

Abstract
Genome-wide association (GWA) studies have identified multiple new genomic loci at which common variants modestly but reproducibly influence risk of type 2 diabetes (T2D)1-11. Established associations to common and rare variants explain only a small proportion of the heritability of T2D. As previously published analyses had limited power to discover loci at which common alleles have modest effects, we performed meta-analysis of three T2D GWA scans encompassing 10,128 individuals of European-descent and ~2.2 million SNPs (directly genotyped and imputed). Replication testing was performed in an independent sample with an effective sample size of up to 53,975. At least six new loci with robust evidence for association were detected, including the JAZF1 (p=5.0×10−14), CDC123/CAMK1D (p=1.2×10−10), TSPAN8/LGR5 (p=1.1×10−9), THADA (p=1.1×10−9), ADAMTS9 (p=1.2×10−8), and NOTCH2 (p=4.1×10−8) gene regions. The large number of loci with relatively small effects indicates the value of large discovery and follow-up samples in identifying additional clues about the inherited basis of T2D.
Genome-wide association studies are unbiased by previous hypotheses concerning candidate genes and pathways, but challenged by the modest effect sizes of individual common susceptibility variants and the need for stringent statistical thresholds. For example, the largest allelic odds ratio of any established common variant for T2D is ~1.35 (TCF7L2), with the nine other validated associations to common variants (excluding FTO, which has its primary effect through obesity) having allelic odds ratios between 1.1 and 1.21-6,11,12. To augment power to detect additional loci of similar and/or smaller effect, we increased sample size by combining three previously published GWA studies (Diabetes Genetics Initiative [DGI], Finland-United States Investigation of NIDDM Genetics [FUSION], and Wellcome Trust Case Control Consortium [WTCCC])1-4, and extended SNP coverage by imputing untyped SNPs based on patterns of haplotype variation from the HapMap dataset13 (Table 1).
Table 1.
Overview of study design.
| Study | n cases# | n controls# | effective sample size# |
n directly genotyped SNPs* |
n imputed SNPs* |
|---|---|---|---|---|---|
| Stage 1 | |||||
|
| |||||
| DGI | 1,464 | 1,467 | 2,521 | 378,860 | 1,853,222 |
| WTCCC | 1,924 | 2,938 | 4,706 | 393,143 | 1,915,393 |
| FUSION | 1,161 | 1,174 | 2,335 | 306,222 | 2,110,199 |
|
| |||||
| Stage 2 | |||||
|
| |||||
| DGI stage 2 | 5,065 | 5,785 | 9,874 | 63 | - |
| FUSION stage 2 | 1,215 | 1,258 | 2,473 | 59 | - |
| UK stage 2 | 3,757 | 5,346 | 9,114 | 66 | - |
|
| |||||
| Stage 3 | |||||
|
| |||||
| deCODE | 1,520 (1,422) | 25,235 (3,455) | 4,280 (3,130) | 11 | - |
| KORA | 1,241 | 1,458 | 2,684 | 6 | - |
| Danish | 4,089 | 5,043 | 8,690 | 11 | - |
| HUNT | 1,004 | 1,503 | 2,412 | 11 | - |
| NHS | 1,506 | 2,014 | 3,468 | 10 | - |
| CCC | 547 | 533 | 1,070 | 11 | - |
| EPIC | 388 | 774 | 1,036 | 10 | - |
| ADDITION/Ely | 892 | 1,610 | 2,288 | 11 | - |
| Norfolk | 2,311 | 2,400 | 4,450 | 11 | - |
| METSIM | 659 | 2,639 | 2,136 | 11 | - |
Autosomal SNPs passing quality control, as defined for directly genotyped and imputed SNPs in each study (QC criteria: SNPTEST information measure≥0.5; r2hat≥0.3; MAF>0.01). For the stage 1 meta-analysis, we combined results for 2,168,847 directly genotyped and imputed SNPs passing QC in all three studies (Methods; Supplementary Methods).
Sample sizes presented here are the maximum available for each study. For the deCODE stage 3 study, we used genotype data from the Icelandic GWA scan5 for rs2641348, rs7578597 and rs9472138, and a perfect proxy (rs2793831, based on HapMap) for rs10923931. The remaining SNPs had not been directly typed as part of this scan and were therefore genotyped separately, in a subset of the GWA scan samples (numbers indicated in parentheses) (Supplementary Methods).
We started with a set of genotyped autosomal SNPs that passed quality control (QC) filters in each study: in WTCCC, 393,143 SNPs from the Affymetrix 500k chip (MAF>0.01; 1,924 cases and 2,938 population-based controls from the Wellcome Trust Case Control Consortium3,4); in DGI, 378,860 SNPs from the Affymetrix 500k chip (minor allele frequency [MAF]>0.01; Swedish and Finnish sample of 1,464 T2D cases and 1,467 normoglycaemic controls, including 326 discordant sibships1); and in FUSION, 306,222 SNPs from the Illumina 317k chip (MAF>0.01, 1,161 T2D cases and 1,174 normal glucose tolerant controls from Finland2) (Supplementary Table 1). There were 44,750 SNPs (MAF>0.01) directly genotyped in all three studies across the two platforms. We used data from the GWA studies and phased chromosomes from the HapMap CEU sample to impute autosomal SNPs with MAF>0.01(14 and Y.L., C.J.W., J.D, P.S., G.R.A. Markov model for rapid haplotyping and genotype imputation in genome wide studies. Submitted, 2007; http://www.sph.umich.edu/csg/abecasis/MaCH/download/). We based our further analyses on 2,168,847 SNPs that met imputation and genotyping QC criteria across all studies (Methods; Supplementary Methods).
