

Abstract
Binding properties of lignin peroxidase (LiP) from the basidiomycete Phanerochaete chrysosporium against a synthetic lignin (dehydrogenated polymerizate, DHP) were studied with a resonant mirror biosensor. Among several ligninolytic enzymes, only LiP specifically binds to DHP. Kinetic analysis revealed that the binding was reversible, and that the dissociation equilibrium constant was 330 μM. The LiP–DHP interaction was controlled by the ionization group with a pKa of 5.3, strongly suggesting that a specific amino acid residue plays a role in lignin binding. A one-electron transfer from DHP to oxidized intermediates LiP compounds I and II (LiPI and LiPII) was characterized by using a stopped-flow technique, showing that binding interactions of DHP with LiPI and LiPII led to saturation kinetics. The dissociation equilibrium constants for LiPI–DHP and LiPII–DHP interactions were calculated to be 350 and 250 μM, and the first-order rate constants for electron transfer from DHP to LiPI and to LiPII were calculated to be 46 and 16 s−1, respectively. These kinetic and spectral studies strongly suggest that LiP is capable of oxidizing lignin directly at the protein surface by a long-range electron transfer process. A close look at the crystal structure suggested that LiP possesses His-239 as a possible lignin-binding site on the surface, which is linked to Asp-238. This Asp residue is hydrogen-bonded to the proximal His-176. This His–Asp⋅⋅⋅proximal-His motif would be a possible electron transfer route to oxidize polymeric lignin.
Lignin is the most abundant renewable aromatic polymer and is known as one of the most recalcitrant biomaterials on earth (1, 2). Its degradation plays a key role in the carbon cycle of the biosphere (2–7). Only white-rot basidiomycetes are responsible for the complete mineralization of this polymer. Phanerochaete chrysosporium, the best studied white-rot fungus, secretes two heme peroxidases, lignin peroxidase (LiP) and manganese peroxidase (MnP) under ligninolytic conditions (3–8). Thus, these enzymes have been believed to be involved in triggering lignin biodegradation. MnP oxidizes MnII to MnIII, and the latter acts as a freely diffusible one-electron oxidizer, nonspecifically reacting with terminal organic substrates such as phenols, thiols, and lignin (3, 8–12). This nonspecific manner is advantageous for lignin degradation because lignin is such a heterogeneous polymer.
LiP is another unique heme peroxidase secreted by P. chrysosporium. It catalyzes a one-electron oxidation of nonphenolic aromatic compounds, forming the aryl cation radical (13, 14), suggesting that oxidized intermediates of the enzyme possess a very high redox potential. The mechanism of LiP catalytic action on lignin is still uncertain, because it has not been clear whether LiP can oxidize lignin through a direct interaction or through radical mediation. Veratryl (3,4-dimethoxybenzyl) alcohol (VA), a preferred substrate for LiP, is synthesized de novo by P. chrysosporium under ligninolytic conditions (15). The VA cation radical has been proposed as a radical mediator to oxidize polymeric substrates with which LiP presumably cannot interact directly (16–20). However, it has been shown that ferrocytochrome c is oxidized by LiP/H2O2 in the absence of VA (21), suggesting that LiP is capable of oxidizing polymeric substrates at the protein surface by a long-range electron transfer mechanism (19, 21). Also, a long-range electron transfer mechanism in proteins has been proposed for cytochrome c peroxidase (CCP) (22, 23).
To better understand the LiP reaction mechanism, we have utilized a resonant mirror biosensor to observe the direct interaction of lignin (dehydrogenated polymerizate, DHP) and LiP. Furthermore, a direct one-electron transfer from DHP to LiPI and LiPII was confirmed by using a stopped-flow technique. Transient state kinetic analysis also showed the binding interaction of LiPI and LiPII with lignin.
MATERIALS AND METHODS
Enzymes.
LiP (isozyme 2) and MnP (isozyme 1) were isolated from the extracellular culture medium of P. chrysosporium (ATCC 34541) and purified as previously described except for using perfusion chromatography (Poros HQ/M, PerSeptive Biosystems) (24, 25). Laccase was isolated from the extracellular medium of Fomitella fraxinea and purified by using DEAE-Sepharose (Pharmacia) and perfusion chromatography (Poros HQ/M). Horseradish peroxidase (HRP) (grade III, Toyobo) was purchased and further purified by using perfusion chromatography (Poros HS/M). All the purified proteins were electrophoretically homogeneous. LiP, MnP, and HRP had RZ (Reinheitszahl, ASoret/A275) values of 5.1, 5.9, and 3.2, respectively.
Resonant Mirror Biosensor.
