

Summary
Among methyltransferases, KsgA and the reaction it catalyzes are conserved throughout evolution. However, the specifics of substrate recognition by the enzyme remain unknown. Here, we report structures of Aquifex aeolicus KsgA, in its ligand-free form, in complex with RNA and in complex with both RNA and S-adenosylhomocysteine (SAH, reaction product of cofactor S-adenosylmethionine), providing the first pieces of structural information on KsgA-RNA and KsgA-SAH interactions. Moreover, the structures show how conformational changes that occur upon RNA binding create the cofactor-binding site. There are nine conserved functional motifs (motifs I-VIII and X) in KsgA. Prior to RNA binding, motifs I and VIII are flexible, each exhibiting two distinct conformations. Upon RNA binding, the two motifs become stabilized in one of these conformations, which is compatible with the binding of SAH. Motif X, which is also stabilized upon RNA binding, is directly involved in the binding of SAH.
Introduction
Ribosome biogenesis has been a subject of great interest for decades. Many nucleotide modifications have been found in ribosomal RNA (rRNA), among which the dimethylation of residues A1518 and A1519 in helix 45, the terminal helix near the 3′ end of Escherichia coli 16S rRNA, by KsgA is universally conserved throughout evolution (Xu et al., 2008). KsgA, first identified in E. coli (Helser et al., 1972), has been found in almost all species. Helix 45, essential for both ribosomal subunit association (Mitchell et al., 1992; Poldermans et al., 1980) and protein synthesis initiation (Poldermans et al., 1979b), is one of the most conserved regions in the small-subunit rRNA (Van Knippenberg et al., 1984). Recently, direct evidence for the interaction of KsgA with small-subunit rRNA near its functional center has been reported by Xu et al (2008). KsgA methylates the two adjacent adenosines through the transfer of four methyl groups from four molecules of the S-adenosylmethionine (SAM) cofactor to atom N6 of each nucleotide while giving rise to four molecules of S-adenosylhomocysteine (SAH). Mutations in KsgA which result in the loss of dimethylation at A1518 and A1519 are the most common cause of resistance to the aminoglycoside antibiotic kasugamycin in E. coli and other bacteria (Helser et al., 1971; Helser et al., 1972; Van Buul et al., 1983).
Ligand-free KsgA from E. coli (Ec-KsgA) is not able to bind cofactor until it binds to RNA (Poldermans et al., 1979a; Thammana and Held, 1974). It was hypothesized that the interaction of KsgA with a part of the 30S subunit separate from helix 45 caused a conformational change, allowing SAM to bind in the binding pocket (O’Farrell et al., 2004). Although KsgA is able to bind naked 16S rRNA (van Gemen et al., 1989), it will not methylate 16S rRNA (Thammana and Held, 1974). A recent study showed that KsgA can only methylate 30S subunits in a translationally inactive form (Desai and Rife, 2006). KsgA shares a high level of sequence homology with the Erm MTases (van Buul and van Knippenberg, 1985), which methylate the large-subunit 23S rRNA in bacterial ribosomes and thus mediate resistance to the macrolide-lincosamide-streptogramin B (MLS-B) group of antibiotics (Skinner et al., 1983). Unlike KsgA, ErmC′ from Bacillus subtilis (Bs-ErmC′) can methylate a fragment of 23S RNA as small as 32 nucleotides (Schluckebier et al., 1999; Weisblum, 1995).
KsgA orthologs from non-bacterial sources are represented by Dim1 from Saccharomyces cerevisiae (Sc-Dim1) (Lafontaine et al., 1995; Lafontaine et al., 1998) and from human (Hs-Dim1) (Law et al., 1998; Oh et al., 2005), Pfc1 from Arabidopsis thaliana (Tokuhisa et al., 1998) and mitochondria mtTFB from human (Hs-mtTFB) (Cotney and Shadel, 2006; McCulloch et al., 2002; Seidel-Rogol et al., 2003). The mtTFB from yeast (Sc-mtTFB), however, has lost its MTase activity (Klootwijk et al., 1975). Orthologs from archaebacteria, eubacteria and eukaryotes have been shown to compensate for the loss of KsgA function in bacteria (Lafontaine et al., 1994; O’Farrell et al., 2006; Seidel-Rogol et al., 2003). Unlike Ec-KsgA, however, Hs-Dim1 appears to be able to bind the cofactor in the absence of RNA (PDB entry 1ZQ9, Structural Genomics Consortium).