Using these directly measured and imputed genotypes, we tested for association of each SNP with T2D in each study separately, corrected each study for residual population stratification, cryptic relatedness or technical artifacts using genomic control, and then combined these results in a genome-wide meta-analysis across a total of 10,128 samples (4,549 cases, 5,579 controls) (Methods; Supplementary Methods). We calculated that this sample size provides reasonable power to detect additional variants with properties similar to those previously identified by less formal data combination efforts1,2,4 (Supplementary Table 2). Unless otherwise indicated, results presented are derived from individually genomic control-adjusted stage 1 results. Meta-analysis OR and confidence intervals are obtained from a fixed-effects model and p values from a weighted z-statistic-based meta-analysis (Methods; Supplementary Methods). As expected, the most significant result was obtained for rs7903146 in TCF7L2. We also observed evidence for association (p<10−3) at eight of the ten established T2D loci (as well as at the FTO obesity locus)12 (Supplementary Table 3). This is unsurprising, as these same data supported discovery of many of these loci. Since our goal was to identify new loci, we excluded 1,981 SNPs in the immediate vicinity of these T2D susceptibility loci from further analysis (with the exception of a signal near PPARG, which was followed-up), and examined the remainder of the autosomal genome (Methods; Supplementary Methods). Even after excluding known loci, we saw a strong enrichment of highly associated variants: 426 with p values <10−4, compared to 217 under the null.
Before proceeding to follow-up, we explored the individual studies and combined data for potential errors and biases. We found a genomic control λ value of 1.04 for the combined results (based on 10,128 samples), which, given the relationship between λ and sample size15, suggests little residual confounding (Supplementary Figure 1; Supplementary Note). We also used genome-wide genotype data to estimate the principal components (PC) of the identity-by-state relationships in each stage 1 sample. For the SNPs presented in Table 2, adjustment for principal components in stage 1 T2D association analysis did not diminish the association in the WTCCC (2 PCs), FUSION (10 PCs), or DGI (10 PCs) sample (Supplementary Note). Additionally, we found no evidence for association between UK population ancestry informative markers3 and disease status in the UK replication sets (Supplementary Note). To ensure that the observed stage 1 associations taken forward to follow-up were not due to imputation errors, we directly genotyped originally imputed variants in the stage 1 samples (Methods; Supplementary Methods). We found strong agreement between the genotype-based and imputed p values (in 38 of 43 cases where a direct genotype-based result was obtained, the p value was within one order of magnitude of that from imputation, and in the remaining 5 cases p values were less than 2 orders of magnitude different) (Supplementary Table 4).
Table 2.
Eleven T2D-associated SNPs taken forward to stages 2 and 3.
| Stage 1 (DGI, FUSION, WTCCC) |
Stage 2 (DGI, FUSION, UKT2D) |
Stage3 (deCODE, KORA, Steno, HUNT, NHS, CCC, EPIC ADDITION/Ely, Norfolk, METSIM) |
All data | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||||||||
| SNP | Chr | Position NCBI35 (bp) | non- risk allele# |
risk allele# |
risk allele frequency# |
nearest gene(s) |
OR (95%CI) | P value | OR (95%CI) | P value | OR (95%CI) | P value | neff | OR (95%CI) | P value | P het | n samples for 80% power## |
| rs864745 | 7 | 27,953,796 | C | T | 0.501 | JAZF1 | 1.14 (1.07-1.20) |
1.5E-04 | 1.08 (1.04-1.12) |
8.1E-05 | 1.10 (1.06-1.15) |
1.3E-07 | 59,617 | 1.10 (1.07-1.13) |
5.0E-14 | 0.70 | 10,610 |
| rs12779790 | 10 | 12,368,016 | A | G | 0.183 | CDC123/CAMK1D | 1.15 (1.06-1.24) |
4.2E-04 | 1.11 (1.06-1.16) |
5.4E-05 | 1.09 (1.04-1.14) |
1.5E-04 | 62,366 | 1.11 (1.07-1.14) |
1.2E-10 | 0.67 | 9334 |
| rs7961581 | 12 | 69,949,369 | T | C | 0.269 | TSPAN8/LGR5 | 1.18 (1.10-1.26) |
1.8E-05 | 1.06 (1.02-1.11) |
9.8E-03 | 1.09 (1.04-1.13) |
4.3E-05 | 62,301 | 1.09 (1.06-1.12) |
1.1E-09 | 0.20 | 23,206 |
| rs7578597 | 2 | 43,644,474 | C | T | 0.902 | THADA | 1.25 (1.12-1.40) |
1.8E-04 | 1.15 (1.07-1.22) |
1.6E-03 | 1.12 (1.05-1.20) |
9.2E-05 | 60,832 | 1.15 (1.10-1.20) |
1.1E-09 | 0.008 | 9,624 |
| rs4607103 | 3 | 64,686,944 | T | C | 0.761 | ADAMTS9 | 1.13 (1.06-1.22) |
5.4E-04 | 1.10 (1.05-1.15) |
1.0E-04 | 1.06 (1.01-1.11) |
3.5E-03 | 62,387 | 1.09 (1.06-1.12) |
1.2E-08 | 0.17 | 9,748 |
| rs10923931* | 1 | 120,230,001 | G | T | 0.106 | NOTCH2 | 1.30 (1.17-1.43) |
1.1E-04 | 1.09 (1.03-1.16) |
2.9E-03 | 1.11 (1.05-1.18) |
1.9E-03 | 58,667 | 1.13 (1.08-1.17) |
4.1E-08 | 0.004 | 21,568 |
| rs1153188 | 12 | 53,385,263 | T | A | 0.733 | DCD | 1.15 (1.08-1.23) |
3.2E-05 | 1.07 (1.03-1.12) |
3.1E-03 | 1.06 (1.02-1.10) |
8.8E-03 | 62,301 | 1.08 (1.05-1.11) |
1.8E-07 | 0.79 | 17,808 |
| rs17036101** | 3 | 12,252,845 | A | G | 0.927 | SYN2/PPARG | 1.33 (1.18-1.50) |
1.0E-05 | 1.13 (1.04-1.22) |
4.5E-03 | 1.11 (1.02-1.20) |
1.2E-02 | 59,682 | 1.15 (1.10-1.21) |
2.0E-07 | 0.19 | 16,370 |
| rs2641348* | 1 | 120,149,926 | A | G | 0.107 | ADAM30 | 1.14 (1.05-1.25) |
1.4E-03 | 1.10 (1.03-1.17) |
1.2E-03 | 1.09 (1.03-1.16) |
7.8E-03 | 60,048 | 1.10 (1.06-1.15) |
4.0E-07 | 0.08 | 17,428 |
| rs9472138 | 6 | 43,919,740 | C | T | 0.282 | VEGFA | 1.13 (1.06-1.21) |
5.4E-05 | 1.07 (1.02-1.12) |
1.5E-03 | 1.03 (1.00-1.07) |
9.5E-02 | 63,537 | 1.06 (1.04-1.09) |
4.0E-06 | 0.43 | 16,696 |
| rs10490072 | 2 | 60,581,582 | C | T | 0.724 | BCL11A | 1.17 (1.10-1.26) |
3.4E-05 | 1.08 (1.03-1.13) |
1.4E-03 | 1.00 (0.97-1.04) |
6.5E-01 | 59,682 | 1.05 (1.03-1.08) |
1.0E-04 | 0.0035 | 13,502 |
|
| |||||||||||||||||
| Maximum available effective sample size | 9,562 | 21,461 | 32,514 | ||||||||||||||
Table 2 presents results from the analysis of directly genotyped data only, except for FUSION stage 1 results for rs7961581 (Supplementary Methods). Combined estimates of odds ratio (OR) were calculated using a fixed effects, inverse variance meta-analysis; DGI discordant sibling pairs were not included in OR estimates. P values were combined using a weighted z score-based meta-analysis including DGI sibships; p values for the three stage 1 studies were individually corrected by genomic control before meta analysis.