Binding studies were conducted with the interaction analysis system (IAsys, Affinity Sensors) in which a laser biosensor measured the change in the refractive index (arc seconds) upon binding of an analyte to its partner immobilized on the sample cuvette. All the proteins were immobilized on cuvettes coated with carboxymethylated dextran by means of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide at 0.8–1.1 pH unit below the pI of each protein. Amounts of immobilized proteins were estimated from a calibration curve prepared with human serum albumin, indicating the immobilized amount, in ng/mm2: LiP, 1.5; MnP, 1.4; laccase, 2.9; HRP, 2.6. LiP was also immobilized on the cuvette coated with aminosilane by bis(sulfosuccinimidyl)suberate (BS3; Pierce): 2.4 ng/mm2. The activities of immobilized enzymes were measured in the resonance cuvette. Recovery of the activities based on the immobilized amount reached 85–92%, indicating that the alteration occurring during immobilization was minimal. A baseline and a resonant scan were stable and intact for at least 10 repeated uses. Control experiments were performed either using the cuvette without the immobilization of proteins or by adding a DHP-omitted solution to the enzyme-immobilized cuvette. Each cuvette contained 200 μl of reaction mixture.
Stopped-Flow Measurement.
Kinetic measurements were conducted by using the Photal RA 401S Rapid Reaction Analyzer (Otsuka Electronics) equipped with a 1-cm observation cell at 25 ± 0.1°C. The reduction rates of LiPI and LiPII by DHP were determined. LiPI was freshly prepared for each experiment by addition of 0.9 eq of H2O2 to the native LiP. LiPII was freshly prepared by addition of 1.1 eq of ferrocyanide to LiPI. One reservoir contained LiPI or LiPII (≈3.6 μM) in water. The other reservoir contained DHP (100–800 μM) in 20 mM succinate buffer. The reduction of LiPI was followed at 417.5 nm, the isosbestic point between LiPII and native LiP. The reduction of LiPII was followed at 407 nm. The pseudo-first-order rate constants were determined by a nonlinear least-squares computer analysis of exponential traces. To examine the effect of DHP on LiPI formation rate, one reservoir contained LiP (2.0 μM) and DHP (100–600 μM) in water, the other reservoir contained H2O2 (20–80 μM) in 20 mM succinate, pH 4.5. Thus, LiP–DHP complex was formed prior to the addition of H2O2. After mixing, LiPI formation was followed at 397 nm, the isosbestic point between LiPI and LiPII. Electronic absorption spectra were recorded on a Perkin–Elmer Lambda 19 spectrophotometer at room temperature.
Substrates.
DHP was synthesized from coniferyl alcohol (Aldrich) as previously described (26). DHP was dissolved in dimethylformamide (DMF) to prepare a stock solution (2.0 mg/ml), thus, when 800 μM DHP was used, the concentration of DMF in the final reaction system was 3.58%. In this study, [DHP] was expressed as M coniferyl alcohol residue. The molecular weight of DHP was roughly estimated to be 3,000 with monodispersity by using gel permeation chromatography on Sephadex LH-60. All chemicals were reagent grade. The concentration of H2O2 was determined daily as previously described (27). Solutions were prepared with deionized water obtained from a Milli Q (Millipore) system.
Protein Structure and Sequence Analysis.
All the structural data were obtained from the Protein Data Bank (PDB) and visualized by using Protein Advisor 2.5 (Fujistu). Amino acid sequence analyses were performed with the blast search program.
RESULTS
Direct Interaction of LiP with DHP.
Fig. 1 shows the binding profile of LiP and DHP obtained by the use of the resonant mirror biosensor. Upon the addition of 5 μg of DHP in 2.0 μl of 50% DMF, DHP bound to immobilized LiP within minutes (Fig. 1A, trace 1). The injection of 2.0 μl of 50% DMF caused the change in refractive index in the first 5 s (Fig. 1A, trace 2). When DHP was injected into the cuvette with no LiP immobilization, the same trace was obtained as when DMF alone was injected (data not shown). The trace of DMF injection (trace 2) was subtracted from the trace of the complete reaction system (trace 1), and the outcome trace is shown in Fig. 1B. This curve exhibits a single-exponential character, supporting the occurrence of the second-order association of LiP with DHP. To evaluate whether DHP interaction was specific to LiP, several related enzymes were applied to the interactive analysis. Fig. 2 clearly shows that only LiP specifically binds to DHP.
Figure 1.

Direct interaction of LiP with DHP. (A) Interaction traces obtained upon the addition of 5 μg of DHP in 50% DMF (2.0 μl) (trace 1, ○) and of 2.0 μl of 50% DMF (trace 2, ▵). (B) Trace 1 minus trace 2, thus showing the binding profile for LiP and DHP. A resonant biosensor was used to monitor the molecular interaction in 10 mM succinate, pH 3.0.
