

Abstract
Summary. It is widely believed that risks of many complex diseases are determined by genetic susceptibilities, environmental exposures, and their interaction. Chatterjee and Carroll (2005, Biometrika 92, 399–418) developed an efficient retrospective maximum-likelihood method for analysis of case–control studies that exploits an assumption of gene–environment independence and leaves the distribution of the environmental covariates to be completely nonparametric. Spinka, Carroll, and Chatterjee (2005, Genetic Epidemiology 29, 108–127) extended this approach to studies where certain types of genetic information, such as haplotype phases, may be missing on some subjects. We further extend this approach to situations when some of the environmental exposures are measured with error. Using a polychotomous logistic regression model, we allow disease status to have K + 1 levels. We propose use of a pseudolikelihood and a related EM algorithm for parameter estimation. We prove consistency and derive the resulting asymptotic covariance matrix of parameter estimates when the variance of the measurement error is known and when it is estimated using replications. Inferences with measurement error corrections are complicated by the fact that the Wald test often behaves poorly in the presence of large amounts of measurement error. The likelihood-ratio (LR) techniques are known to be a good alternative. However, the LR tests are not technically correct in this setting because the likelihood function is based on an incorrect model, i.e., a prospective model in a retrospective sampling scheme. We corrected standard asymptotic results to account for the fact that the LR test is based on a likelihood-type function. The performance of the proposed method is illustrated using simulation studies emphasizing the case when genetic information is in the form of haplotypes and missing data arises from haplotype-phase ambiguity. An application of our method is illustrated using a population-based case–control study of the association between calcium intake and the risk of colorectal adenoma.
Keywords: EM algorithm, Errors in variables, Gene-environment independence, Gene-environment interactions, Likelihood-ratio tests in misspecified models, Inferences in measurement error models, Profile likelihood, Semiparametric methods
1. Introduction
With the advent of modern genotyping technologies, epidemiologists have been increasingly interested in identifying genetically defined subgroups within a population with unusual resistance or susceptibility to environmental exposures, both because such interactions may yield insight into mechanisms of action of exposures and because they may suggest disease prevention strategies. Case–control studies are often used to detect such gene–environment interactions. Traditionally, case–control data are analyzed using prospective logistic regression ignoring the fact that under this design subjects are sampled retrospectively conditional on their disease status. The validity of this approach relies on the classic results by Cornfield (1956) who showed the equivalence of prospective and retrospective odds ratios. The efficiency of the approach was established in two other classic papers by Andersen (1970) and Prentice and Pyke (1979) who showed that standard prospective analysis of case–control data yields the proper maximum-likelihood estimates of the odds-ratio parameters under the retrospective design as long as the distribution of the underlying covariates are allowed to remain completely unrestricted (nonparametric).
A number of researchers have noted that in studies of genetic epidemiology, the efficiency of the standard analysis for case–control data can be improved by exploiting certain natural model assumptions for the underlying genetic and the environmental covariates. In the context of haplotype-based analysis of case–control studies, Epstein and Satten (2003) and Satten and Epstein (2004) noted that retrospective maximum-likelihood methods can be more efficient than analogous prospective methods by taking full advantage of an assumption of Hardy–Weinberg equilibrium (HWE) for the underlying population. Chatterjee and Carroll (2005) exploited an assumption of gene–environment independence to yield more precise maximum-likelihood estimates of the odds-ratio parameters than those obtained from standard logistic regression analysis. Spinka, Carroll, and Chatterjee (2005) extended the results of Chatterjee and Carroll to allow for missing genetic information and haplotype-phase ambiguity.
In this article, we propose to extend earlier methods for analysis of case–control data under gene–environment independence and possibly HWE to account for measurement error in environmental exposures. Our work was motivated by a case–control study of colorectal adenoma (Peters et al., 2004) designed to investigate the interactions of dietary calcium intake and genetic variants in the calcium-sensing receptor (CASR) region. In this study, a total of 772 cases and 778 controls were sampled from the screening arm of the prostate, lung, colorectal, and ovarian (PLCO) cancer screening trial. Information on dietary food intake of the participants were available from a baseline food-frequency questionnaire (FFQ). Genotype data were available on three nonsynonymous single nucleotide polymorphisms (SNP) in the CASR region. One of the major goals of the study was to investigate the interaction of dietary calcium and the CASR gene based on “haplotypes,” which is a combinations of alleles at three different CASR loci along individual chromosomes. Two technical problems arose. First, as is typical, only locus-specific genotype data are available to provide information on two alleles that a subject carries on the pair of homologous chromosome, at each locus separately. Such genotype data lack the phase information, i.e., which combinations of allele arise together on the individual chromosomes, thus giving rise to an interesting missing data problem. Second, it is well known that FFQ as an instrument for measuring dietary intake is prone to both bias and random error, as illustrated in the OPEN study (Subar et al., 2003). We will use data from an external study (Potischman et al., 2002) to form estimates of the bias and variance of the measurement error for calcium intake. The availability of such external data gives rise to the opportunity for studying the calcium–CASR interaction after correcting for measurement error due to use of FFQ. Development of new methods, however, were needed to allow for such adjustment, which is the main contribution of this article.
Further, in our setting it is undesirable to conduct inferences using the Wald-type procedure. Schafer and Purdy (1996) advocated likelihood analysis for regression models with errors in explanatory variables, for data problems in which the relevant distributions can be adequately modeled. They point out that the likelihood ratio (LR) tests and confidence intervals can be substantially better than tests and confidence intervals based on estimates and standard errors, because the sampling distribution of measurement-error-corrected estimators are very often skewed, especially if the measurement errors are large. The fact that the data are collected using a case–control sampling scheme and are analyzed as if they were a random sample means that LR tests are not technically correct. We correct standard asymptotic results and propose a LR procedure that can be used successfully in this setting, and demonstrate its power in simulations.
An outline of this article is as follows. In Section 2, we give the technical formulation of the problem, describe our methodology, and state the main distributional results. Section 3 gives the results of simulation studies, where we show that our methodology overcomes the bias caused by measurement error. Section 4 analyzes the example discussed above. Section 5 gives concluding remarks. All technical derivations are given in the Web Appendix, along with many more simulation results.