Ancestral allele is denoted in bold, based on Entrez SNP and derived by comparison against chimpanzee sequence. The risk allele frequencies presented are sample size-weighted risk allele frequencies across the stage 2 studies.
SNPs rs10923931 and rs2641348 appear to represent the same signal (r2=0.92 in HapMap CEU)13. Results for rs2934381 and rs2793831 (perfect proxies for rs10923931) are presented for UK (stage 1,2) and deCODE (stage 3) respectively.
The signal at SNP rs17036101 is indistinguishable from rs1801282, the established P12A variant in PPARG.
Sample size (sum of case and control samples) required for 80% power (to achieve nominal replication at α=0.05) is calculated based on the stage 2 OR estimate, sample size-weighted risk allele frequency across the stage 2 studies and assuming an equal number of cases and controls (Supplementary Methods).
We selected SNPs for replication based principally on the statistical evidence for association in stage 1, excluding SNPs with evidence for heterogeneity of ORs (p<10−4) across studies (Methods; Supplementary Methods). Sixty nine SNPs were taken forward to an initial round of replication (stage 2) in up to 22,426 additional samples of European descent (Table 1, Supplementary Table 1). The distribution of association p values in stage 2 was highly inconsistent with a null distribution. Of the 69 signals selected for follow-up, a total of 65 were successfully genotyped in stage 2, and represented loci that were independent of each other and of previously established susceptibility loci. Nine of these had a p value ≤0.01 with association in the same direction as the original signal, far in excess of 0.33 expected under the null (p=1.4×10−12, binomial test; Supplementary Methods), and two SNPs had p<10−4 as compared to an expectation of 0.0033 (p=5.2×10−6) (Supplementary Methods; Supplementary Table 5).
We identified eleven SNPs (ten separate signals, nine of which represent novel loci) with p<0.005 in stage 2, for which the combined stage 1 and stage 2 data (based on direct genotyping of stage 1 samples, where previously imputed) generated p<10−5. These eleven SNPs were further genotyped in up to 57,366 additional samples (14,157 cases and 43,209 controls) of European descent in stage 3 (Table 1; Supplementary Table 1; Methods; Supplementary Methods). The distribution of p values for these 11 SNPs was again inconsistent with a null distribution: all nine new and independent SNPs had effects in the same direction as in the stage 1 + 2 meta-analysis (p=0.002), and seven had p<0.05 in the direction of the original association (p=2.1×10−10) (Table 2).
Based on the combined stage 1-3 analyses, six signals reach compelling levels of evidence (p=5.0×10−8 or better) for T2D association (Table 2). As in all LD-mapping approaches, characterization of the causal variants responsible, their effect sizes, and the genes through which they act will require extensive resequencing and fine-mapping. However, on current evidence, the most associated variants in each of these signals map to intron 1 of JAZF1, between CDC123 and CAMK1D, between TSPAN8 and LGR5, in exon 24 of THADA, near ADAMTS9, and in intron 5 of NOTCH2.
The strongest statistical evidence for a novel association signal was with rs864745 in intron 1 of the JAZF1 gene (Figure 1), one of a cluster of associated SNPs with strong evidence for association in the stage 1 meta-analysis, and across each replication sample set (Table 2; Supplementary Table 6). The overall estimate of effect was an OR[95%CI] of 1.10[1.07-1.13] (p=5.0×10−14 under an additive model), based on 68,042 individuals. The JAZF1 (juxtaposed with another zinc finger gene 1) gene encodes a transcriptional repressor of NR2C2 (nuclear receptor subfamily 2, group C, member 2)16. Mice deficient in Nr2c2 exhibit growth retardation, low IGF1 serum levels, and perinatal and early postnatal hypoglycaemia17. While this paper was in review, a SNP in JAZF1 was identified as associated with prostate cancer18; this is particularly interesting given the recent finding that SNPs in TCF2 are also associated both with T2D and prostate cancer19,20.
Figure 1.