Structures of KsgA and Erm proteins have been reported for E. coli KsgA (Ec-KsgA, PDB entry 1QYR) (O’Farrell et al., 2004), Hs-Dim1 (PDB entry 1ZQ9) and Sc-mtTFB (PDB entry 1I4W) (Schubot et al., 2001), Bs-ErmC′ (PDB entries 2ERC, 1QAM, 1QAN, 1QAO and 1QAQ) (Bussiere et al., 1998; Schluckebier et al., 1999) and Streptococcus pneumoniae ErmAM (PDB entry 1YUB) (Yu et al., 1997). However, none of these structures is bound to RNA. Here, we present the crystal structures of Aquifex aeolicus KsgA (Aa-KsgA) in complex with RNA with and without a bound SAH molecule, along with two high-resolution ligand-free structures, providing insights into the molecular details of ligand recognition by KsgA and the relationship between RNA binding and cofactor binding.
Results and Discussion
We report four crystal structures of Aa-KsgA: two in its ligand-free form (KsgA1 and KsgA2), one in complex with RNA (KsgA-RNA) and one in complex with both RNA and SAH (KsgA-RNA-SAH). We did not observe SAM in the structure, for which the reason is not clear, and our crystallization trials with SAH, in the presence or absence of RNA, did not yield any crystals. The Aa-KsgA sequence contains 248 amino acid residues. A non-native Ser at the N-terminus from cloning is not visible in any of the four structures. KsgA1 contains residues 7-246 and 287 water oxygen atoms; KsgA2 contains residues 12-246 and 180 water oxygens; KsgA-RNA contains residues 14-246 and 44 nucleotide residues; and KsgA-RNA-SAH contains residues 1-246, 44 nucleotide residues, an SAH molecule and 37 water oxygens. X-ray data collection and refinement statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics
| Crystal | KsgA1 | KsgA2 | KsgA-RNA | KsgA-RNA-SAH |
|---|---|---|---|---|
| X-ray Diffraction Data | ||||
| Space group | P212121 | P212121 | C2 | P21 |
| Unit cell parameters | ||||
| a (Å) | 43.1 | 42.9 | 85.2 | 43.1 |
| b (Å) | 57.4 | 53.6 | 58.3 | 58.1 |
| c (Å) | 87.6 | 101.0 | 91.1 | 69.0 |
| α (°) | 90.0 | 90.0 | 90.0 | 90.0 |
| β (°) | 90.0 | 90.0 | 107.4 | 109.8 |
| γ (°) | 90.0 | 90.0 | 90.0 | 90.0 |
| Resolution (Å) | 30.0 - 1.44 (1.44 - 1.48)a | 30.0 - 1.68 (1.68 - 1.74) | 30.0 - 3.0 (3.0 - 3.11) | 30.0 - 2.78 (2.78 - 2.88) |
| Measured reflections | 265573 | 173774 | 38284 | 26715 |
| Unique reflections | 38509 | 25134 | 7560 | 6848 |
| Completeness (%) | 96.9 (80.1) | 92.3 (54.8) | 88.5 (52.9) | 84.2 (49.9) |
| Multiplicity | 6.9 (2.7) | 6.9 (2.9) | 5.1 (2.9) | 3.9 (2.1) |
| Rmerge (%)b | 6.8 (44.6) | 6.3 (50.0) | 10.3 (43.0) | 14.4 (39.7) |
| I/σ(I) | 24.6 (1.9) | 25.7 (1.7) | 13.4 (1.9) | 9.0 (2.4) |
| Crystal Structure | ||||
| Effective resolution (Å)c | 1.44 | 1.72 | 3.16 | 3.05 |
| No. of non-H atoms | ||||
| Protein | 2071 | 1947 | 1876 | 1986 |
| Nucleic acid | N/A | N/A | 950 | 950 |
| Water | 287 | 180 | 0 | 37 |
| Rfactor (%)d | 15.9 (29.3) | 20.6 (35.3) | 23.3 (31.0) | 23.9 (34.4) |
| Rfree (%)d | 20.0 (33.6) | 24.6 (40.9) | 28.0 (44.0) | 28.8 (60.3) |
| RMSDe bonds (Å) | 0.005 | 0.009 | 0.006 | 0.005 |
| RMSD angles (°) | 0.9 | 1.2 | 1.1 | 1.0 |
| Overall B-factor (Å2) | 22.0 | 31.3 | 83.4 | 28.7 |
| B-factor for protein (Å2) | 20.3 | 30.0 | 85.5 | 30.6 |
| B-factor for RNA (Å2) | – | – | 79.4 | 25.1 |
| B-factor for SAH (Å2) | – | – | – | 26.3 |
| B-factor for solvent (Å2) | 34.4 | 42.9 | – | 22.1 |
| Ramachandran statistics (%) | ||||
| Most favored ϕ/ψ | 94.4 | 94.3 | 86.1 | 84.0 |
| Additionally allowed ϕ/ψ | 5.6 | 5.7 | 13.0 | 14.2 |
| Disallowed ϕ/ψ | 0.0 | 0.0 | 0.0 | 0.0 |
Values in parentheses are for the highest resolution shell.