2. Methodology and Main Theoretical Results
2.1 Model and Notation
Let D be the categorical indicator of disease status. We allow D to have K + 1 levels with the possibility of K ≥ 1 to accommodate different subtypes of a disease. Let D = 0 denote the disease-free (control) subjects and D = k, k ≥ 1 denote the diseased (case) subjects of the kth subtype. Suppose there are M loci of interest within a genomic region. Let Hdip = (H1, H2) denote the corresponding diplotype status for an individual, i.e., the two haplotypes that the individual carries in his/her pair of homologous chromosomes. Let (X, Z) denote all of the environmental (nongenetic) covariates of interest with X denoting the factors susceptible to measurement error. Given the environmental covariates X and Z and diplotype data Hdip, the risk of the disease in the underlying population is given by the polytomous logistic regression model
| (1) |
Here m(·) is a known function parameterizing the joint risk of the disease from Hdip, X, and Z in terms of the odds-ratio parameters β.
The model (1) cannot be used directly for analysis due to two reasons. First, the diplotype information Hdip is not measurable using standard genotyping technology. Typically, multilocus genotype information, denoted by G = (G1, G2, …, GM), is available. Due to lack of haplotype-phase information, the same genotype data can be consistent with multiple configuration of haplotypes for a given subject. For example, if A/a and B/b denote the major/minor alleles in two bi-allelic loci, then subjects with genotypes (Aa) and (Bb) at the first and the second locus, respectively, are considered “phase ambiguous”: their genotypes could arise from either the haplotype-pair (A-B, a-b) or the haplotype-pair (A-B, ab). Let ℋdip denote the set of all possible diplotypes in the underlying population. Analogously, let denote the set of all possible diplotypes that are consistent with a particular genotype vector G. We assume independence of Hdip and (X, Z) in the underlying population. Moreover, we assume a parametric model of the form pr(Hdip) = Q(Hdip, θ). Note, however, that our method can be readily extended to a general parametric model for Hdip given (X, Z) that could account for gene–environment association (Chatterjee et al., 2006). For our numerical examples, we assume HWE so that the distribution of the diplotypes can be specified in terms of the frequency of the haplotypes. Our general framework, however, allows use of more flexible models than HWE (see, e.g., Satten and Epstein, 2004; Lin and Zeng, 2006).
A second problem is that in our motivating example, the covariate X is measured with error. Let W denote the error-prone version of X. We assume a parametric model of the form fmem(w|X, Hdip, Z, D; ξ) for the conditional distribution of W given the true exposure X, additional environmental factors Z, and disease-status D. Measurement error can be modeled both as differential and nondifferential. If measurement error can be assumed to be nondifferential by disease status, then one can simplify the model as fmem(w|X, Hdip, Z, D; ξ) = fmem(w|X, Hdip, Z; ξ). We assume that the joint distribution of the environmental factors in the underlying population can be specified according to a semiparametric model of the form fX,Z(x, z) = fX(x|z; η)fZ(z), where fZ (z) is left completely unspecified.
2.2 Semiparametric Inference Based on a Pseudolikelihood
For d ≥ 1, define nd to be the number of subjects in the sample with disease at stage d, πd = pr(D = d), κd = β0d + log(nd/n0) − log(πd/π0), and κ̃ = (κ1, …, κK)T. Define κ0 = β00. Let β̃0 = (β01, …, β0K)T. Let , ℬ =(ΩT, ηT)T and υ = (ηT, ξT)T. Make the definition
| (2) |
Consider a sampling scenario where each subject from the underlying population is selected into the case–control study using a Bernoulli sampling scheme, where the selection probability for a subject given his/her disease status D = d is proportional to μd = nd/pr(D = d). Let R = 1 denote the indicator of whether a subject is selected in the case–control sample under the above Bernoulli sampling scheme. We propose parameter estimation using a pseudolikelihood of the form
Calculations given in the Appendix show that
| (3) |
Observe that conditioning on Z in L* allows it to be free of the nonparametric density function fZ (z), thus avoiding the need of estimating potentially high-dimensional nuisance parameters. In the absence of measurement error in environmental exposures, Chatterjee and Carroll (2005) and Spinka et al. (2005) used a profile-likelihood technique to show the equivalence of pseudolikelihood of the form L* with the proper retrospective likelihood of case–control data given by . In the presence of measurement error, although we do not have such a general theorem, a simulation study in a simple scenario shows that L* has a similar efficiency as the corresponding retrospective likelihood, see Section 3.1.
2.2.1 Rare disease approximation
In the case of rare disease, the denominator of (2) is approximately = 1, in which case
does not depend on β0.
2.3 Estimation with Known Measurement Error Distribution
In this section, we assume that the parameter ξ controlling the distribution of the measurement error is known. We show that maximization of L*, although it is not the actual retrospective likelihood for case–control data, leads to consistent and asymptotically normal parameter estimates. Recall that ℬ = (ΩT, ηT)T. Let Ψ(d, g, w, z, Ω, η, ξ) be the derivative of log{L(d, g, w, z, Ω, η, ξ)} with respect to ℬ. Then define
where all expectations are taken with respect to the case–control sampling design. We propose to estimate ℬ as the solution to
| (4) |
calling the solution ℬ̂ = (Ω̂T, η̂T)T. Our main technical result, the proof of which is given in the Web Appendix H, concerns the limiting properties of ℬ̂.
Theorem 1
The estimating function ℒn(Ω, η, ξ) is unbiased, i.e., has mean zero when evaluated at the true parameter values. In addition, under suitable regulatory conditions, there is a consistent sequence of solutions to (4), with the property that
| (5) |
Remark 1
It is easy to obtain consistent estimates of both ℐ and Λ. For example, to get an estimate Λ̂, in the definition of Λ, we can estimate E{Ψ(D, G, W, Z, Ω, η, ξ)|D = d} by . Similarly, n−1∂{ℒn(ℬ̂, ξ)}/∂ℬT is a consistent estimate of ℐ. Alternatively, if Σ̂ is the sample covariance matrix of the terms Ψ(Di, Gi, Wi, Zi, ℬ̂, ξ), then Σ̂ + Λ̂ consistently estimates ℐ.
Remark 2
An EM-algorithm for computation, based along the lines of Spinka et al. (2005) is given in the Appendix.
Remark 3
Similar to the settings of Chatterjee and Carroll (2005) and Spinka et al. (2005), here, the intercept parameters (β0d, d ≥ 1) of the polytomous logistic regression model are theoretically identifiable from the pseudolikelihood L*, even though the sampling is retrospective. For rare diseases, however, in formula (2) and so L* is expected to contain very little information about βd. If information on Pr (D = d) is available externally, as could be the situation for population-based case–control studies, then πd, d ≥ 1 could be treated as fixed known parameters in the definition of κd allowing estimation of β0d to be much more tractable. If Pr (D = d) is not known, one could employ the rare disease assumption under which β0d’s disappear from the likelihood. Alternatively, one can estimate parameters (Ω, η, ξ) by maximizing the likelihood function for the values of πd fixed on a grid and then performing a grid-search method to identify the value of πd that maximizes the profile likelihood ℒn{Ω(πd), η (πd), ξ}.