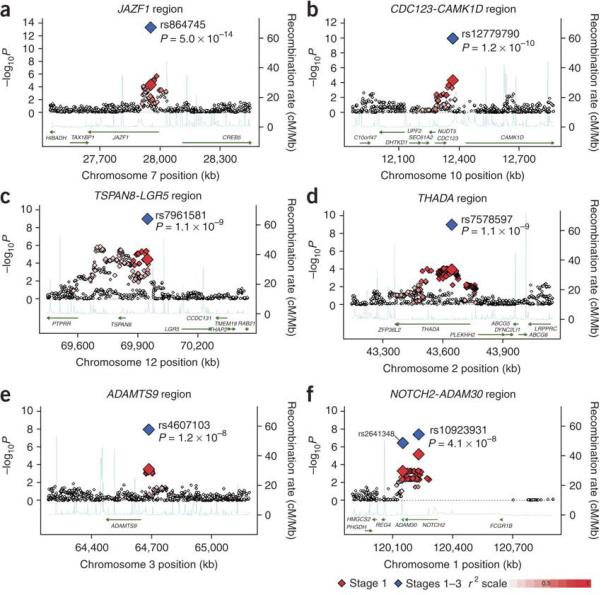
Regional plots of six confirmed associations. For each of the (A) JAZF1, (B) CDC123/CAMK1D, (C) TSPAN8/LGR5, (D) THADA, (E) ADAMTS9 and (F) NOTCH2/ADAM30 regions, genotyped and imputed SNPs passing QC across all three stage 1 studies are plotted with their meta-analysis p values (as −log10 values) as a function of genomic position (with NCBI Build 35). In each panel, the SNP taken forward to stages 2 and 3 is represented by a blue diamond (meta-analysis p value across stages 1-3), and its initial p value in stage 1 data is denoted by a red diamond. Estimated recombination rates (taken from HapMap)13 are plotted to reflect the local LD structure around the associated SNPs and their correlated proxies (according to a white to red scale from r2=0 to r2=1; based on pairwise r2 values from HapMap CEU)13. Gene annotations were taken from the University of California-Santa Cruz genome browser.
The second strongest new statistical signal (rs12779790, combined OR[95%CI] of 1.11[1.07-1.14], p=1.2×10−10) lies in an intergenic region ~90 kb from the CDC123 (cell division cycle 123 homolog [S. cerevisiae]) and ~63.5 kb from the CAMK1D (calcium/calmodulin-dependent protein kinase ID) genes (Figure 1; Table 2; Supplementary Table 6). CDC123 is regulated by nutrient availability in S. cerevisiae and plays a role in cell cycle regulation21. Evidence from previous GWA studies implicating variants in CDKAL1 and near CDKN2A/B in T2D predisposition suggests that cell cycle dysregulation may be a common pathogenetic mechanism in T2D1,2,4.
The third strongest statistical signal resides upstream of the TSPAN8 (tetraspanin 8) gene (rs7961581; combined OR[95%CI]: 1.09[1.06-1.12], p=1.1×10−9) (Figure 1; Table 2; Supplementary Table 6). Tetraspanin 8 is a cell-surface glycoprotein expressed in carcinomas of the colon, liver and pancreas.
The fourth strongest novel association signal (rs7578597, a non-synonymous SNP [T1187A]; combined OR[95%CI] of 1.15[1.10-1.20], p=1.1×10−9) resides in exon 24 of the widely-expressed THADA (thyroid adenoma associated) gene (Figure 1; Table 2; Supplementary Table 6). Disruption of THADA by chromosomal rearrangements (including fusion with intronic sequence from PPARG) is observed in thyroid adenomas22. The function of THADA has not been well-characterized, but there is some evidence to suggest it may be involved in the death receptor pathway and apoptosis23.
Rs4607103 (combined OR[95%CI]: 1.09[1.06-1.12], p=1.2×10−8), representing a cluster of associated SNPs, resides ~38 kb upstream of the ADAMTS9 (ADAM metallopeptidase with thrombospondin type 1 motif, 9) gene, and is the fifth strongest new signal (Figure 1; Table 2, Supplementary Table 6). ADAMTS9 is a secreted metalloprotease that cleaves the proteoglycans versican and aggrecan, and is expressed widely, including in skeletal muscle and pancreas.
The sixth strongest signal, marked by rs10923931, resides within intron 5 of the NOTCH2 (Notch homolog 2 [Drosophila]) gene (combined OR[95%CI]: 1.13[1.08-1.17], p=4.1×10−8) (Figure 1; Table 2; Supplementary Table 6). Rs2641348, a non-synonymous SNP (L359P) within the neighboring ADAM30 (ADAM metallopeptidase domain 30) gene represents the same signal (r2=0.92 based on HapMap CEU data) and was also followed-up, but its overall signal (combined OR[95%CI]: 1.10[1.06-1.15], p=4.0×10−7; Table 2) was slightly less strong. NOTCH-2 is a type 1 transmembrane receptor; in mice, the Notch-2 receptor is expressed in embryonic ductal cells of branching pancreatic buds during pancreatic organogenesis, the likely source of endocrine and exocrine stem cells24.
The strength of the association evidence for the remaining four variants taken into stage 3 does not meet our prespecified threshold of p≤5.0×10−8. However, it is likely (based on individual significance values and their overall distribution) that several of these also represent genuine association signals. In all, three of these additional SNPs showed p values <10−5 across the combined data (Table 2), and two demonstrated p<0.05 in stage 3 in the same direction as in stages 1 and 2. Variants near DCD (dermcidin) showed evidence for association (rs1153188; overall p=1.8×10−7) (Supplementary Figure 2). A signal in VEGFA had previously been noted in the UK GWA scan4, but displays inconsistent evidence for replication: further studies will be required to establish its status. We also found association at rs17036101, ~44 kb downstream of SYN2 (synapsin II) and 115.3 kb upstream of the established T2D susceptibility variant rs1801282 (Pro12Ala) in the PPARG gene (r2=0.54 in HapMap CEU) (Supplementary Figure 3). Conditional analyses in stage 1 + 2 samples could not differentiate between the effect of these two SNPs (Supplementary Note; Supplementary Table 7).
