by RT-PCR. This microarray study revealed a novel association between LPLUNC1 and cholera and generated the biological underpinnings to support the study of LPLUNC1 as a candidate gene for susceptibility to cholera.
The study of the role of host genetics in susceptibility to enteric infections is in its infancy. Immune response genes, particularly those in the HLA locus, have been the most frequently identified host susceptibility factors. 8 In a study of North American travelers to Mexico, a variant in exon 15 of lactoferrin was found to be associated with travelers’ diarrhea. 9 Similar to LPLUNC1, lactoferrin is a component of the innate immune system that has antimicrobial and immunoregulatory properties. Taken together with the present results, this finding indicates that further studies of the relationship between variants in host innate immunity genes and the outcome of exposure to enteric organisms may be productive.
The variant in LPLUNC1 that we identified as associated with cholera, rs11906665, may not be the causal variant. Our haplotype analysis revealed a haplotype containing rs11906665 to be more significantly associated with cholera than the variant itself. The variant is located in the promoter region of the gene, and comparative sequence data indicate that this is not a conserved region of the genome across mammals (http://genome.ucsc.edu/). A sequence comparison with the rhesus monkey indicates that the G allele is the ancestral allele at this locus (http://genome.ucsc.edu/). Further genotyping data may help shed light on variants that are in linkage disequilibrium with rs11906665.
Our study has strengths and limitations. The study population was comprised of nuclear families sharing water, cooking and hygiene facilities; thus, all individuals within the household were highly likely to have been exposed to V. cholerae along with the index case. By prospectively following family members for three weeks following the presentation of the case, we were able to obtain accurate phenotype information on these individuals for the current exposure. Nevertheless, individuals who were classified as unaffected for this analysis may have been infected with V. cholerae in the past; such misclassification may have limited the power of our analysis. Although we were able to detect evidence of an association between cholera and LPLUNC1, we cannot exclude additional effects of the four other genes evaluated. Validation of the association with LPLUNC1 should be carried out in other populations with exposure to V. cholerae, and further biological data delineating the mechanism of the observed association between LPLUNC1 and cholera are needed. As it is likely that more than one host genetic factor might contribute to susceptibility to cholera, a genome-wide search for additional associated loci is planned, with the aim of identifying other chromosomal regions related to this important global disease.
MATERIALS AND METHODS
Study subjects
Samples were collected as part of a household-based prospective observational study aimed at identifying immunologic and host factors associated with the risk of cholera in an endemic setting. The study design has been described previously. 3, 10 Briefly, index cases presenting to the ICDDR,B hospital with severe acute watery diarrhea were eligible for inclusion if their stool cultures were subsequently positive for V. cholerae, if they were older than 6 months, and if they were without significant comorbid conditions. All index cases included in this analysis were infected with V. cholerae O1 El Tor. Upon presentation of the index case, a field team discussed enrollment with family members sharing a household with the index case, and consenting individuals were enrolled into the study immediately upon culture confirmation of the index case. Enrollment of family members typically occurred within 24 hours of presentation of the index case. All enrolled family members shared the same household and cooking pot as the index cases for at least the three preceding days. Blood specimens were collected from index cases and consenting relatives upon enrollment and over the course of follow-up. The field team visited households on each of the next six days, and again on days 14 and 21. During these visits, relatives were questioned about diarrheal symptoms, and rectal swabs were obtained for V. cholerae culture. Diarrhea was defined as 3 or more loose stools within a 24 hour period. Among relatives in this study, cholera was defined as a positive rectal swab culture for V. cholerae or symptoms of diarrhea during the 21 days of follow up.
Informed consent for participation in this research was obtained from participants or their guardians. The human experimentation guidelines of the U.S. Department of Health and Human Services were followed in the conduct of this research. Approval for this human study was obtained from the Institutional Review Board of the Massachusetts General Hospital and the Research and the Ethical Review Committees of the ICDDR,B.
DNA extraction
Peripheral blood mononuclear cells were isolated from venous blood by gradient centrifugation on Ficoll-Isopaque (Pharmacia, Uppsala, Sweden). Cells were transferred to a cell lysis solution, and DNA was extracted according to the manufacturer’s protocol, including treatment with RNAse A (Gentra Puregene, Qiagen, Valencia, California). DNA concentration was determined by spectrophotometry.
Genotyping
Five genes were selected for inclusion in the candidate gene association study. SNPs located within these genes or within 10 kilobases in either direction from the gene were selected using data from dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/). Selection focused on the inclusion of validated, coding and tagging SNPs (Tagger). 11 Genotyping for selected SNPs was performed using the iPlex Gold genotyping system (Sequonom, Newton, Massachusetts). Primers for each evaluated SNP were designed using PrimerExpress (Applied Biosystems, Foster City, California) and checked using Sequonom RealSNP Assay Design software (Sequonom, Newton, Massachusetts). Genotyping was performed on the study subjects as well as on a plate of individuals from the Caucasian (CEU) population of the International HapMap Project for validation. Only SNPs with genotyping percentages ≥ 90% in the CEU and study populations, as well as minor allele frequencies ≥ 0.01 and Hardy-Weinberg p-values ≥ 0.01 in the study population, were included in the analysis. Any marker genotype inconsistencies were evaluated and the genotypes were zeroed for those select pedigrees.