Remark 4
Rarely the practical identifiability of intercept parameters β0d and κd, or equivalently, πd may be a problem. But as illustrated in our example where the practical identifiability is a problem, the other parameter estimates are not much affected. Hence a practical method is to constrain the probability of disease to a wide range and perform a grid-search method to find an estimate.
2.4 Estimated Measurement Error Distribution
In practice, the parameter ξ controlling the measurement error distribution will be unknown, and typically additional data are necessary to estimate it. Here we consider the case of additive mean-zero measurement error with replications of W.
Our convention is that there are at most M replications of the W for any individual. Let Wi denote this ensemble of the M replicates, and let mi be the number of replicates we actually observe. Let fmem(w | d, hdip, x, z, m, ξ) be the joint density of the first m replicates for m = 1, …, M; Ψ(D, G, W, Z, Ω, η, ξ, j),ℐj, and Λj be matrices defined in the Section 2.3 for the case with exactly m = j replicates for each individual. Assume that mi is independent of (Di, W i, Zi, Gi, Xi, ) and that pr(mi = j) = p(j). Further, define . It is shown in the Appendix that the estimating function for ℬ = (Ω T, ηT, ξT)T can be written in the form
| (6) |
Theorem 2
The estimating function (6) is unbiased, i.e., has mean zero when evaluated at the true parameter values. In addition, under suitable regulatory conditions, there is a consistent sequence of solutions to (6), with the property that
| (7) |
Remark 5
Consistent estimates of ℐ and Λj can be obtained by applying formulas that are analogous to those outlined in the Remark 1. Web Appendix H contains the proof of Theorem 2.
2.5 Covariates Z Measured Exactly, Full Retrospective Likelihood
Let fZ (z) be the marginal density of Z. Abusing notation slightly and using a generic h, define ℋ(d, h, x, z, β0d, β) = [H{β0d + m(h, x, z, β)}]d[1 − H{β0d + m(h, x, z, β)}]1−d.
Then the retrospective likelihood is
| (8) |
It is clear then that if we want to compute the full retrospective likelihood, we need a parametric model for the marginal distribution of Z.
However, instead of (8), suppose that we construct another retrospective likelihood:
| (9) |
Note how (9) is a legitimate conditional likelihood function, and it does not involve the marginal distribution of Z.
We are thus led to think about the comparison of (3) and (9). Both make the same assumptions, and both are explicit. The obvious question is: why would we bother with (3) when we already have (9) available?
The key here is that
| (10) |
cannot be identified merely from (β0, β1, θ, η), as it could when there was no Z. On the other hand, pr(D = 1) is identifiable when we use our likelihood (3). Thus, (3) and (9) cannot be made to be the same thing.
The advantage of our method (3) lies in the very identifiability of pr(D = 1).
If pr(D = 1) is known in the population, then our method automatically uses this information, whereas (9) does not. Thus, one assumes that when pr(D = 1), the semi-parametric likelihood formulation will be more efficient than attempting to implement (9).
Probably even more crucially, we know that pr(D = 1) and β0 are probably not very precisely estimated. However, in our semiparametric formulation, we can easily place quite realistic bounds on pr(D = 1), and we know from the no-measurement error case that this can improve performance. Thus, the conjecture is that the semi-parametric method will also be more efficient than (9) as long as reasonable bounds are placed on pr(D = 1).
2.6 Construction of Test Statistic for Case–Control Data
The fact that the data are collected using a case–control sampling scheme and are analyzed as if they were a random sample means that LR tests are not technically correct. The main objective of this section is hence to propose a LR procedure that can be used in this setting.
Recall that the standard LR procedure for testing
| (11) |
is based on the following statistic
| (12) |
Under the assumption of a correct model, Wilks (1938) and Roy (1957) derived the limiting chi-square distribution of −2log(λn) using consistency and asymptotic normality of the maximum-likelihood estimates. Kent (1982) examined the distribution of the LR statistic when the data do not come from the specified parametric model, but when the “nearest” member of the parametric family still satisfies the null hypothesis. Foutz and Srivastava (1977) studied the asymptotic performance of the LR test when the probability distribution of the data is not a member of the model from which the LR test is constructed.
In this section, we describe a LR test procedure for testing simple and composite hypotheses based on the likelihood function (3). The critical technical part is that the asymptotic distribution of the LR test statistic needs to be adjusted to take the retrospective sampling plan into account. Web Appendix H gives the proofs of our results.
2.6.1 Simple hypothesis
First consider the null hypothesis of the form H0: ℬ = ℬ0. If the second derivative of ℒn(•) is given as ℒℬℬ(•), then the estimate ℬ̂ satisfies
Our main technical result, the proof of which is given in Web Appendix H, is a limiting property of the test (11) based on a likelihood-type function (3).
Theorem 3
Define 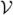 = Normal{0, ℐ−1(ℐ − Λ)ℐ−1}. Using Cholesky decomposition the covariance matrix can be factored as ℐ−1(ℐ − Λ)ℐ−1 = LLT, where L is a lower-triangular matrix. Let λi, i = 1, …, k be eigenvalues of the matrix LℐLT. Let
denote independent
random variables. Then when H0 is true, the adjusted retrospective likelihood ratio (ARLR) test statistic based on the pseudolikelihood (3) has the limiting distribution that is the same as
= Normal{0, ℐ−1(ℐ − Λ)ℐ−1}. Using Cholesky decomposition the covariance matrix can be factored as ℐ−1(ℐ − Λ)ℐ−1 = LLT, where L is a lower-triangular matrix. Let λi, i = 1, …, k be eigenvalues of the matrix LℐLT. Let
denote independent
random variables. Then when H0 is true, the adjusted retrospective likelihood ratio (ARLR) test statistic based on the pseudolikelihood (3) has the limiting distribution that is the same as
| (13) |
Remark 6
To estimate the λi’s, apply Cholesky decomposition to ℐ̂−1(ℐ − Λ̂) ℐ̂−1 = L̂L̂T and obtain the λ̂’s as the eigenvalues of L̂ℐ̂L̂T. Percentiles of this weighted sum of chi-squared random variables are easily computed via simulation. Interestingly, in our numerical work we found that ℐ̂−1 Λ̂ℐ̂−1 was very close to zero, and ordinary LR tests also had good coverage.