None of the 11 SNPs (Table 2) were convincingly associated with body mass index (BMI) (Supplementary Table 8) or other T2D-related traits (with p<10−3) (Supplementary Table 9). The largest fold-change in T2D association p values before and after adjusting for BMI was for rs17036101 (p=8.1×10−8 before adjustment and p=7.5×10−6 after adjustment for BMI; Supplementary Table 10). Conditioning on the associated SNP that was taken forward to stages 2 and 3 in each region revealed no additional independent association signals (p<10−4) in stage 1 data (Supplementary Figure 4; Supplementary Note).
By combining three GWA scans involving 10,128 samples (enhanced through imputation approaches) and undertaking large-scale replication in up to 79,792 additional samples, we have identified six additional loci from the human genome that apparently harbor common genetic variants that influence susceptibility to T2D. These findings are consistent with a model in which the preponderance of loci detectable through the GWA approach (using current arrays and indirect, LD mapping, at least) have modest effects (ORs between 1.1 and 1.2). Given such a model, our study (in which we followed up only 69 signals out of over 2 million meta-analysed SNPs) would be expected to recover only a subset of the loci with similar characteristics (that is, those that managed to reach our stage 1 selection criteria). Further efforts to expand GWA meta-analyses and to extend the number of SNPs taken forward to large-scale replication should confirm additional genomic loci, as should targeted analysis of copy number variation. However, estimates based on the first common SNP marker in a region are certainly underestimates. The effect of the actual common causal mutation(s) will typically be larger, since effect sizes are now estimated through LD. Moreover, the same genes likely carry rare mutations of larger effect: three genes with common variants that influence risk of T2D were first discovered based on rare Mendelian mutations (KCNJ11, WSF1 and TCF2). Regardless of effect size, these loci provide important clues to the processes involved in the maintenance of normal glucose homeostasis and in the pathogenesis of type 2 diabetes.
Methods
Stage 1 samples, genome-wide genotyping and quality control (expanded in Supplementary Methods)
UK
The WTCCC stage 1 sample consists of 1,924 T2D cases and 2,938 population controls from the UK3,4. These samples were genotyped on the Affymetrix GeneChip Human Mapping 500k Array Set. The call frequency of included samples was >0.97. 393,143 autosomal SNPs passed quality control (QC) criteria: Hardy-Weinberg equilibrium [HWE] p>10−4 in T2D cases and controls, call frequency >0.95, minor allele frequency (MAF)>0.01, and good clustering, as defined in3,4.
DGI
The DGI stage 1 Swedish and Finnish sample consists of 1,464 T2D cases and 1,467 normoglycemic controls. Of these, 2,097 are population-based T2D cases and controls matched for BMI, gender, and geographic origin, and 834 are T2D cases and controls in 326 sibships discordant for T2D1. These samples were genotyped on the Affymetrix GeneChip Human Mapping 500k Array Set, and all included samples had a genotype call rate >0.95. 378,860 autosomal SNPs passed QC criteria (call frequency >0.95, HWE p>10−6 in controls and MAF>0.01 in both population and familial components)1.
FUSION
The FUSION stage 1 sample consists of 1,161 Finnish T2D cases and 1,174 Finnish normal glucose tolerant controls2. In addition, 122 FUSION offspring with genotyped parents were included for quality control purposes and quantitative trait analysis. Samples were genotyped with the Illumina HumanHap300 BeadChip (version 1.1). All samples included had a call frequency >0.975. 306,222 autosomal SNPs passed QC2 and had a HWE p≥10−6 in the total sample, ≤3 combined duplicate or non-Mendelian inheritance errors (out of 79 duplicate samples and 122 parent-offspring sets), call frequency ≥0.90, and MAF>0.01.
Analysis of stage 1 genotype data
In combining data across the three studies, we did not attempt, given differences in study design and implementation, to harmonize every aspect of individual study analysis and QC. For the UK, DGI and FUSION studies respectively, 393,143, 378,860 and 306,222 SNPs were analyzed under an additive model. The genomic control values for these directly genotyped SNPs were 1.08 (UK), 1.06 (DGI) and 1.03 (FUSION) (Supplementary Methods).
Stage 1 imputation and T2D analysis
For each stage 1 sample set, we imputed genotypes for autosomal SNPs that were present in HapMap Phase II but were not present in the genome-wide chip or did not pass direct genotyping QC. In each sample, genotypes were imputed using the genotype data from the GWA chips and phased HapMap II genotype data from the 60 CEU HapMap founders. We retained SNPs that had an estimated MAF>0.01 in the control samples. Imputed SNPs were then tested for T2D association. The genomic control values for these imputed SNPs were 1.08 (UK), 1.07 (DGI) and 1.04 (FUSION) (Supplementary Methods).
Stage 1 meta-analysis (expanded in Supplementary Methods)
We used meta-analysis to combine the T2D association results for the stage 1 WTCCC, DGI and FUSION samples. The combined stage 1 data are comprised of 10,128 samples: 4,549 T2D cases and 5,579 controls. We used association results from directly genotyped SNPs, where available, and imputed genotype association results at all other positions. 2,168,847 genotyped and imputed autosomal SNPs passed QC and had MAF>0.01 in each of the three samples (44,750 were genotyped in all three samples, 308,628 were genotyped in two samples, 245,158 were genotyped in one sample, 1,570,311 were imputed in all samples). All association results were expressed relative to the forward strand of the reference genome based on dbSNP125. For our initial analysis, which was used to select signals for stage 2 genotyping, for each SNP we combined the ORs for a given reference allele weighted by the confidence intervals using a fixed effects model. We investigated evidence for heterogeneity of ORs using two commonly used statistics: Cochrans's Q statistic and I2 (25).
We repeated the meta-analysis combining evidence for association based solely on the p value. Specifically, for each study we converted the two-sided p value to a z-statistic which was signed to reflect the direction of the association given the reference allele. Each z-score was then weighted; the squared weights were chosen to sum to 1 and each sample-specific weight was proportional to the square root of the effective number of individuals in the sample. Weighted z-statistics were summed across studies and the summary z-score converted to a two-sided p value.