Rmerge = Σ|(I-<I>)|/σ(I), where I is the observed intensity.
At the effective resolution of a structure, the completeness of X-ray data > 93% and the observable data > 70% for the highest resolution shell according to the Notes for Authors 2008, Acta Crystallographica Section D, Biological Crystallography.
Rfactor and Rfree = Σ||Fobs|-|Fcalc||/Σ|Fobs| where Rfree was calculated over 5% of the amplitude chosen at random and not used in the refinement. Both values were calculated for the resolution range of data collection.
RMSD, root-mean-square deviation.
Figure 1 depicts a structure-based sequence alignment of Aa-KsgA with four related proteins: Ec-KsgA, Bs-ErmC′, Hs-Dim1 and Hs-mtTFB. In amino (N4-cytosine or N6-adenine) MTases, there are nine common SAM-dependent MTase functional motifs, motifs I-VIII and X (Malone et al., 1995). Motifs I, II, III, IV and VIII are highly conserved, whereas motifs V, VI and VII are less conserved. Motifs I, II and III contain amino acid residues important for cofactor binding while motifs IV and VIII contains residues important for catalysis (Goedecke et al., 2001; O’Farrell et al., 2004; Schluckebier et al., 1999). The location of motif X in the primary sequence is one of the major differences between C5-cytosine MTases and amino MTases. In the C5-cytosine MTases, this motif is located in the carboxy-terminus, whereas in the amino MTases, it is always to the amino side of motif I (Malone et al., 1995). Motif X and the upstream N-terminal residues are not visible in any of the previously reported N6-adenine RNA MTase structures, including Ec-KsgA (PDB entry 1QYR), Hs-Dim1 (PDB entry 1ZQ9) and ErmC′ (PDB entries 2ERC, 1QAM, 1QAN and 1QAO). In our KsgA-RNA-SAH structure, motif X and the entire N-terminus of the protein are visible.
Figure 1. Structure-based sequence alignment.
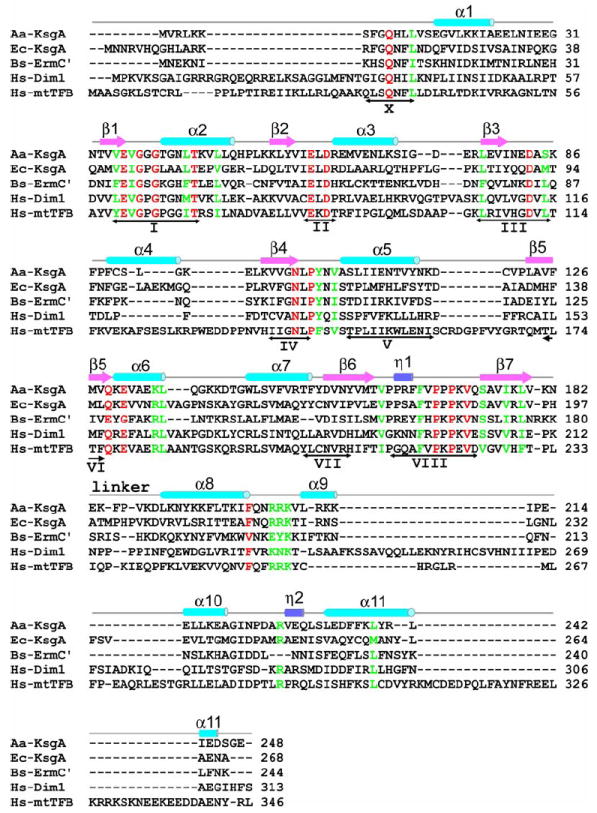
The sequences of Aa-KsgA, Ec-KsgA, Bs-ErmC′ and Hs-Dim1 (GenBank entries NP_214246, P06992, P13956 and NP_055288, respectively) were aligned based on their structures, whereas the Hs-mtTFB sequence (GenBank entry NP_057104) was aligned based on sequence homology. Secondary structure elements and the position of the linker between the NTD and CTD are indicated above the sequences. The double-headed arrows under the sequences indicate the nine conserved functional motifs common for SAM-dependent MTases. Absolutely and highly conserved residues in KsgA sequences are indicated in red and green, respectively, most of which are also conversed in Bs-ErmC′ although it is not a KsgA protein.