2.6.2 Composite hypothesis
Let ℬ = (δ, γ), where δ is an r-dimensional vector of interest and γ is (k − r)-dimensional nuisance vector. Let the null hypothesis be H0: δ= δ0 whatever γ may be. Here we investigate the LR test for (11) based on a likelihood (3).
Define S11 and S22 to be diagonal blocks of the matrix 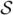 = ℐ (ℐ − Λ)−1ℐ that correspond to parameters of interest and nuisance parameters, respectively. Similarly, the corresponding blocks of ℐ are ℐ11 and ℐ22. Let
and using Frobenius formula it can be easily seen that
= ℐ (ℐ − Λ)−1ℐ that correspond to parameters of interest and nuisance parameters, respectively. Similarly, the corresponding blocks of ℐ are ℐ11 and ℐ22. Let
and using Frobenius formula it can be easily seen that
The following theorem is an analog of Theorem 3 for the case of a composite hypothesis.
Theorem 4
Define
1 = Normal(0, 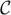 −1). Using Cholesky decomposition the covariance matrix can be factored as −1 = LLT, where L is a lower-triangular matrix. Let λi, i = 1, …, r be eigenvalues of the matrix L
−1). Using Cholesky decomposition the covariance matrix can be factored as −1 = LLT, where L is a lower-triangular matrix. Let λi, i = 1, …, r be eigenvalues of the matrix L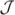 LT. Let
denote independent
random variables. Then under H0 the ARLR test statistic based on the pseudolikelihood (3) has the limiting distribution that is the same as
LT. Let
denote independent
random variables. Then under H0 the ARLR test statistic based on the pseudolikelihood (3) has the limiting distribution that is the same as
| (14) |
Remark 7
To estimate the λi’s, apply a procedure analogous to the one described in Remark 6.
3. Simulations
3.1 The Binary Case
When all variables are binary, it is possible to compute the retrospective likelihood of case–control data. In this section, we compare of our pseudolikelihood method with those based on the full retrospective likelihood, in the case that the genetic factor of interest is a directly observable binary variable, such as the carrier status for a variant allele in a specific genetic locus.
In this case, there are no covariates Z; the retrospective likelihood is given as follows. Define H{β0 + m(hdip, x, β)} = pr(D = 1|X, Hdip) and ℋ(d, hdip, x, β0, β) = [H{β0 + m(hdip, x, β)}]d[1 − H{β0 + m(hdip, x, β)}]1−d. Then
Because we have specified a distribution for X, all variables are binary, and there is no Z, the parameters (β0, β, θ, η) are sufficient to identify pr(D = 1), i.e.,
| (15) |
Because of this, κ is identified from (β0, β, θ, η) as well. Hence, simply using (3) as a likelihood function directly will be unstable because of overparametrization. To overcome this issue, we parameterized in terms of pr(D = 1), and let κ and β0, the latter through (15), be functions of it. The obvious solution is to replace both β0 and κ in (3) by the appropriate functions of pr(D = 1) as given in (15) and the definition of κ, which is what we did.
We did a small simulation experiment in order to illustrate our approach in this simple case. We assumed that environmental variables (X, W), genetic variant (G), and disease status (D) are binary. Given the values of (G, X) we generated a binary disease outcome D from the logistic model logit {pr(D | G, X)} = β0 + βx X + βg G + βxg X *G, with parameters (βx, βg, βxh) = (1.099, 0.693, 0.693). The misclassification probabilities were pr(W = 0 | X = 1) = 0.20 and pr(W = 1 | X = 0) = 0.10. Here we assume that the misclassification model and relevant parameters are known.
We estimated parameters using the foregoing algorithm and investigated the effect of knowing the probability of disease. We found that our proposed method yielded estimates that were numerically identical to those based on the full retrospective likelihood: we believe but have not been able to show that this is true in general. Our method showed no noticeable bias in the parameter estimates, either in the risk parameters or in the genotype probabilities, whereas the naive analysis that ignores measurement error resulted in large biases (Table 1).
Table 1.
Biases and root mean squared errors (RMSEs) for ordinary logistic regression, retrospective maximum likelihood, and our approach, where disease status (D), the genetic variant (G), and the environmental covariate (X) are binary. The environmental variable is measured with error, with misclassification probabilities being 0.20 for exposed and 0.10 for nonexposed subjects. The results are based on a simulation study with 500 replications for 1000 cases and 1000 controls. Results are given when pr(D = 1) is known and when it is unknown.
| Logistic |
Retrospective |
Pseudo-likelihood |
||||||
|---|---|---|---|---|---|---|---|---|
| pr(D = 1) | Parameter | True value | Bias | RMSE | Bias | RMSE | Bias | RMSE |
| Known | β0 | −5.000 | 4.294 | 4.295 | −0.006 | 0.108 | −0.006 | 0.108 |
| βg | 0.693 | 0.239 | 0.323 | −0.005 | 0.305 | −0.004 | 0.305 | |
| βx | 1.099 | −0.327 | 0.344 | 0.005 | 0.155 | 0.005 | 0.155 | |
| βxg | 0.693 | −0.284 | 0.395 | 0.001 | 0.327 | 0.001 | 0.327 | |
| pr(X = 1) | 0.100 | 0.002 | 0.021 | 0.002 | 0.022 | |||
| pr(G = 1) | 0.100 | 0.000 | 0.009 | 0.000 | 0.008 | |||
| Unknown | β0 | −5.000 | 4.294 | 4.295 | −1.016 | 2.042 | −1.016 | 2.042 |
| βg | 0.693 | 0.239 | 0.323 | −0.009 | 0.306 | −0.009 | 0.306 | |
| βx | 1.099 | −0.327 | 0.344 | 0.004 | 0.155 | 0.004 | 0.155 | |
| βxg | 0.693 | −0.284 | 0.395 | 0.013 | 0.333 | 0.013 | 0.333 | |
| pr(X = 1) | 0.100 | 0.023 | 0.022 | 0.002 | 0.022 | |||
| pr(G = 1) | 0.100 | 0.000 | 0.009 | 0.000 | 0.009 | |||
| pr(D = 1) | 0.016 | 0.002 | 0.019 | 0.002 | 0.019 | |||
Further, we performed inferences based on the ARLR test and Wald procedures for small (n = 400) and moderate (n = 2000) sample sizes. The results are presented in the Web Table 13. We found that the proposed method closely achieves the nominal coverage, while Wald test resulted in rather elevated error rates, thus causing undercoverage. The sampling distribution of the parameter estimates is slightly skewed, and skewness is more pronounced for small sample sizes. Hence the variance estimate is larger than the mean variance estimate and it is undesirable to use Wald-type confidence intervals, because they are based on asymptotic normality.