SNP prioritisation for stage 2 genotyping
We prioritized 69 SNPs for replication in stage 2 based on the results from the three-study stage 1 meta-analysis, using a set of criteria we developed as part of a heuristic approach to the prioritization of loci for follow-up (Supplementary Methods). Briefly, we considered SNPs with a meta-analysis p value <10−4 and a meta-analysis heterogeneity p value >10−4. These selections were largely made using the initial OR-based version of the meta-analysis. We allowed some exceptions to the above follow-up criteria.
Five SNPs were selected for replication genotyping on the basis of their strong association with T2D in the DGI GWA study (2 SNPs), association with T2D and with insulinogenic index in the DGI study (1 SNP), and overlap with FUSION or WTCCC (p<0.05 in DGI and one or both studies; 2 SNPs). For known T2D loci (TCF7L2, CDKAL1, IGF2BP2, KCNJ11, HHEX/IDE, SLC30A8, CDKN2A/2B region, WFS1, TCF2, and FTO) we excluded from follow-up all SNPs that resided within the surrounding region, with region boundaries defined by the furthest neighboring SNPs with p values remaining ~0.01 (n=1,981). For the PPARG region, we identified a SNP, rs17036101, with a p value two orders of magnitude lower than the established Pro12Ala susceptibility variant, rs1801282, and took this signal forward to replication. A total of 69 SNPs were taken forward to stage 2 genotyping.
Stage 2 samples, genotyping and analysis
UK
We genotyped the prioritized SNPs in cases and controls from three UK replication sets (RS1, RS2 and RS3, described in4; Supplementary Table 1; Supplementary Methods). Genotyping of prioritized SNPs in RS1, RS2 and RS3 was performed by Kbiosciences (Herts., UK). All assays were validated prior to use, using a standard 96-well validation plate used by Kbiosciences and up to 296 samples from the WTCCC study (see Comparison of genotypes from imputation and direct genotyping; Supplementary Methods). Concordance rates between the Affymetrix and KASPar/TaqMan genotypes (based on up to 296 replicate stage 1 samples) were 97.5% on average. All genotyped SNPs had genotype call frequency rates >94% in the replication sets and no SNPs had HWE p value<0.001 in cases or controls. We tested for association with T2D using the Cochran-Armitage test for trend. Results from the 3 replication sets were combined in a Cochran-Mantel-Haenszel meta-analysis framework.
DGI
We genotyped the prioritized SNPs in three stage 2 case-control samples1 (Supplementary Table 1; Supplementary Methods). The prioritized SNPs were genotyped in all DGI stage 1 and 2 samples using the iPLEX Sequenom MassARRAY platform (http://www.sequenom.com/Assets/pdfs/appnotes/8876-006.pdf). 63 SNPs passing QC (>94% call rate, MAF>0.01 and HWE p value >0.001) were used for association testing. We tested for T2D association in each DGI stage 2 case-control set using a chi-squared analysis (assuming an additive genetic model). Results from the three DGI stage 2 samples were combined using Cochran-Mantel-Haenszel meta-analysis.
FUSION
We genotyped the prioritized SNPs in a Finnish case-control sample (Supplementary Table 1; Supplementary Methods) using the Sequenom Homogeneous Mass EXTEND or iPLEX Gold SBE assays, carried out at the National Human Genome Research Institute (NHGRI). 59 SNPs had genotype call frequency >94% and HWE p value >0.001. The genotype consistency rate among 56 duplicate samples was 100% and the average call frequency of successfully genotyped SNPs was 97.3%. SNPs were analyzed using logistic regression with adjustment for sex, 5-year age category and birth province and an additive model for the genetic effect.
Comparison of genotypes from imputation and direct genotyping
A proportion of the prioritized imputed signals was genotyped in the stage 1 samples of the three studies and respective concordance rates were calculated (Supplementary Methods; Supplementary Table 4). All results presented in the main manuscript text are based on directly-typed stage 1 data.
Combined meta-analysis for stages 1 and 2
We combined stage 1 and stage 2 data using both the OR-based and the weighted z score-based meta-analysis approaches described above (Stage 1 meta-analysis). We also assessed our results using random effects meta-analysis to better account for any heterogeneity between the studies (Supplementary Table 6). Locus-specific and combined sibling relative risk estimates were calculated using sample size-weighted estimates of the effect size and risk-allele frequency derived from stage 2 replication samples only, and under the assumption of allelic and locus independence, as described by26,27.
Stage 3 sample, genotyping and association analysis
Eleven SNPs (rs2641348, rs10490072, rs7578597, rs17036101, rs4607103, rs9472138, rs864745, rs12779790, rs1153188, rs10923931, and rs7961581) were followed up in the stage 3 samples, from the deCODE, KORA, Danish, HUNT, NHS, GEM Consortium (CCC, EPIC, ADDITION/Ely, Norfolk) and METSIM studies (Supplementary Table 1; Supplementary Methods).
Combined meta-analysis for stages 1, 2, and 3
We combined stage 1, 2 and 3 data using both meta-analysis approaches (fixed-effects model to combine ORs and weighted p value-based z-statistic combination across all sample sets) described above (Stage 1 meta-analysis). We also assessed our results using random effects meta-analysis (Supplementary Table 6). We observed some evidence for heterogeneity across studies (the I2 statistic ranged from 0 to 57.8% depending on SNP), with rs7578597 and rs10923931 displaying the largest fold differences in association p value between the fixed- and random-effects model analyses. Differences in strength of association across studies (leading to evidence for heterogeneity) could reflect interesting biological associations that vary from study to study depending on subject ascertainment scheme.
Genomic control (expanded in Supplementary Methods)
We have adopted two strategies in reporting the findings from this study. In the first, we performed GC-correction of data from DGI, FUSION and WTCCC prior to stage 1 meta-analysis. We corrected each individual study for the GC inflation observed (directly genotyped and imputed data separately), and combined results across studies. We present the genome-wide distribution of association statistics in Supplementary Figure 1. We note that, after study-specific genomic control adjustment, the estimated inflation factor for the stage-1 meta-analysis test statistic was 1.04.