Structures of ligand-free Aa-KsgA
The KsgA1 structure is shown in Figure 2A. The N-terminal domain (NTD) exhibits the SAM-dependent MTase fold. This fold, reminiscent of the Rossmann fold often encountered in mono- or di-nucleotide-binding domains, consists of alternating α-helices and β-strands, with a central seven-stranded β-sheet sandwiched between a variable number of α-helices. Strands 1-6 of the sheet are parallel, with strand 7 inserted antiparallel between strands 5 and 6. Aa-KsgA has three α-helices on one side of the sheet and four on the other, and an additional 310-helix in the NTD. Also notable in the NTD is a disulfide bond between residue C90 in α4 and C120 in the α5-β5 turn, which is observed in all four Aa-KsgA structures. This disulfide bond may be unique for Aa-KsgA because the C90 and C120 residues are not conserved in other members of the family (Figure 1). The C-terminal domain (CTD) of Aa-KsgA, consisting of four α-helices and one 310-helix, forms a cleft with the NTD (Figure 2A).
Figure 2. Ligand-free Aa-KsgA structures.
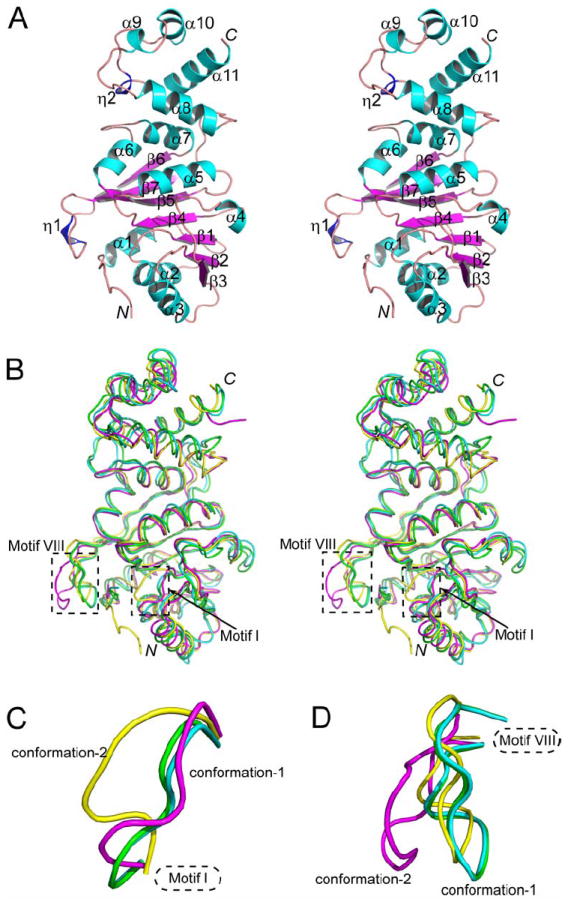
(A) Stereoview showing the ribbon diagram of the KsgA1 structure. The α-helices, β-strands and 310-helices are numbered and shown in cyan, magenta and blue, respectively.
(B) Cα superposition of KsgA1 (yellow), KsgA2 (magenta), Ec-KsgA-chain A (cyan, PDB entry 1QYR) and Ec-KsgA-chain B (green, PDB entry 1QYR).
(C) Two distinct conformations of motif I.
(D) Two distinct conformations of motif VIII.
For Aa-KsgA, there is one molecule in the asymmetric unit, while for Ec-KsgA (PDB entry 1QYR), there are two polypeptide chains (A and B) in the asymmetric unit. Thus, we have four ligand-free KsgA structures to analyze. Despite the relatively low sequence identity between Aa- and Ec-KsgA, they share very similar three-dimensional structures (Figure 2B). The overall root-mean-square deviation (RMSD) for the Cα positions between KsgA1 and Ec-KsgA chain A is 1.8 Å and the RMSD between KsgA1 and Ec-KsgA chain B is 1.6 Å. The most noticeable differences among the four structures are in the conformation of motifs I and VIII (Figure 2B). For each motif, three conformations are similar and the fourth is distinct. Thus, each motif exhibits two distinct conformations. The three similar conformations of each motif will be referred to as conformation-1 while the fourth will be termed as conformation-2 (Figures 2C and 2D).
Structure of RNA
Although KsgA does not methylate free 16S rRNA, it binds to a small fragment of rRNA containing the target adenosine bases (van Gemen et al., 1989). The RNA used in our study contains the 24 contiguous nucleotide residues of helix 45 (Figure 3A, middle panel) found in the 16S rRNA from E. coli. In the structure of the E. coli 30S subunit (PDB entry 2AW7) (Schuwirth et al., 2005), helix 45 forms a hairpin structure capped by a central GGAA tetraloop (Figure 3A, left panel). In our protein-RNA structures, however, two RNA molecules form a duplex with four mismatched G-A pairs in the middle (Figure 3A, right panel). None of the mismatched nucleotides flips out of the duplex. The RNA duplex in both KsgA-RNA and KsgA-RNA-SAH are well defined (For an example, see Figure 3B).