Figure 2.

DHP binding profiles for several ligninolytic enzymes and HRP. Traces were obtained as described for Fig. 1. Reactions were performed in 10 mM succinate, pH 3.0 (LiP, ●), and 4.5 (MnP, ▵; laccase, ▴; HRP, ○).
Kinetics of LiP–DHP Interaction.
For kinetic study, LiP was immobilized to the cuvette coated with aminosilane via BS3 to avoid nonspecific binding of DHP to the carboxymethylated dextran layer (28) and to increase the amount of LiP immobilized. After subtraction of the DMF trace from the complete trace as shown in Fig. 1, the exponential trace was fitted to Eq. 1 (28) shown below by using a nonlinear least-squares fit method:
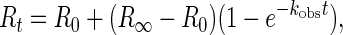 |
1 |
where Rt, R0, and R∞ are responses to the change in the refractive index (arc s) at time t, time zero, and infinity, respectively, kobs is a pseudo-first-order association constant (s−1), and t is time (s). A series of calculated kobs was replotted against [DHP], indicating the linear relationship (r = 0.99) between [DHP] and kobs (Fig. 3). This behavior can be described by a simple binding interaction according to Eqs. 2 and 3:
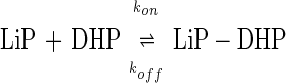 |
2 |
and
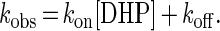 |
3 |
Figure 3.

Kinitics of LiP binding to DHP. LiP was immobilized on an aminosilane-coated cuvette and buffered at pH 3.0 with 10 mM succinate. kobs was determined from the exponential change in refractive index as shown in Fig. 1B. Each determination of kobs is the mean of at least five traces.
From the data and Eq. 3, the second-order association rate constant, kon, and the first-order dissociation constant, koff, were calculated to be (1.6 ± 0.1) × 102 M−1⋅s−1 and (5.3 ± 0.7) × 10−2 s−1, respectively. The dissociation equilibrium constant, Kd, was then estimated to be 330 ± 40 μM.
pH Dependence of LiP–DHP Interaction.
A series of kobs at [DHP] of 1.4 × 10−5 M were plotted against pH (Fig. 4). As the pH increased, the rate was decreased. Since DHP is almost completely protonated over the pH range of 3.0–6.0, the enzyme-derived acid dissociation appears to affect the value of kobs. This acid dissociation constant (Ka) was calculated from Eq. 4,
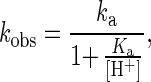 |
4 |
where ka is a pH-independent first-order rate constant. Then a pKa of 5.3 was obtained from the nonlinear least-squares fit analysis (Fig. 4).
Figure 4.

pH dependence of the binding of LiP and DHP. The pseudo-first order rate constants obtained at [DHP] of 1.4 × 10−5 M were plotted against pH. The ionic strength was adjusted at 25 mM using KNO3. Each determination is the mean of 5 traces.
Effect of VA on LiP–DHP Interaction.
kobs at [DHP] of 2.8 × 10−6 M in the presence of VA was measured. The rates were almost unchanged at (5.8–6.0) × 10−2 s−1, over a [VA] range of 0–2.0 mM.
Reaction of LiPI and LiPII with DHP.
Fig. 5 shows the time course of the change in absorbance at 417.5 nm, which occurs after the addition of DHP to LiPI. The change was monitored at the isosbestic points between LiPII and native LiP (29); thus, a possible conversion of LiPII back to the native enzyme would not contribute any error to the rate constant determination. However, trace 1 shown in Fig. 5A exhibited a biphasic character. The addition of DHP to native LiP caused a slow increase in absorbance (Fig. 5A, trace 2). Trace 2 was subtracted from trace 1, and the outcome trace, showing a single-exponential character, is shown in Fig. 5B. Actually, the slow increase in absorbance was observed when DHP solution (3.58% DMF) and water were mixed, suggesting that it might be caused by the change in a refractive index but not by the formation of DHP precipitate. From the single-exponential curves, pseudo-first-order rate constants (k1obs) were determined. The plot of k1obs versus [DHP] is hyperbolic, leveling off at high DHP concentration (Fig. 6A). This behavior can be described by a simple binding interaction between reactants according to Eqs. 5 and 6,
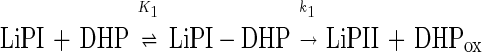 |
5 |
and
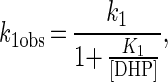 |
6 |
where k1 is a first-order rate constant (s−1) and K1 is an apparent dissociation constant (M) given by Eq. 7.
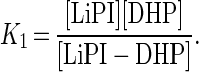 |
7 |
The constants k1 and K1 were calculated from Eq. 6 by using a nonlinear least-squares fit to the data. k1 and K1 were determined to be (46 ± 4) s−1 and (350 ± 60) μM, respectively.