In the second, we combined GC-uncorrected data from DGI, FUSION and WTCCC for stage 1 meta-analysis and did not correct the meta-analysis test statistics for the overall GC (to guard against over-conservativeness in the estimate of strength of association for interesting signals). We also present the genome-wide distribution of these statistics in Supplementary Figure 1.
For the combination of data across stages 1, 2 and 3, we also adopted these two strategies (of using GC-corrected and GC-uncorrected stage 1 data). In the first, we performed individual GC-correction of DGI, FUSION and WTCCC stage 1 data prior to meta-analysis with stage 2 and stage 3 data (an approach which may be over-conservative where, as here, none of the T2D-associated SNPs has particular hallmarks of stratification) (Supplementary Note). In the second, we combined only uncorrected data (except for the deCODE data, where we have applied GC correction given a more marked genomic control inflation [GC ~1.3] in that sample). We present the resulting data from both approaches (of using GC-corrected and GC-uncorrected stage 1 data for stage 1-3 meta-analysis) in Supplementary Table 6 and a comparison of results (showing very small differences) in the Supplementary Note. All data presented elsewhere in the manuscript reflect the GC-corrected analysis strategy outcome.
Conditional analysis of T2D signals
For each SNP in Table 2, we assessed the additive SNP association in the stage 1 and 2 samples before and after including body mass index in the logistic regression model. For each genotyped and imputed SNP surrounding a specific T2D signal we assessed the additive SNP association in the stage 1 sample before and after including the Table 2 SNP from the same region in the model. We analyzed the data and adjusted for covariates for the stage 1 and stage 2 analysis of each sample. Data were combined across studies as described above. The ORs and CIs were calculated using a fixed-effects model and p values were calculated using the weighted z-score method. For the UK stage 1 samples, we did not have BMI information available for ~1,500 of the population-based controls. We therefore carried out the conditional BMI analyses by using all T2D cases and only those controls for whom BMI data were available.
Quantitative trait analyses
Quantitative trait analyses were carried out in the UK, DGI and FUSION samples for the 11 SNPs taken forward to stage 3. We tested BMI, quantitative glycemic traits (fasting and 2 hour glucose and insulin, HOMA-IR), lipid traits (total, HDL and LDL cholesterol, and serum triglycerides) and blood pressure (systolic and diastolic), where available, for association using an additive genetic model (Supplementary Methods).
Supplementary Material
Acknowledgements
UK: Collection of the UK type 2 diabetes cases was supported by Diabetes UK, BDA Research and the UK Medical Research Council (Biomedical Collections Strategic Grant G0000649). The UK Type 2 Diabetes Genetics Consortium collection was supported by the Wellcome Trust (Biomedical Collections Grant GR072960). The GWA genotyping was supported by the Wellcome Trust (076113) and replication genotyping by the European Commission (EURODIA LSHG-CT-2004- 518153), MRC (Project Grant G016121), Wellcome Trust, Peninsula Medical School, and Diabetes UK. EZ is a Wellcome Trust Research Career Development Fellow. We acknowledge the contribution of Dr Michael Sampson, and our team of research nurses. We acknowledge the efforts of Jane Collier, Phil Robinson, Steven Asquith and others at Kbiosciences (http://www.kbioscience.co.uk/) for their rapid and accurate large-scale genotyping.
DGI: We thank the study participants who made this research possible. We thank colleagues in the Broad Genetic Analysis and Biological Samples Platforms for their expertise and contributions to genotyping, data and sample management, and analysis. The initial GWAS genotyping was supported by Novartis (to DA); support for additional analysis and genotyping in this report was provided by funding from the Broad Institute of Harvard and MIT, by The Richard and Susan Smith Family Foundation / American Diabetes Association Pinnacle Program Project Award (to DA), and by a Freedom to Discovery award of the Foundation of Bristol Myers Squibb (to DA). PIWdB, MJD, DA, acknowledge support from NIH/ NHLBI grant (U01 HG004171). DA was a Burroughs Wellcome Fund Clinical Scholar in Translational Research, and is a Distinguished Clinical Scholar of the Doris Duke Charitable Foundation. LG, TT, BI, MRT and the Botnia Study are principally supported by the Sigrid Juselius Foundation, the Finnish Diabetes Research Foundation, The Folkhalsan Research Foundation and Clinical Research Institute HUCH Ltd; work in Malmö, Sweden was also funded by a Linné grant from the Swedish Research Council (349-2006-237). We thank the Botnia and Skara research teams for clinical contributions, and colleagues at MGH, Harvard, Broad, Novartis and Lund for helpful discussions throughout the course of this work.
FUSION: We thank the Finnish citizens who generously participated in this study, and Ryan Welch for bioinformatics support. Support for this research was provided by NIH grants DK062370 (M.B.), DK072193 (K.L.M.), HL084729(G.R.A.), HG002651 (G.R.A.), and U54 DA021519; National Human Genome Research Institute intramural project number 1 Z01 HG000024 (F.S.C.); and a post-doctoral fellowship award from the American Diabetes Association (C.J.W.). Genome-wide genotyping was performed by the Johns Hopkins University Genetic Resources Core Facility (GRCF) SNP Center at the Center for Inherited Disease Research (CIDR) with support from CIDR NIH Contract Number N01-HG-65403 and the GRCF SNP Center.
deCODE: We thank the Icelandic study participants whose contribution made this work possible. We also thank the nurses at Noatun (deCODE's sample recruitment center) and personnel at the deCODE core facilities for their hard work and enthusiasm.
KORA study: We thank Christian Gieger and Guido Fischer for expert data handling. The MONICA/KORA Augsburg studies were financed by the GSF-National Research Center for Environment and Health, Neuherberg, Germany and supported by grants from the German Federal Ministry of Education and Research (BMBF). Part of this work was financed by the German National Genome Research Network (NGFN). Our research was also supported within the Munich Center of Health Sciences (MC Health) as part of LMUinnovativ. We thank all members of field staffs who were involved in the planning and conduct of the MONICA/KORA Augsburg studies.