Figure 3. Structure of RNA and of the KsgA-RNA complex.
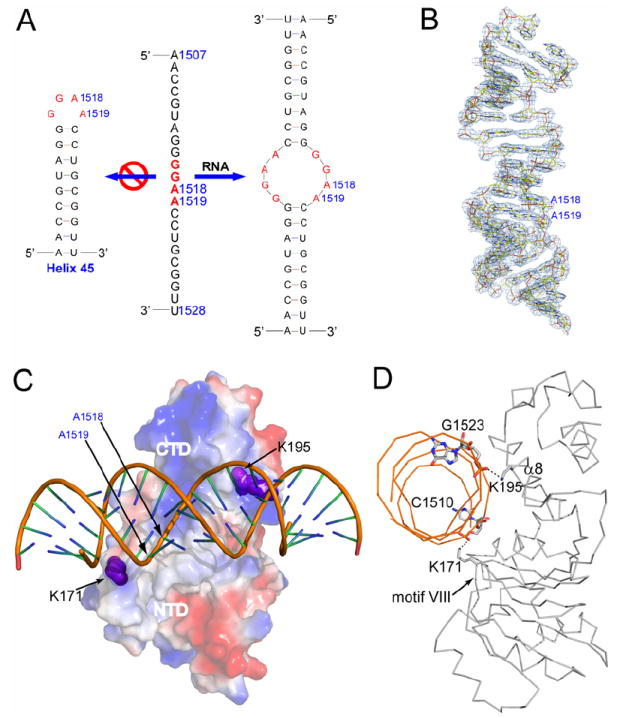
(A) The sequence of the substrate RNA (1507-1528, E. coli numbering) is shown in the middle, while its presumed and observed secondary structures are shown on the left and right, respectively. The presumed tetraloop GGAA are highlighted in red.
(B) Structure of the RNA as observed in the ternary KsgA-RNA-SAH complex. The RNA is shown as a stick model in atomic colors (carbon in yellow, nitrogen in blue, oxygen in red and phosphorous in orange) and outlined with the final 2Fo − Fc electron density map (contoured at 1.0 σ in light blue).
(C) Surface electrostatic potential for the protein in the binary KsgA-RNA complex. Positively charged area is indicated in blue and negatively charged in red. RNA is shown as a tube-and-stick model. Residues K171 (from motif VIII) and K195 (from α8) are highlighted with sphere representation in magenta.
(D) Ribbon diagram of the binary KsgA-RNA complex viewed along the bound RNA. Shown as stick models are the amino acid or nucleotide residues involved in the two hydrogen bonds (dashed lines in black) between the protein and the RNA.
Assuming that the substrate RNA has a stem-loop conformation, the hairpin RNA could be aligned with the duplex RNA in two possible ways based on the sequences shown in Figure 3A. Our structures suggest, however, only one way of the hairpin positioning, which brings the tetraloop of the hairpin into contact with K171/motif VIII, is likely to be catalytically compatible. A hypothetical model for the protein-substrate complex will be proposed in the section of implications for catalysis.
Structure of the binary KsgA-RNA complex
The cleft between the NTD and CTD is positively charged, providing a platform for the enzyme to bind RNA (Figure 3C). Only residues on the NTD side of the cleft have been previously shown to be important in the binding of Bs-ErmC′ to its substrate RNA (Maravic et al., 2003a). We show here that positively charged side chains on both sides of the cleft are involved in double-stranded (ds) RNA binding by Aa-KsgA. In the KsgA-RNA structure, K171 (in motif VIII) and K195 (in helix α8) each forms a strong hydrogen bond with a phosphate O2P of RNA (Figure 3D). As a result of RNA binding, the enzyme undergoes conformational changes in the relative positioning of the NTD and the CTD as well as within the two domains, among which the most significant is the stabilization of motifs I and VIII. In ligand-free KsgA, each motif exhibits two distinct conformations (Figures 2C and 2D). In the protein-RNA complexes, however, both motifs are stabilized in conformation-1 (Figure 4A).
Figure 4. Structure of the KsgA-RNA-SAH complex.