Figure 5.

Kinetic traces for the reduction of LiPI by DHP. (A) Kinetic traces obtained upon the addition of 225 μM DHP to LiPI (trace 1) and to native LiP (trace 2) in 10 mM succinate, pH 3.0. (B) Trace 1 minus trace 2 and its computer-fit exponential curve, from which kobs was calculated.
Figure 6.

Reaction of LiPI and LiPII with DHP. (A) Reduction of LiPI by DHP. The plot of k1obs versus DHP concentration was a nonlinear least-squares fit on the data. (B) Reduction of LiPII by DHP. The plot of k2obs versus DHP concentration was a nonlinear least-squares fit on the data. Pseudo-first-order rate constants kobs were obtained as shown in Fig. 5 and in the text. Eqs. 6 and 9 were used for the curve fit. Each determination of kobs is the mean of at least five traces.
Similarly, pseudo-first-order rate constants k2obs of the LiPII reduction by DHP were determined. Single-exponential traces were also obtained in LiPII reduction by DHP (data not shown). The plot of k2obs vs. [DHP] is hyperbolic (Fig. 6B). This behavior can be explained by Eqs. 8 and 9.
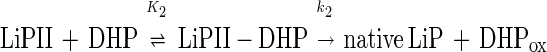 |
8 |
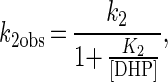 |
9 |
where k2 is a first-order rate constant (s−1) and K2 is an apparent dissociation constant (M) given by Eq. 10.
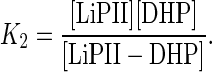 |
10 |
k2 and K2 were determined to be (16 ± 1) s−1 and (250 ± 50) μM, respectively.
The formation rate of LiPI from LiP when excess H2O2 was added was also measured in the presence of excess DHP. Each kinetic trace again exhibited a biphasic character, as shown in Fig. 5A. After subtraction by the trace obtained upon the addition of DHP to native LiP, a single-exponential curve was observed, from which the pseudo-first-order rate constants were calculated. These pseudo-first-order rate constants were linearly proportional to [H2O2] from 10 to 40 times in excess. The plot of the constants vs. [H2O2] passed through the origin within experimental error (data not shown). The second-order rate constants in the presence of various concentrations of DHP were obtained from each slope. The presence of DHP did not complicate the kinetic traces for the reaction of LiP with H2O2. These second-order rate constants were plotted against DHP concentration (Fig. 7). LiPI formation rate was not affected by the addition of DHP (0–300 μM final concentration).
Figure 7.

Effect of DHP on LiPI formation rate. The second-order rate constants kapp (M−1⋅s−1) for LiPI formation from native LiP and H2O2 were measured in the presence of various concentrations of DHP.
pH Dependence on the Reduction of LiPI by DHP.
The pH dependence of LiPI–DHP reaction was examined. At pH 4.0, 5.0, and 6.0, saturation kinetics were observed as seen at pH 3.0 (Fig. 6A). Therefore, the dissociation constant K1 and the first-order rate constant k1 at each pH were calculated by using Eq. 6 (Table 1).
Table 1.
pH dependence of LiPI reduction by DHP
| pH | K1, μM | k1, s−1 |
|---|---|---|
| 3.0 | 400 ± 100 | 90 ± 20 |
| 4.0 | 400 ± 100 | 80 ± 10 |
| 5.0 | 320 ± 10 | 35 ± 1 |
| 6.0 | 1,400 ± 400 | 38 ± 9 |
All experiments were performed in 20 mM sodium succinate.
DISCUSSION
LiP shares many structural and mechanistic features with other peroxidases, but it has several unique properties (3–8, 30–32). The enzyme catalyzes the one-electron oxidation of nonphenolic aromatic compounds with high redox potentials via the formation of a substrate cation radical (13, 14). The most controversial argument was found in the LiP reaction mechanism for the oxidation of recalcitrant substrates (16–20). Once widely accepted, VA cation radical mediation mechanisms are now questioned (33–35).
If the radical mediation does not play the main role in the oxidation of recalcitrant and polymeric substrates such as lignin, what is the real oxidant? A crystallographic study on the LiP structure revealed that the heme was totally buried in the LiP protein, strongly suggesting that the heme cannot interact directly with polymeric substrates (36–38). Recently, however, Gold and coworkers (21) have reported that ferrocytochrome c can be oxidized by LiP even in the absence of VA, suggesting that the long-range electron transfer mechanism may be involved in LiP catalysis. In this report, the possible interaction of lignin and LiP was studied with a resonant mirror biosensor, showing the occurrence of a direct interaction. Furthermore, using the stopped-flow technique, we also showed the occurrence of a one-electron transfer from lignin to the oxidized intermediates of the enzyme. Because DHP has been the most widely accepted polymeric lignin model and it is much more homogenous than isolated lignin, this synthetic polymer was thought to be more suitable for kinetic studies.