Danish study: This work was supported by the European Union (EUGENE2, grant no. LSHM-CT-2004-512013), Lundbeck Foundation centre of Applied Medical Genomics in Personalized Disease Prediction, Prevention and Care and The Danish Medical Research Council.
HUNT: The Nord-Trøndelag Health Study (The HUNT Study) is a collaboration between The HUNT Research Centre, Faculty of Medicine, Norwegian University of Science and Technology (NTNU), The National Institute of Public Health, The National Screening Service of Norway and The Nord-Trøndelag County Council.
NHS: The Nurses' Health Study is funded by National Cancer Institute grant CA87969. L.Q. is supported by an American Heart Association Scientist Development Grant. F.B.H. is supported by NIH grants DK58845 and U01 HG004399.
GEM Consortium: We thank all study participants. The work on the Cambridgeshire case-control, Ely, ADDITION and EPIC-Norfolk studies was funded by support from the Wellcome Trust and MRC. The Norfolk Diabetes study is funded by the MRC with support from NHS Research & Development and the Wellcome Trust. We are grateful to Dr Simon Griffin, MRC Epidemiology Unit, for assistance with the ADDITION study and Dr Mike Sampson and Dr Elizabeth Young for help with the Norfolk Diabetes Study. We thank Suzannah Bumpstead, William E Bottomley and Amy Chaney for rapid and accurate genotyping and Jilur Ghori for assay design and informatics support. We are grateful to Panos Deloukas for overall genotyping support. F.P. and I.B. are funded by the Wellcome Trust.
METSIM: The METSIM study has received grant support from the Academy of Finland (no. 124243).
References
- 1.Diabetes Genetics Initiative Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 2.Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeggini E, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinthorsdottir V, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 2007;39:770–775. doi: 10.1038/ng2043. [DOI] [PubMed] [Google Scholar]
- 6.Sladek R, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 7.Florez JC, et al. A 100K genome-wide association scan for diabetes and related traits in the Framingham Heart Study: replication and integration with other genome-wide datasets. Diabetes. 2007;56:3063–3074. doi: 10.2337/db07-0451. [DOI] [PubMed] [Google Scholar]
- 8.Rampersaud E, et al. Identification of novel candidate genes for type 2 diabetes from a genome-wide association scan in the Old Order Amish: evidence for replication from diabetes-related quantitative traits and from independent populations. Diabetes. 2007;56:3053–3062. doi: 10.2337/db07-0457. [DOI] [PubMed] [Google Scholar]
- 9.Hanson RL, et al. A search for variants associated with young-onset type 2 diabetes in American Indians in a 100K genotyping array. Diabetes. 2007;56:3045–52. doi: 10.2337/db07-0462. [DOI] [PubMed] [Google Scholar]; information valid in the study of a chronic disease such as diabetes? The Nord-Trøndelag diabetes study. J Epidemiol Community Health. 1992;46:537–542. doi: 10.1136/jech.46.5.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayes MG, et al. Identification of type 2 diabetes genes in Mexican Americans through genome-wide association studies. Diabetes. 2007;56:3033–3044. doi: 10.2337/db07-0482. [DOI] [PubMed] [Google Scholar]
- 11.Salonen J, et al. Type 2 diabetes whole-genome association study in four populations: the DiaGen consortium. Am. J. Hum. Genet. 2007;81:338–345. doi: 10.1086/520599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarthy MI, Zeggini E. Genome-wide association scans for Type 2 diabetes: new insights into biology and therapy. Trends Pharmacol. Sci. 2007;28:598–601. doi: 10.1016/j.tips.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 13.International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 15.Freedman ML, et al. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 16.Nakajima T, Fujino S, Nakanishi G, Kim YS, Jetten AM. TIP27: a novel repressor of the nuclear orphan receptor TAK1/TR4. Nucleic Acids Res. 2004;32:4194–4204. doi: 10.1093/nar/gkh741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins LL, et al. Growth retardation and abnormal maternal behavior in mice lacking testicular orphan nuclear receptor 4. Proc. Natl. Acad. Sci. USA. 2004;101:15058–15063. doi: 10.1073/pnas.0405700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas G, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet. doi: 10.1038/ng.91. 10 February 2008 | doi:10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 19.Gudmundsson J, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat. Genet. 2007;39:977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 20.Winckler W, et al. Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes. 2007;56:685–693. doi: 10.2337/db06-0202. [DOI] [PubMed] [Google Scholar]
- 21.Bieganowski P, Shilinski K, Tsichlis PN, Brenner C. Cdc123 and checkpoint forkhead associated with RING proteins control the cell cycle by controlling eIF2gamma abundance. J. Biol. Chem. 2004;273:44656–44666. doi: 10.1074/jbc.M406151200. [DOI] [PubMed] [Google Scholar]
- 22.Drieschner N, et al. Evidence for a 3p25 breakpoint hot spot region in thyroid tumors of follicular origin. Thyroid. 2006;16:1091–1096. doi: 10.1089/thy.2006.16.1091. [DOI] [PubMed] [Google Scholar]
- 23.Drieschner N, et al. A domain of the thyroid adenoma associated gene (THADA) conserved in vertebrates becomes destroyed by chromosomal rearrangements observed in thyroid adenomas. Gene. 2007;403:110–117. doi: 10.1016/j.gene.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 24.Lammert E, Brown J, Melton DA. Notch gene expression during pancreatic organogenesis. Mech. Dev. 2000;94:199–203. doi: 10.1016/s0925-4773(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 25.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 27.Lin S, Chakravarti A, Cutler DJ. Exhaustive allelic transmission disequilibrium tests as a new approach to genome-wide association studies. Nat. Genet. 2004;36:1181–1188. doi: 10.1038/ng1457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.