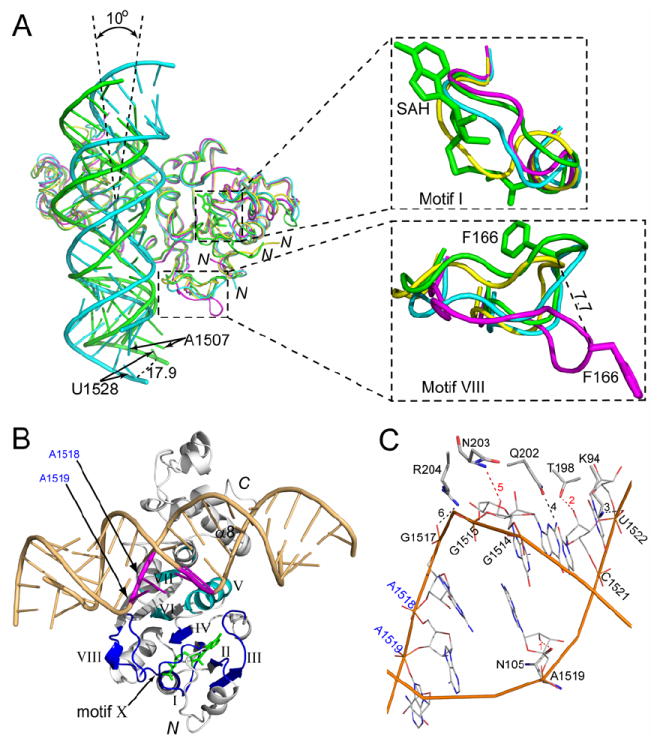
(A) Cα superposition of KsgA1 (yellow), KsgA2 (magenta), KsgA-RNA (cyan) and KsgA-RNA-SAH (green). Protein is shown as a ribbon diagram, RNA as tube-and-stick models and cofactor SAH as a stick model. The rotation of RNA (~10°) and the distance of the U1528 shift (17.9 Å) relative to the RNAs in the two complexes are indicated. Close-up views of motifs I and VIII are shown in the upper and lower right panels. The distance (7.7 Å) of the Cα shift of the highly conserved residue F166 between KsgA2 and KsgA-RNA-SAH is also indicated. The side chain of F166 is shown as a stick model.
(B) Front view of the KsgA-RNA-SAH structure: protein as a ribbon diagram in white; substrate RNA as a tube-and-stick model in light orange with the adjacent A1518 and A1519 highlighted in magenta; cofactor SAH as a stick model in green. Roman numerals indicate the conserved structural motifs of the protein: well conserved motifs I-IV, VIII and X in blue; less conserved motifs V-VII in cyan.
(C) Six strong hydrogen bonds (dashed lines) are observed between the protein and the RNA in the KsgA-RNA-SAH structure. Amino acid residues interacting with the bound RNA are shown as thicker sticks while the nucleotide residues are shown as thinner sticks. Hydrogen bonds 1, 2 and 5 (colored in red) involve the O2′ hydroxyl groups of nucleotide residues.
Structure of the ternary KsgA-RNA-SAH complex
Ec-KsgA does not bind cofactor until it binds to RNA (O’Farrell et al., 2004; Poldermans et al., 1979a; Thammana and Held, 1974). We have observed that Aa-KsgA does not crystallize in the presence of SAM alone, but crystallizes in the presence of either RNA alone or both RNA and SAH, suggesting that once KsgA binds the RNA in the cleft between the NTD and CTD, it can then bind the cofactor. RNA binding in the cleft stabilizes motifs I and VIII in conformation-1 (Figure 4A). In conformation-1, motif VIII is pointing toward the cofactor-binding site, whereas in conformation-2, it is pointing away from the cofactor-binding site. In conformation-1, motif I is not in the way of the bound SAH, whereas in conformation-2, it blocks the cofactor-binding site. Thus, for both motifs, conformation-1 is compatible with cofactor binding and conformation-2 is not.
Previously, motif X was thought to be non-conserved and to comprise residues in the N-terminal α-helix (O’Farrell et al., 2004). We show here that motif X contains conserved residues Q10 and L13, but does not include α1 residues (Figure 1). It appears that the N-terminal loop and motif X of KsgA become ordered only after binding both the RNA and the cofactor although the functional role of the N-terminal loop is not clear. Among the better conserved motifs, motifs I-IV and X are located in proximity to the cofactor, while motif VIII interacts with both the cofactor and the RNA. Less conserved motifs V-VII do not contribute directly to the binding of RNA or cofactor (Figure 4B).
The structures of the bound RNA in the binary and the ternary complexes are similar. However, the binding of the SAH occurs in concert with a change of the relative positioning between the protein and the RNA, which is equivalent to a ~10° counterclockwise rotation of the RNA duplex (Figure 4A). When the cofactor-binding site is occupied, the interaction between the protein and the RNA is significantly enhanced. Compared to the binary complex, a total of 12 residues make contacts with the RNA, including N105, K140, K171, V172, Q173, K194, K195, T198, K199, Q202, N203 and R204. Among the contacts between the protein and the RNA, six strong hydrogen bonds are observed, three interacting with the O2′ hydroxyls of G1515, A1519 and C1521, two with phosphate oxygen O2P of G1517 and U1522, and one with atom N2 of G1514 (Figure 4C). Apparently, the three strong hydrogen bonds from N105, T198 and N203 to the O2′ hydroxyls of RNA distinguish the dsRNA from dsDNA.