Interaction of LiP with Lignin.
Figs. 1 and 2 clearly show that among several ligninolytic enzymes, only LiP binds to DHP in a specific manner. Kinetic analysis revealed that the binding occurred reversibly (Fig. 3). Ferrocytochrome c has been reported to be oxidized by LiP without help of VA, leading to the proposal that a one-electron transfer from cytochrome c heme to LiP heme occurred via a long-range electron transfer through a surface protein–protein interaction (21). Lignin may also bind to the surface of the LiP protein and is oxidized by long-range electron transfer. To better understand the location of the lignin-binding site of LiP, we studied the pH dependence of LiP-lignin interaction.
pH Dependence of LiP–Lignin Interaction.
The pH dependency on the interaction of LiP with DHP indicated the involvement of an ionization group with a pKa of 5.3 (Fig. 4), strongly suggesting that the imidazole group of histidine or the carboxylic group of glutamate might control the binding. The pKa was obtained by the computer fit to the data of each kobs for each pH. Thus, it could be a weighted average of pKa for kon and koff. But the simple binding Eq. 2 was well fit by the data, suggesting that only one ionization group is most likely involved in either association or dissociation.
The pH dependency on the reduction of LiPI and LiPII by VA was extensively studied (29). Either VA or DHP exhibited better binding to LiP at lower pH. The effect of VA on LiP–DHP interaction was investigated; however, no effect was shown, which suggests that VA and lignin bind to different sites of LiP.
Electron Transfer from Lignin to Heme.
One of our major concerns was whether electron transfer occurred from lignin to the LiP heme through a protein surface interaction. The reaction of LiPI and LiPII with DHP was investigated with a stopped-flow apparatus. LiPII and native LiP formation rate depended on [DHP] (Fig. 6), clearly indicating that the electron transfer occurs between both LiP oxidized intermediates and DHP. Transient formation of LiPII during the reaction of LiPI with DHP was confirmed by rapid scan analysis (data not shown). These results combined with binding data shown above strongly suggest that the one-electron transfer occurs by the long-range electron transfer mechanism. Furthermore, the transient state kinetic analysis revealed the occurrence of the binding interaction of LiPI and LiPII with DHP. LiPI– or LiPII–DHP complexes were kinetically detectable (Fig. 6). The K1 of 350 μM for LiPI–DHP and the K2 of 250 μM for LiPII–DHP binding interaction are about the same as the Kd of 330 μM for native LiP–DHP binding (Fig. 3). Structural change during the catalytic cycle caused minimal changes in dissociation equilibrium constants, supporting the idea that the lignin-binding site most likely exists on the protein surface but not in the heme distal pocket. On the other hand, a large shift of binding constants for native LiP and LiPII against VA has been reported (29).
The k1 increased as the pH was decreased in the range of 3–6 (Table 1). This behavior can be explained by the higher redox potential at the lower pH, and it may support the involvement of Asp or Glu in the electron transfer reaction between LiPI and DHP. The possible involvement of Asp in the electron transfer from VA to LiPI and to LiPII has been reported (29). The reduction rate of LiPII (k2) was  that of LiPI. This behavior could be attributed the fact that reduction of LiPII involves a proton transfer step and spin state change of the heme iron (39).
that of LiPI. This behavior could be attributed the fact that reduction of LiPII involves a proton transfer step and spin state change of the heme iron (39).
Possible Lignin-Binding Site of LiP.
Two long-range electron transfer routes for LiP mechanism (isozyme 2) have been proposed. One is His-Pro-Asn, residues 82–84, and Asn-84 is hydrogen bonded to the distal His-47 (19, 39). The other is Trp-Leu residues 171 and 172 to heme (40, 41). His-82 has been shown to be located near the cleft (36–38), which could be the substrate channel. Since LiP is oxidized by bulky organic peroxides such as m-chloroperoxybenzoic acid, this cleft should be the only channel accessible to the heme distal pocket. If His-82 is a part of the lignin-binding site, this cleft is concealed upon lignin binding, reducing the reactivity with peroxide. However, the LiPI formation rate was unchanged in the presence of DHP (Fig. 7). LiP oxidation of VA in the presence of DHP was also examined. VA oxidation was negligibly inhibited in the presence of 300 μM DHP (data not shown). The fact that VA oxidation was not noncompetitively inhibited by DHP, combined with the observation that VA addition did not affect DHP binding to LiP, suggests that VA and lignin are oxidized at different sites.