3.2 Continuous Simulations
In this simulation, we considered continuous environmental variables and assumed that the genetic risk depends on the number of copies of a putative haplotype. We simulated the true environmental covariate (X) from a Normal distribution with zero mean and variance 0.1. To simulate observed environmental variables we used additive model of the form W = X + U, where U is generated from the normal distribution with zero mean and variance ξ = 0.25. Note that we are simulating a case of large measurement error, such as would occur for dietary measurements. This gives a stern test for our methodology.
Following the simulation setup of Spinka et al. (2005), given the following haplotype frequencies (h1, h2, h3, h4, h5, h6) = (0.25, 0.15, 0.25, 0.1, 0.1, 0.15) we directly generated diplotypes for each subject under the assumption of HWE. Then we coded haplotype h3 as 1 and all the rest as 0. Given the diplotype information Hdip and environmental covariate X we generated binary disease status according to the following model
where N3(Hdip) is the number of copies of h3 in Hdip. In this setting we are interested in estimating the relative risk parameters and the frequency of haplotype h3. To estimate the probability of disease we used a grid-search method on the interval (0.001, 0.051) with step 0.005 by maximizing the pseudolikelihood function for values of probability of disease fixed on a grid and then performing a grid search to identify the value of probability of disease that maximized the likelihood.
3.2.1 Measurement error distribution is known
Within this simulation setup we suppose that the measurement error distribution is known. Moreover, we assessed the effect of missing data by assuming that 50% of subjects were not genotyped and for those who were genotyped linkage phase is unknown.
We found that for our method there is no noticeable bias in parameter estimates, whereas the naive approach that ignores existence of the measurement error results in substantial bias, as illustrated in the Table 2. It is somewhat remarkable that even with 50% of the genotypes being missing, and with such large measurement error, our method still remains largely unbiased.
Table 2.
Biases and (RMSEs) for the naive approach that ignores existence of measurement error and our proposed method. The results are based on a simulation study with 500 replications for 1000 cases and 1000 controls, where disease status (D) is binary, environmental variables (X, W) are continuous, and the genetic variant h3 is in the form of diplotype with a multiplicative interaction. The environmental variable is measured with error and the error variance is 0.25. Two cases are considered: (a) genotype known for all subjects and (b) genotype missing for 50% of the subjects.
| Naive approach |
Proposed method |
|||||
|---|---|---|---|---|---|---|
| Parameter | True value | Bias | RMSE | Bias | RMSE | |
| Complete data | β0 | −5.000 | 1.207 | 1.459 | 0.230 | 0.086 |
| βg | 0.693 | 0.080 | 0.011 | −0.001 | 0.007 | |
| βx | 1.099 | −0.797 | 0.645 | 0.001 | 0.137 | |
| βxg | 0.693 | −0.478 | 0.235 | 0.006 | 0.088 | |
| pr(h3) | 0.250 | 0.005 | 0.000 | 0.000 | 0.000 | |
| pr(D = 1) | 0.046 | −0.032 | 0.001 | 0.008 | 0.000 | |
| η1 | 0.000 | 0.003 | 0.001 | |||
| η2 | 0.100 | −0.001 | 0.000 | |||
| 50% of genetic information is missing | β0 | −5.000 | 1.206 | 1.460 | 0.228 | 0.084 |
| βg | 0.693 | 0.082 | 0.015 | −0.002 | 0.007 | |
| βx | 1.099 | −0.794 | 0.647 | 0.013 | 0.161 | |
| βxg | 0.693 | −0.477 | 0.243 | 0.011 | 0.102 | |
| pr(h3) | 0.250 | 0.004 | 0.000 | 0.000 | 0.000 | |
| pr(D = 1) | 0.046 | −0.032 | 0.001 | 0.008 | 0.000 | |
| η1 | 0.000 | 0.003 | 0.001 | |||
| η2 | 0.100 | −0.002 | 0.000 | |||
Additionally, we constructed Wald and ARLR confidence intervals. The results of this simulation study are presented in Table 3. We found that the ARLR confidence interval achieved coverage close to nominal. The Wald confidence interval performed poorly in this situation, despite the fact that there is essentially no bias in the parameter estimates.
Table 3.
Coverage probabilities of Wald and ARLR confidence intervals. The results are based on a simulation study with 750 replications for 1000 cases and 1000 controls, where disease status (D) is binary, environmental variables (X, W) are continuous, and the genetic variant h3 is in the form of diplotype with a multiplicative interaction. The environmental variable is measured with error. The probability of disease is known in the population, and the genotype is missing for 50% of the subjects.
| Measurement error variance ξ | 0.10 | 0.15 | 0.20 | 0.25 |
|---|---|---|---|---|
| True value of βxg | 0.693 | 0.693 | 0.693 | 0.693 |
| Mean of β̂xg | 0.704 | 0.708 | 0.700 | 0.678 |
| Median of β̂xg | 0.694 | 0.691 | 0.683 | 0.700 |
| Coverage of the Wald test | 0.793 | 0.727 | 0.707 | 0.697 |
| Coverage of the LR test | 0.957 | 0.947 | 0.952 | 0.941 |
We advocate the use of the ARLR procedure as opposed to Wald-type inferences in situations when large amounts of measurement error are present in environmental covariates.
3.2.2 Distribution of the environmental covariates is mis-specified
The aim of this simulation is to investigate robustness of our procedure to mild misspecification of the environmental covariate distribution. Here we calculated our method under the assumption that the environmental covariate had a normal distribution, while we simulated the environmental covariate from a t-distribution with 10 degrees of freedom.
The results presented in Web Table 1 illustrate that the proposed procedure results in parameter estimates that are nearly unbiased, which illustrates robustness of our methodology to mild misspecification of the environmental covariate distribution.
3.2.3 Measurement error distribution is estimated using replications
In Section 2.4 we developed a method for the case when measurement error distribution is estimated using repeated measurements. The goal of this simulation study is to investigate the performance of the proposed method in the case when measurement error distribution is estimated by replicating 50 randomly selected individuals in order to estimate the measurement error variance. Define to be a random variable distributed as chi square with df degrees of freedom. We generated the estimated measurement error variance ξ̂ as , where ξ = 0.25 is the true measurement error variance.
Simulation results presented in Web Table 2 illustrate that in this setting the proposed methodology resulted in parameter estimates that are nearly unbiased.