Cofactor binding
Five out of the nine common motifs are involved in the binding of SAH. Motif X (8FGQHLL13) makes extensive contacts with the bound SAH (Figures 4B and 5A). The distance between the sulfur of SAH and residue Q10 or H11 is 3.7 Å, suggesting that these two residues may assist in orienting the methyl group of SAM for catalysis. Motif I (36VEVGGGTGNLT45), i.e., the G-loop, forms part of the cofactor-binding pocket (Figure 5A). Motif II (59LELD62) contains two absolutely conserved residues, E60 and D62, among which E60 forms a hydrogen bond with the O2′ hydroxyl on the ribose moiety of SAH. Motif III (78LEVINEDAS85) contains one absolutely conserved residue D83, which forms a hydrogen bond with the exocyclic N6 amine group of the SAH adenine moiety. Motif IV (98VVGNLP103) contains residues that are important for both cofactor binding and catalysis. One of the absolutely conserved residues, N101, is hydrogen bonded with atom N of homocysteine (Figure 5A).
Figure 5. The cofactor-binding site in Aa-KsgA and Bs-ErmC′.
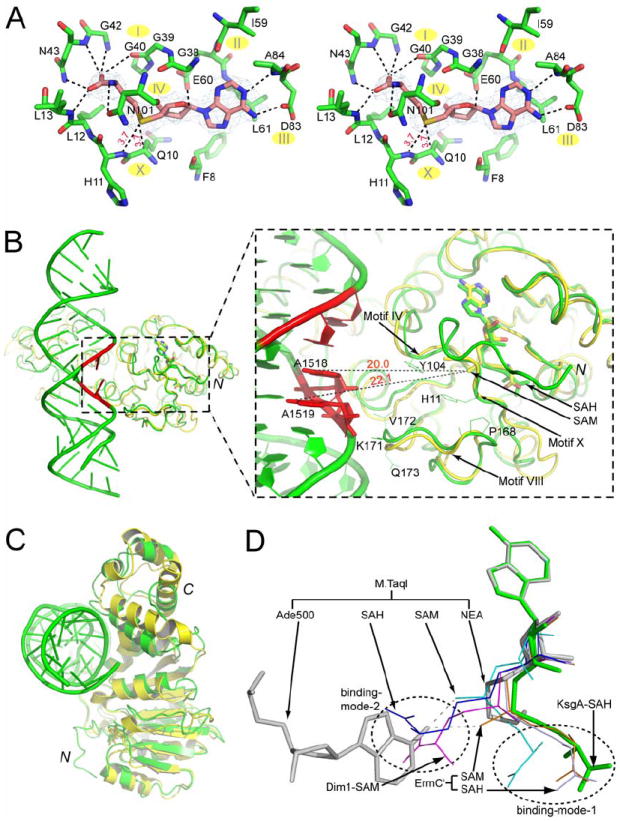
(A) Stereoview of the SAH-binding site in the ternary KsgA-RNA-SAH complex. The SAH cofactor and the surrounding amino acid residues within hydrogen bonding distance (3.5 Å) are shown as stick models in atomic colors (carbon in magenta in the SAH and in green in the surrounding amino acids, nitrogen in blue, oxygen in red and sulfur in yellow). The cofactor is outlined with the final 2Fo − Fc electron density map contoured at 1.0 σ and in blue. Hydrogen bonds between SAH and nearby amino acid residues are indicated with dashed lines in black, and the distance between the SAH sulfur and the Cα of Q10 and the nitrogen of H11 are indicated in red. The locations of motifs I-IV and X are indicated.
(B) Cα-trace superposition between the KsgA in the ternary KsgA-RNA-SAH complex (green) and the Bs-ErmC′ in the binary ErmC′-SAM complex (PDB entry 1QAO). Proteins are illustrated as ribbon diagrams and RNA as a tube-and-stick model. The adjacent adenosines are highlighted in red. Residues A1518 and A1519, which are closer to the putative catalytic center, are shown as stick models. The co-factors are shown as stick models in atomic colors (carbon in green in SAH and in yellow in SAM, nitrogen in blue, oxygen in red and sulfur in yellow). Conserved residues in KsgA which appear to play important roles in either the binding of cofactor/RNA or the stabilization of target adenosines are shown as thinner sticks in green for KsgA. The distances between atom S of SAM and the N6 of highlighted adenosines are indicated with dashed lines in black.