Very recently, it was reported that Cβ of Trp-171 in LiP is autocatalytically hydroxylated, and W171F and W171S LiP mutants expressed in Escherichia coli showed no activity for VA oxidation even though compound I formation rates were unchanged (40, 41). If this amino acid residue plays a key role in lignin binding, part of our results can be explained. Glu-250 and Glu-168 and hydrophobic amino acid residues, Leu-167, Phe-267, Leu-270, and Ala-271 are located in the vicinity of Trp-171 of LiP isozyme 2 (37). Protonation of these glutamates under acidic conditions, which would form a large hydrophobic surface region surrounding Trp-171, may support hydrophobic interaction of LiP with lignin. However, if Trp-171 is involved in the oxidation of either VA or DHP, the lack of effect of VA on LiP–DHP interaction shown in this study cannot be explained. Most likely, Trp-171 is a key residue for VA oxidation but not for lignin.
On the basis of our observations, an alternative lignin-binding site of LiP can be proposed, as shown in Fig. 8: that is, His-239 and some hydrophobic amino acid residues surrounding this His such as Val-225 and Phe-215 (37). The pKa shift of His-239 to 5.3 (Fig. 4) could be explained by the hydrophobic environment of this residue. The imidazole of His-239 is located on the surface of LiP protein and faces outside (37). A possible electron transfer route is His-239–Asp-238⋅⋅⋅proximal- His-176–heme, where Asp is hydrogen-bonded to proximal His. Recently, Musah and Goodin (42) proposed that Tyr-236 of CCP is the minority radical center alternate to Trp-191, the well characterized radical center. Utilizing the W191F mutant of CCP, Tyr-236 radical was identified, suggesting that Tyr–proximal-His via hydrogen-bonded Asp could provide an effective electron transfer route if the real radical center, Trp, is lost. The electron transfer route Tyr-236–Asp-235⋅⋅⋅proximal-His–heme is very suggestive, since both Tyr-236 of CCP and His-239 of LiP are located next to Asp that is hydrogen-bonded to proximal His in each primary structure (Table 2). These findings support the involvement of His. Our pH-dependency study also supports the involvement of His and Asp (Fig. 4 and Table 1). The His–Asp⋅⋅⋅proximal-His motif was searched for in other peroxidases. An Asp residue that is hydrogen-bonded to the proximal His is conserved in almost all heme peroxidases. In contrast, the amino acid residue next to the Asp at the possible lignin-binding site is not conserved. Among the peroxidases registered in the Protein Data Bank, only in LiP was His observed (Table 2). This His is well conserved among LiP isozymes. With P. chrysosporium LiP isozymes, only one LiP cDNA, encoding H10 isozyme, contains Phe instead of His. However, this isozyme has been reported to be kinetically different from other LiP isozymes, including one we used in this study. H10 oxidizes VA and a lignin model dimeric compound. Km and kcat of H10 for VA were very similar to those of LiP isozyme 2 (H8) (50). On the other hand, those parameters of H10 for the dimeric compound were completely different from those of LiP isozyme 2, indicating that the dimeric model is not a preferred substrate for H10 isozyme (50). H10 may exhibit different behavior toward lignin. Both MnP isozymes 1 and 2 from P. chrysosporium do not have this motif.
Figure 8.

Proposed lignin-binding site of LiP and the possible electron transfer pathway. Broken line indicates the hydrogen bond (35, 36).
Table 2.
Xaa-Asp⋅⋅⋅His (proximal) motif of various peroxidases
| Enzyme | Xaa | Ref. |
|---|---|---|
| LiP (H8) | His-239 | 37 |
| CCP | Tyr-236 | 43 |
| MnP | Phe-243 | 44 |
| HRP | Gln-248 | 45 |
| APX | Lys-209 | 46 |
| ARP | Ala-247 | 47 |
| PNP | Lys-240 | 48 |
| BP | Gln-251 | 49 |
Xaa indicates the amino acid residue next to the conserved Asp that is hydrogen-bonded to proximal His. All residues were found on the surface of the proteins. APX, ascorbate peroxidase; ARP, Arthromyces ramosus peroxidase; PNP, peanut peroxidase; BP, barly peroxidase.
It may be still too early to conclude that the His–Asp⋅⋅⋅proximal-His motif plays a role in lignin oxidation. However, most importantly, in this study we have demonstrated the direct interaction of LiP and lignin by using a surface plasmon resonant spectral technique. Our results strongly support that LiP may possess two (or more) substrate interaction sites (41). Further investigations, including site-directed mutagenesis, are necessary to test this possibility.
Acknowledgments
We acknowledge support from Kyushu University Interdisciplinary Programs in Education and Projects in Research Development (to H.W.) and Research Fellowships of the Japan Society for the Promotion of Science for Young Scientists (to T.J.).