3.2.4 With environmental covariates measured exactly
For the setting described above we simulated the error-prone covariate X from the normal distribution with mean a1Z with a1 = 0.25 and variance . The environmental covariate Z measured exactly is simulated from a Normal distribution with mean zero and variance 0.10. Further, we introduced a main effect of Z in the risk model with risk coefficient βz = log(2.5). The probability of disease is assumed to be known in the population.
The results of this simulation are presented in Web Table 3. The naive approach that ignores existence of the measurement error resulted in estimates of environment and interaction risk parameters that are largely biased. The proposed analysis produced estimates that are nearly unbiased and less variable.
4. Colorectal Adenoma Study Data Analysis
4.1 Modeling
Here we analyze the colorectal adenoma study data described in the introduction. To recap, there were 772 cases and 778 controls, the response D was colorectal adenoma status, the genetic data observed were three SNPs in the calcium receptor gene CaSR, the environmental variable X measured with error was log(1+calcium intake), which was measured by W, the result of a FFQ. The variables Z measured without error were age, sex, and race. The possible haplotypes in the data were ACG, ACT, AGG, GCG, AGT, GGG, and GCT. Because haplotypes AGT, GGG, GCT are rare, we pooled them with the next most common haplotype AGG. A few subjects do not have measurements of calcium intake and we eliminated them from the analysis.
Given calcium intake (X) and diplotype information (Hdip) we considered the following risk model
where N2(Hdip) is the number of haplotypes ACT observed in a diplotype, N4(Hdip) is the number of haplotypes GCG observed in a diplotype and N5(Hdip) is number of haplotypes AGG, AGT, GGG, or GCT observed in a diplotype.
Unfortunately, there is no direct information in the study to assess the measurement error properties of calcium intake W. We used a combination of outside data and sensitivity analysis instead. The outside data come from The Women’s Interview Study of Health (WISH; Potischman et al., 2002). There were ≈400 women in this study, which used the same FFQ as in the colorectal adenoma study and also included the results of six 24-hour recall measurements, which we denote by Tij for the ith individual and jth replicate. The models for these data are that
where Ui = normal (0, ) and Vij = normal (0, ). Using variance components analysis, we estimated (α0, α1, ), and took these as fixed and known in the colorectal adenoma study, although we also varied . The distribution of X was taken to be Gaussian with mean linear in Z and variance ξ. We used the method of Fuller (1987, Chapters 2, 5) and found estimates α̂0 = 0.22, α̂1 = 0.75, . To assess sensitivity to the measurement error model specification we considered several scenarios by imposing measurement error structure estimated using WISH data and varying it through .
4.2 Results
The probability of disease was constrained to be on the interval (0.001, 0.5), but the likelihood function was flat either as a function of the probability of disease, or, equivalently, as a function of the intercept parameter β0. However, estimates of the risk parameters are unchanged for different values of probability of disease. This result is illustrated in Web Appendix D.
The four sets of parameter estimates presented in Table 4 correspond to different values of the measurement error variance. These results illustrate sensitivity of parameter estimates to the measurement error variance specification and the importance of assessing the measurement error process, as its incorrect specification results in substantial biases.
Table 4.
Estimates of risk parameters for the colorectal adenoma study assuming different variances (ξ) of the measurement error. The estimated measurement error variance is ξ = 0.65.
| Parameter | Naive | ξ = 0.10 | ξ = 0.60 | ξ = 0.65 | ξ = 0.70 |
|---|---|---|---|---|---|
| βh2 | −0.2087 | −0.1866 | −0.1606 | −0.1770 | −0.1365 |
| βh4 | −0.1663 | −0.1908 | −0.3710 | −0.4289 | −0.5377 |
| βh5 | −0.2770 | −0.3670 | −0.6609 | −0.7584 | −0.9379 |
| βx | −0.0852 | −0.0683 | −0.1402 | −0.1507 | −0.1850 |
| βxh2 | 0.0398 | 0.0394 | 0.1296 | 0.1044 | 0.2224 |
| βxh4 | −0.1886 | −0.1749 | −0.5192 | −0.5817 | −0.8124 |
| βxh5 | −0.2804 | −0.2361 | −0.7136 | −0.8885 | −1.1234 |
Additionally, the Wald and Bootstrap standard error estimates are presented in Web Tables 9 and 10. Also, 95% model-based and bootstrap confidence intervals are reported in Web Tables 11 and 12. Inspection of these results reveals that for large measurement error the model-based confidence intervals are narrower than those computed using the bootstrap. This phenomena is due to the fact that when large amounts of error in measurement is present in the data, the sampling distribution of parameter estimates can be skewed and thus model-based confidence intervals that are built using the asymptotic normality assumptions inherent in the Wald method have elevated error rates resulting in undercoverage.
Recall that the estimated error variance is ξ = 0.65. Inspection of the interaction parameter estimates β̂xh4 = −0.58 and β̂xh5 = −0.89 and corresponding 95% confidence intervals based on the estimated standard errors βxh4: (−0.98, −0.18) and βxh5: (−1.31, −0.47) suggests that at significance level 0.05 there is sufficient evidence to indicate that among carriers of h4 and h5 haplotypes, increased calcium intake is associated with decrease in risk of colorectal tumor development. Additionally, we computed bootstrap; standard errors and confidence intervals based on 300 samples. 95% bootstrap confidence interval for βxh4 is (−1.22, −0.02) and for βxh5 is (−2.11, −0.18). Note that the bootstrap method produced confidence intervals that are wider than those based on the Normality assumption. This effect is due to the fact that sampling distribution of parameter estimates is oftentimes skewed when measurement error is present and skewness is more pronounced for large measurement errors, which is the case in our situation.
Further, we performed inference based on the ARLR and Wald procedures. For the majority of cases λ was very close to 1 and there was no noticeable difference between the ARLR confidence interval and the one based on the standard asymptotics. Both ARLR and Wald procedures announced βxh2 to be not significant, for all measurement error models we considered, so we based our analysis on a reduced model by setting βxh2 to be 0. Analysis of the reduced model showed that βxh5 is significantly different from 0 for all measurement error model specifications we considered. The Wald test announced βxh4 as significant for measurement error variance 0.5 and greater, while the LR test showed it is significantly different from 0 for measurement error variance of 0.4 and larger.
Because the measurement error distribution was estimated using the WISH study that included female subjects only, we performed the analysis on 451 female cases and 459 female controls. The pattern shown in Table 5 reveals that the ARLR CIs are substantially wider than Wald CI and the difference is more pronounced for large measurement errors. A similar effect is present in the simulation study, namely the coverage probability of the Wald CI is considerably less than nominal. It appears that a symmetric CI centered at the estimate tends to perform poorly, because even for fairly large samples, with large error in measurement and missing data the sampling distribution of parameters can be substantially skewed.