Statistical analyses
Quality assessment of the genotyping results was performed using Haploview version 4.1. 12 Transmission disequilibrium testing was performed with Haploview version 4.1. Familial-based association testing was performed using the FBAT statistical package version 2.0.2c (http://www.biostat.harvard.edu/~fbat/default.html). Haplotypes were estimated using an E-M algorithm in Haploview version 4.1; haplotype blocks were defined using a solid spine of linkage disequilibrium (D’ > 0.9) unless otherwise indicated. Permutation testing was done within Haploview using 100,000 permutations for single markers and haplotypes; both nominal and empirical p-values are reported. Descriptive statistics were performed using SPSS version 10.1. Continuous variables were compared using one-way ANOVA and dichotomous variables were compared by two-sided Pearson ChiSquare.
Supplementary Material
Supplemental Table 1: Results of transmission disequilibrium testing for all SNPs in candidate genes
Table 6. Results of transmission disequilibrium testing for a haplotype block in LPLUNC1.
| Haplotype | Frequency | Transmission: non-transmissions to affected offspring | Chi Square | Nominal p value | Permutation p value * |
|---|---|---|---|---|---|
| TAGAG | 0.847 | 32.8 : 10.8 | 11.108 | 0.0009 | 0.001 |
| TGGAG | 0.075 | 4.1 : 17.1 | 7.963 | 0.0048 | 0.012 |
| TAAAG | 0.047 | 5.0 : 5.0 | 0 | 1 | 1 |
| GAGGT | 0.026 | 2.8 : 11.7 | 5.381 | 0.0204 | 0.068 |
Results of 100,000 cycles of permutation testing performed in Haploview v. 4.1
ACKNOWLEDGEMENTS
The authors wish to thank Cherylyn Smith and Liuda Ziagra of the Broad Institute for their work on the genotyping. We are grateful to Elinor Karlsson for a critical reading of the manuscript. This research was supported by a Claflin Distinguished Scholar Award from the Massachusetts General Hospital (R.C.L.), K01-TW07144 from the Fogarty International Center (R.C.L.), a Physician Scientist Early Career Award from the Howard Hughes Medical Institute (R.C.L.), the Intramural Program of the National Human Genome Research Institute of the NIH (P.D.), K01-TW07409 from the Fogarty International Center (J.B.H.), U01-AI58935 from the National Institute of Allergy and Infectious Diseases (S.B.C.), U01-AI077883 from the National Institute of Allergy and Infectious Diseases (E.T.R), R03-AI063079 from the National Institute of Allergy and Infectious Diseases (F.Q.), and the International Centre for Diarrhoeal Disease Research, Bangladesh (F.Q.).
REFERENCES
- (1).Glass RI, Holmgren J, Haley CE, Khan MR, Svennerholm AM, Stoll BJ, et al. Predisposition for cholera of individuals with O blood group: possible evolutionary significance. Am.J.Epidemiol. 1985 Jun;121(6):791–796. doi: 10.1093/oxfordjournals.aje.a114050. [DOI] [PubMed] [Google Scholar]
- (2).Mourant AE. Blood relations: blood groups and anthropology. Oxford University Press; London: 1983. [Google Scholar]
- (3).Harris JB, LaRocque RC, Chowdhury F, Khan AI, Logvinenko T, Faruque AS, et al. Susceptibility to Vibrio cholerae Infection in a Cohort of Household Contacts of Patients with Cholera in Bangladesh. PLoS Negl Trop.Dis. 2008 Apr 9;2(4):e221. doi: 10.1371/journal.pntd.0000221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science. 1994 Oct 7;266(5182):107–109. doi: 10.1126/science.7524148. [DOI] [PubMed] [Google Scholar]
- (5).Rodman DM, Zamudio S. The cystic fibrosis heterozygote--advantage in surviving cholera? Med.Hypotheses. 1991 Nov;36(3):253–258. doi: 10.1016/0306-9877(91)90144-n. [DOI] [PubMed] [Google Scholar]
- (6).Flach CF, Qadri F, Bhuiyan TR, Alam NH, Jennische E, Lonnroth I, et al. Broad up-regulation of innate defense factors during acute cholera. Infect.Immun. 2007 May;75(5):2343–2350. doi: 10.1128/IAI.01900-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhou HD, Li XL, Li GY, Zhou M, Liu HY, Yang YX, et al. Effect of SPLUNC1 protein on the Pseudomonas aeruginosa and Epstein-Barr virus. Mol.Cell.Biochem. 2008 Feb;309(1-2):191–197. doi: 10.1007/s11010-007-9659-3. [DOI] [PubMed] [Google Scholar]
- (8).Petri WA, Jr, Miller M, Binder HJ, Levine MM, Dillingham R, Guerrant RL. Enteric infections, diarrhea, and their impact on function and development. J.Clin.Invest. 2008 Apr;118(4):1277–1290. doi: 10.1172/JCI34005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mohamed JA, DuPont HL, Jiang ZD, Belkind-Gerson J, Figueroa JF, Armitige LY, et al. A novel single-nucleotide polymorphism in the lactoferrin gene is associated with susceptibility to diarrhea in North American travelers to Mexico. Clin.Infect.Dis. 2007 Apr 1;44(7):945–952. doi: 10.1086/512199. [DOI] [PubMed] [Google Scholar]
- (10).Saha D, LaRocque RC, Khan AI, Harris JB, Begum YA, Akramuzzaman SM, et al. Incomplete correlation of serum vibriocidal antibody titer with protection from Vibrio cholerae infection in urban Bangladesh. J.Infect.Dis. 2004 Jun 15;189(12):2318–2322. doi: 10.1086/421275. [DOI] [PubMed] [Google Scholar]
- (11).de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat.Genet. 2005 Nov;37(11):1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- (12).Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005 Jan 15;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Results of transmission disequilibrium testing for all SNPs in candidate genes