ABBREVIATIONS
- CCP
cytochrome c peroxidase
- DHP
dehydrogenated polymerizate (synthetic lignin)
- DMF
dimethylformamide
- HRP
horseradish peroxidase
- LiP
lignin peroxidase
- LiPI
compound I of LiP
- LiPII
compound II of LiP
- MnP
manganese peroxidase
- VA
veratryl alcohol
References
- 1.Sarkanen K V, Ludwig C H. Lignins: Occurrence, Formation, Structure and Reactions. New York: Wiley; 1971. [Google Scholar]
- 2.Crawford R L. Lignin Biodegradation and Transformation. New York: Wiley; 1981. [Google Scholar]
- 3.Gold M H, Wariishi H, Valli K. In: Biocatalysis in Agricultural Biotechnology, ACS Symposium Series. Whitaker J R, Sonnet P, editors. Vol. 389. Washington, DC: Am. Chem. Soc.; 1989. pp. 127–140. [Google Scholar]
- 4.Kirk T K, Farrell R L. Annu Rev Microbiol. 1987;41:465–505. doi: 10.1146/annurev.mi.41.100187.002341. [DOI] [PubMed] [Google Scholar]
- 5.Tien M. CRC Crit Rev Microbiol. 1987;15:141–168. doi: 10.3109/10408418709104456. [DOI] [PubMed] [Google Scholar]
- 6.Higuchi T. Wood Sci Technol. 1990;24:23–63. [Google Scholar]
- 7.Schoemaker H E. Recl Trav Chim Pays-Bas Belg. 1990;109:255–272. [Google Scholar]
- 8.Gold M H, Mayfield M G, Godfery B, Brown J, Wariishi H, Alic M. In: Plant Peroxidases: Biochemistry and Physiology. Welinder K G, Rasmussen S K, Panel C, Greppin H, editors. Geneva: Univ. of Geneva; 1993. pp. 87–95. [Google Scholar]
- 9.Glenn J K, Gold M H. Arch Biochem Biophys. 1985;242:329–341. doi: 10.1016/0003-9861(85)90217-6. [DOI] [PubMed] [Google Scholar]
- 10.Wariishi H, Valli K, Gold M H. Biochemistry. 1989;28:6017–6023. [Google Scholar]
- 11.Wariishi H, Valli K, Renganathan V, Gold M H. J Biol Chem. 1989;264:14185–14191. [PubMed] [Google Scholar]
- 12.Wariishi H, Valli K, Gold M H. Biochem Biophys Res Commun. 1991;176:269–275. doi: 10.1016/0006-291x(91)90919-x. [DOI] [PubMed] [Google Scholar]
- 13.Kersten P J, Tien M, Kalyanaraman B, Kirk T K. J Biol Chem. 1985;260:2609–2612. [PubMed] [Google Scholar]
- 14.Schoemaker H E, Harvey P J, Bowen R M, Palmer J M. FEBS Lett. 1985;183:7–12. [Google Scholar]
- 15.Lundquist K, Kirk T K. Phytochemistry. 1978;17:1676–1681. [Google Scholar]
- 16.Harvey P J, Schoemaker H E, Bowen R M, Palmer J M. FEBS Lett. 1985;183:13–16. [Google Scholar]
- 17.Leisola M S A, Haemmerli S D, Waldner R, Schoemaker H E, Schmidt H W H, Fiechter A. Cellulose Chem Technol. 1988;22:267–277. [Google Scholar]
- 18.Hammel K E, Moen M A. Enzyme Microb Technol. 1991;13:15–18. [Google Scholar]
- 19.Schoemaker H E, Lundell T K, Hatakka A I, Piontek K. FEMS Microbiol Rev. 1994;13:321–332. [Google Scholar]
- 20.Barr D P, Aust S D. Rev Environ Contam Toxicol. 1994;138:49–72. doi: 10.1007/978-1-4612-2672-7_3. [DOI] [PubMed] [Google Scholar]
- 21.Wariishi H, Sheng D, Gold M H. Biochemistry. 1994;33:5545–5552. doi: 10.1021/bi00184a025. [DOI] [PubMed] [Google Scholar]
- 22.Beratan D N, Onuchic J N, Winkler J R, Gray H B. Science. 1992;258:1740–1471. doi: 10.1126/science.1334572. [DOI] [PubMed] [Google Scholar]
- 23.Moser C C, Keske J M, Warncke K, Farid R S, Dutton L. Nature (London) 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 24.Wariishi H, Gold M H. J Biol Chem. 1990;265:2070–2077. [PubMed] [Google Scholar]