Table 5.
Wald and ARLR confidence intervals for βxh4 for the Colorectal Adenoma Study assuming various measurement error variances. The estimated measurement error variance is 0.65. The analysis is performed for female subjects.
| Wald CI | LR-type CI | |
|---|---|---|
| ξ = 0.10 | (−0.416, −0.090) | (−0.460, 0.119) |
| ξ = 0.60 | (−0.972, 0.175) | (−1.220, 0.388) |
| ξ = 0.65 | (−1.143, 0.198) | (−1.462, 0.420) |
| ξ = 0.70 | (−1.394, 0.230) | (−1.912, 0.583) |
5. Discussion
We have considered the problem of relating risk of a complex disease to genetic susceptibilities, environmental exposures, and their interaction when the environmental covariates are measured with error and some of the genetic information is missing. Utilizing a polychotomous logistic regression model, pseudolikelihood and a model for the distribution of underlying gene information, we constructed a relatively simple yet efficient semiparametric algorithm for parameter estimation. We have shown that the resulting estimates are consistent and derived their asymptotic variance when the distribution of measurement error is known, and when it is estimated from replications.
Our simulation results illustrate that for large studies there is no noticeable bias in our parameter estimates, whereas the naive approach that ignores the existence of the measurement error results in substantial bias.
We also developed an adjusted LR test statistic, the ARLR method, that is appropriate for the sampling design and performs much better in terms of test level than Wald-type inferences.
In our development, we have used a parametric model for the distribution of the environmental covariate measured with error. Our simulations and the example were based upon normal distributions, which seem reasonable in this context, but clearly more general models are possible, e.g., the semi-nonparametric family of Zhang and Davidian (2001). While such parametric assumptions can be wrong, often the resulting inferences are not badly affected by mild misspecification as illustrated in our numerical example, especially for logistic regression. For example, in the running Framingham data example in Carroll et al. (2006), the underlying variable X (transformed systolic blood pressure) appears to be more accurately modeled by a t-distribution with 5 degrees of freedom, but the differences in inference compared to a normal distribution assumption are hardly noticeable.
Issues of testing per se have a different focus than estimation of parameters. As described by Carroll et al. (2006, Chapter 10), generally if one is interested in the global null hypothesis of a null effect for a variable X measured with error, then tests that ignore measurement error are generally valid and often efficient in local power. The reason for this is that under the global null hypothesis, the observed data will also show no effect due to the observed W. If one has a complex model for X, e.g., a quadratic model, or if X is multivariate, then tests for subcomponents of the model associated with X are generally invalid. Tests for factors measured exactly, such as covariates Z or for phase-known haplotypes, are generally invalid, except when X is independent of the relevant covariates.
In models such as that described in Section 4.1, hypothesis testing for interactions or main effects while ignoring measurement error and replacing X by W can have real subtleties. It is clear theoretically that such tests cannot, in general, have nominal test level. Suppose, for example, that one is interested in testing whether there is an interaction between X measured with error and (Z1, Z2) measured without error, where (Z1, Z2) are independent of X but correlated with each other. Suppose the full model is a linear regression with mean β0 + β1X + β2Z1 + β3Z2 + β4XZ 1 + β5XZ 2, and the interest is in testing whether β5 = 0. The observed data model has mean β0 + β1E(X | W) + β2Z1 + β3Z2 + β4E(X | W) Z1 + β5E(X | W) Z2. Thus, if E(X | W) is linear in W, the naive test for interaction that ignores measurement error will be approximately valid, approximately because the observed data model has some heteroscedasticity due to the interaction between X and Z1. However, if E(X | W) = W 2, for example, then in ignoring measurement error and replacing X by W, one is fitting a misspecified model, and it is easy to construct situations where the naive test level is far from nominal. For example, take n = 200, β1 = ··· = β4 = 1, let (W, Z1, Z2) have mean zero and variance one, let the correlation between (Z1, Z2) be 0.6, let var (Y | X, Z1, Z2) = 0.252, let E(X | W) = W2 and var (X | W) = 0.25. Then the naive test for the hypothesis that β5 = 0 has test level exceeding 0.30, not even close to the nominal 0.05. However, what happens in logistic regression is more difficult to ascertain, and replacing linear regression by logistic regression in the example above does not reveal any major problems with test level.
For logistic regression when W is unbiased for X and with normally distributed measurement error, there are possible methods that can in principle avoid the use of distributional assumptions. The most widely used approach aimed at achieving this nonparametric feature is that of Stefanski and Carroll (1987), who use conditioning on sufficient statistics. Unfortunately, this approach will not work in our context, because in gene–environment interaction studies the sufficient statistic includes the underlying genetic variable, and hence cannot be allowed to be missing. Other methods that might be employed are SIMEX (Cook and Stefanski, 1995; Carroll et al., 2006) and Monte Carlo corrected scores (MCCS; Stefanski Novick, and Devanarayan, 2005; Carroll et al., 2006). Neither method results in actual consistent estimation of the parameters, although the latter is generally close to unbiased. However, MCCS requires the use of complex variable calculations, which users may find to be a practical hindrance.
Finally, we have assumed that H and (X, Z) are independently distributed in the underlying population, and we have assumed a parametric model for the distribution of H. Only changes in notation are required if there is a small number of strata, so that H and (X, Z) are independent within strata. More generally, all that we really require is a parametric model for H given (X, Z).
Acknowledgments
Our research was supported by grants from the National Cancer Institute (CA57030, CA90301) and by the Texas A&M Center for Environmental and Rural Health via a grant from the National Institute of Environmental Health Sciences (P30-ES09106). RJC’s work partially occurred during a visit to and with the support of the Department of Mathematics and Statistics at the University of Melbourne.
Appendix
An EM Algorithm
In this section, we describe an EM algorithm for solving the score-equations associated with the pseudolikelihood L*. All technical arguments are given in A.2. To facilitate the calculations, make the following definitions:
Note that neither α(•) nor γ(•) depend on Θ.
We split up the EM calculations into a series of steps.
EM Algorithm for Θ
Under HWE, if θi is the frequency of haplotype hi, if hi = hj and = 2θiθj if hi ≠ hj. Let Nk (Hdip) be the number of copies of hk in Hdip, and note that as in Spinka et al. (2005), Nk(Hdip)/θk = ∂log {pr(Hdip)}/∂θk. Define
Then if ℬ(s) is the current value of ℬ, we update θk to as
| (A.1) |
Further, in each iteration we normalize .