- 25.Wariishi H, Dunford H B, MacDonald I D, Gold M H. J Biol. Chem. 1989;264:3335–3340. [PubMed] [Google Scholar]
- 26.Wayman M, Obiaga T I. Can J Chem. 1974;52:2102–2110. [Google Scholar]
- 27.Cotton M L, Dunford H B. Can J Chem. 1973;51:582–587. [Google Scholar]
- 28.Edwards P R, Gill A, Pollard-Knight D V, Hoare M, Buckle P E, Lowe P A, Leathbarrow R J. Anal Biochem. 1995;231:210–217. doi: 10.1006/abio.1995.1522. [DOI] [PubMed] [Google Scholar]
- 29.Wariishi H, Huang J, Dunford H B, Gold M H. J Biol Chem. 1991;266:20694–20699. [PubMed] [Google Scholar]
- 30.Dunford H B, Stillman J S. Coord Chem Rev. 1976;19:187–251. [Google Scholar]
- 31.Renganathan V, Gold M H. Biochemistry. 1986;25:1626–1631. [Google Scholar]
- 32.Marquez L, Wariishi H, Dunford H B, Gold M H. J Biol Chem. 1988;263:10549–10552. [PubMed] [Google Scholar]
- 33.Candeias L P, Harvey P J. J Biol Chem. 1995;270:16475–16479. doi: 10.1074/jbc.270.28.16745. [DOI] [PubMed] [Google Scholar]
- 34.Khindaria A, Grover T A, Aust S D. Biochemistry. 1995;34:6020–6025. doi: 10.1021/bi00018a003. [DOI] [PubMed] [Google Scholar]
- 35.Harvey P J, Floris R, Lundell T K, Palmer J M, Schoemaker H E, Wever R. Biochem Soc Trans. 1992;20:345–349. doi: 10.1042/bst0200345. [DOI] [PubMed] [Google Scholar]
- 36.Edwards S L, Raag R, Wariishi H, Gold M H, Poulos T L. Proc. Natl Acad Sci USA. 1993;90:750–754. doi: 10.1073/pnas.90.2.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulos T L, Edwards S L, Wariishi H, Gold M H. J Biol Chem. 1993;268:4429–4440. doi: 10.2210/pdb1lga/pdb. [DOI] [PubMed] [Google Scholar]
- 38.Piontek K, Glumoff T, Winterhalter K H. FEBS Lett. 1993;315:119–124. doi: 10.1016/0014-5793(93)81146-q. [DOI] [PubMed] [Google Scholar]
- 39.Schoemaker H E, Piontek K. Pure Appl Chem. 1996;68:2089–2096. [Google Scholar]
- 40.Blodig W, Doyle A, Smith A T, Winterhalter K, Choinowski T, Piontek K. Biochemistry. 1998;37:8832–8838. doi: 10.1021/bi9727186. [DOI] [PubMed] [Google Scholar]
- 41.Doyle W A, Blodig W, Veitch N C, Piontek T, Smith A T. Biochemistry. 1998;37:15097–15105. doi: 10.1021/bi981633h. [DOI] [PubMed] [Google Scholar]
- 42.Musah R A, Goodin D B. Biochemistry. 1997;36:11665–11674. doi: 10.1021/bi9708038. [DOI] [PubMed] [Google Scholar]
- 43.Finzel B C, Poulos T L, Kraut J. J Biol Chem. 1984;259:13027–13036. [PubMed] [Google Scholar]
- 44.Sundaramoorthy M, Kishi K, Gold H M, Poulos T L. J Biol. Chem. 1994;269:32759–32767. [PubMed] [Google Scholar]
- 45.Gajhede M, Schuller J S, Henriksen A, Smith A T, Poulos T L. Nat Struct Biol. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- 46.Patterson W R, Poulos T L. Biochemistry. 1995;34:4331–4341. doi: 10.1021/bi00013a023. [DOI] [PubMed] [Google Scholar]
- 47.Kunishima N, Fukuyama K, Matsubara H, Hatanaka H, Shibano Y, Amachi T. J Mol Biol. 1994;235:331–344. doi: 10.1016/s0022-2836(05)80037-3. [DOI] [PubMed] [Google Scholar]
- 48.Schuller D J, Ban N, van Huystee R B, Mcpherson A, Poulos T L. Structure. 1996;4:311–321. doi: 10.1016/s0969-2126(96)00035-4. [DOI] [PubMed] [Google Scholar]
- 49.Henriksen A, Welinder K G, Gajhede M. J Biol Chem. 1998;273:2241–2248. doi: 10.1074/jbc.273.4.2241. [DOI] [PubMed] [Google Scholar]
- 50.Farrell R L, Murtagh K E, Tien M, Mozuch M D, Kirk T K. Enzyme Microb Technol. 1989;11:322–328. [Google Scholar]