EM Algorithm for κd and β
For j = 1, …, K, we update κj by solving the following equation for κj:
| (A.2) |
To update β, we solve
| (A.3) |
EM Algorithm for β0d and η
The updating schemes for β0d and η are of the form (A.3) with Vβ(•) replaced by Vβ0d(d, hdip, x, z, Ω) = −pr(D = d ≥ 1|hdip, x, z) and Vη (x, z, η) = ∂log{fX(x | z, η)}/∂η for β0d and η, respectively.
EM Calculations
Here we justify the EM algorithm previously described. In what follows, we will need the following identities:
| (A.4) |
| (A.5) |
| (A.6) |
| (A.7) |
Argument for (A.1)
As in Spinka et al. (2005) the estimating equation for θk is
Note that
and
Since , therefore λ = 0. Using (A.4) and (A.5), we arrive at (A.1).
Argument for (A.2)
It is readily seen that the estimating function for κj is
Since ∂log {T(D, Hdip, X, Z, Ω)}/∂ κj = I(D=j)(D), using (A.7), estimation can be performed by iteratively solving (A.2).
Argument for (A.3)
The estimating function for β is
Using (A.6) and (A.7), we arrive at (A.3). The arguments for updating the β0d and η are similar.
Footnotes
6. Supplementary Materials
Proofs mentioned in Section 2 and Web Tables referenced in Sections 3 and 4 are available under the Paper Information link at the Biometrics website http://www.biometrics.tibs.org.
References
- Andersen EB. Asymptotic properties of conditional maximum likelihood estimators. Journal of the Royal Statistical Society, Series B. 1970;32:283–301. [Google Scholar]
- Carroll RJ, Ruppert D, Stefanski LA, Crainiceanu CM. Measurement Error in Nonlinear Models. 2. Boca Raton, FL: Chapman & Hall CRC Press; 2006. [Google Scholar]
- Chatterjee N, Carroll RJ. Semiparametric maximum likelihood estimation in case–control studies of gene–environmental interactions. Biometrika. 2005;92:399–418. [Google Scholar]
- Chatterjee N, Chen J, Spinka C, Carroll RJ. Comment on the paper Likelihood based inference on haplotype effects in genetic association studies by D. J. Lin and D. Zhang. Journal of the American Statistical Association. 2006;102:108–110. [Google Scholar]
- Cook J, Stefanski LA. A simulation extrapolation method for parametric measurement error models. Journal of the American Statistical Association. 1995;89:1314–1328. [Google Scholar]
- Cornfield J. A statistical problem arising from retrospective studies. In: Neyman J, editor. Proc 3rd Berkeley Symp Math Statist Prob. Vol. 4. Berkeley: University of California Press; 1956. pp. 135–148. [Google Scholar]
- Epstein M, Satten G. Inference of haplotype effects in case–control studies using unphased genotype data. American Journal of Human Genetics. 2003;73:1316–1329. doi: 10.1086/380204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foutz RV, Srivastava RC. The performance of the likelihood ratio test when the model is incorrect. Annals of Statistics. 1977;5(6):1183–1194. [Google Scholar]
- Fuller WA. Measurement Error Models. New York: John Wiley & Sons; 1987. [Google Scholar]
- Kent JT. Robust properties of likelihood ratio test. Biometrika. 1982;69:19–27. [Google Scholar]
- Lin DY, Zeng D. Likelihood-based inference on haplotype effects in genetic association studies (with discussion) Journal of the American Statistical Association. 2006;101:89–118. [Google Scholar]
- Peters U, Chatterjee N, Yeager M, Chanock SJ, Schoen RE, McGlynn KA, Church TR, Weissfeld JL, Schatzkin A, Hayes RB. Association of genetic variants in the calcium-sensing receptor with risk of colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2004;13(12):2181–2186. [PubMed] [Google Scholar]
- Potischman N, Coates RJ, Swanson CA, Carroll RJ, Daling JR, Brogan DR, Gammon MD, Midthune D, Curtin J, Brinton LA. Increased risk of early stage breast cancer related to consumption of sweet foods among women less than age 45. Cancer Causes and Control. 2002;13:937–946. doi: 10.1023/a:1021919416101. [DOI] [PubMed] [Google Scholar]
- Prentice RL, Pyke R. Logistic disease incidence models and case–control studies. Biometrika. 1979;66:403–412. [Google Scholar]
- Roy KP. A note on asymptotic distribution of likelihood ratio. Calcutta Statistical Association Bulletin. 1957;1:60–62. [Google Scholar]
- Satten GA, Epstein MP. Comparison of prospective and retrospective methods for haplotype inference in case–control studies. Genetic Epidemiology. 2004;27:192–201. doi: 10.1002/gepi.20020. [DOI] [PubMed] [Google Scholar]
- Schafer DW, Purdy KG. Likelihood analysis for errors-in-variables regression with replicate measurements. Biometrika. 1996;83:813–824. [Google Scholar]
- Spinka C, Carroll RJ, Chatterjee N. Analysis of case–control studies of genetic and environmental factors with missing genetic information and haplotype-phase ambiguity. Genetic Epidemiology. 2005;29:108–127. doi: 10.1002/gepi.20085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanski LA, Carroll RJ. Conditional scores and optimal scores in generalized linear measurement error models. Biometrika. 1987;74:703–716. [Google Scholar]
- Stefanski LA, Novick SJ, Devanarayan V. Estimating a nonlinear function of a normal mean. Biometrika. 2005;92:732–736. [Google Scholar]
- Subar AF, Kipnis V, Troiano RP, Midthune D, Schoeller DA, Bringham S, Sharbaugh CO, Trabusli J, Runswick S, Ballard-Barbash R, Sunshine J, Schatzkin A. Using intake biomarkers to evaluate the extent of dietary misreporting in a large sample of adults: The Observing Protein and Energy Nutrition (OPEN) study. American Journal of Epidemiology. 2003;54:426–485. doi: 10.1093/aje/kwg092. [DOI] [PubMed] [Google Scholar]
- Wilks SS. The large-sample distribution of the likelihood ratio for testing composite hypothesis. Annals of Mathematical Statistics. 1938;7:73–77. [Google Scholar]
- Zhang D, Davidian M. Linear mixed models with flexible distributions of random effects for longitudinal data. Biometrics. 2001;57:795–802. doi: 10.1111/j.0006-341x.2001.00795.x. [DOI] [PubMed] [Google Scholar]