(C) Cα-trace superposition between the ErmC′-SAH complex (yellow, PDB entry 1QAN) and the KsgA-RNA-SAH complex (green).
(D) Superposition of the SAH in KsgA-RNA-SAH (green), SAH in ErmC′-SAH (light blue, PDB entry 1QAN), SAH in M·TaqI-SAH (blue, PDB entry 1AQI) (Schluckebier et al., 1997), SAM in ErmC′-SAM (light orange, PDB entry 1QAO), SAM in Dim1-SAM (magenta, PDB entry 1ZQ9) and SAM in M·TaqI-SAM (cyan, PDB entry 2ADM) (Labahn et al., 1994; Schluckebier et al., 1997). The substrate adenosine and the cofactor analog NEA in M·TaqI-DNA-NEA (grey, PDB entry 1G38) is also aligned. The alignment is based on the superposition of the adenine base of the cofactors. Stick models are shown for KsgA-RNA-SAH and M·TaqI-DNA-NEA, and line models are used for other structures.
Motifs V (residues 107-117), VI (residues 126-128) and VII (residues 151-156) are less conserved (Figure 1). These motifs do not directly interact with SAH (Figure 4B). The highly conserved motif VIII (162PPRFFVPPPKVQ173) may play multiple roles in KsgA catalysis. This motif contains the highly conserved residue F166 (Figure 4A). This residue in DNA N6-adenine MTase M·TaqI is F196, the side chain of which is in an edge-to-face π-stacking arrangement with the target adenine (PDB entry 1G38). There is an extensive network of electrostatic and hydrophobic interactions between residues F166, V167 and P168 in motif VIII and residues H11 and L12 in motif X (not shown). Residue H11 also interacts with motif IV through highly conserved Y104 (Figure 5B). Thus, motifs IV, VIII and X must be functionally dependent on one another.
Cofactor SAM and product SAH assume a common binding mode in Bs-ErmC′ (Schluckebier et al., 1999). This binding mode is also observed in KsgA-RNA-SAH. Based on the Cα positions in the NTD, three structures are aligned and depicted in Figure 5B for the superposition of KsgA-RNA-SAH and ErmC′-SAM (PDB entry 1QAO), and in Figure 5C for the superposition of KsgA-RNA-SAH and ErmC′-SAH (PDB entry 1QAN). The positioning of the adenine and ribose moieties of the SAH in KsgA is virtually the same as that of SAM in EcmC′, and the two molecules show only small conformational differences in their methionine moieties (Figure 5B). This cofactor-binding mode (designated as binding-mode-1) has also been observed for the SAM molecule in the M·TaqI-SAM structure (PDB entry 2ADM).
For N6-adenine MTases, a second cofactor-binding mode has been observed in which the methionine moiety is pointing in the opposite direction (designated as binding-mode-2) as shown in Figure 5D. Among members of the N6-adenine MTase family, molecular details for the methyl group transfer have been revealed for M·TaqI on the basis of the structure of the enzyme in complex with both DNA and a cofactor analog NEA (PDB entry 1G38). The arrangement of the substrate adenine base and the NEA in the M·TaqI-DNA-NEA structure suggests that in binding-mode-1 the activated methyl group of SAM points towards the N6 of the substrate adenine, whereas in binding-mode-2 the methyl group points away from the substrate and consequently the methionine moiety appears to block the proper positioning of the adenine base (Figure 5D). Therefore, cofactor binding-mode-1 is compatible with the direct transfer of the methyl group from SAM to atom N6 of adenosine, whereas binding-mode-2 may initiate the product release.
Implications for catalysis
In KsgA-RNA-SAH, the two RNA molecules form a duplex with four G-A mismatches in the middle (Figure 3A). Although this arrangement is not likely the catalytic assembly, it shows how KsgA binds a dsRNA. In addition, it provides guidance for a comparative analysis of available structural information, suggesting how KsgA may bind a substrate RNA. It has previously been suggested that the methylation of A1518 and A1519 alters the RNA conformation significantly (Rife and Moore, 1998). If the substrate RNA assumes the steo-loop conformation, a structure of dimethylated helix 45 (PDB entry 2AW7), and a model of unmethylated stem loop (PDB entry 1AFX, Butcher et al., 1997) can be superpositioned with the RNA duplex in the KsgA-RNA-SAH structure (Figure 6A). Because the RNA duplex in the structure is symmetric, the two stem-loop structures can also be aligned with the other half of the duplex. However, only the alignment shown in Figure 6A positions the target adenosines at the entrance of the catalytic center and in proximity to motif VIII. Although the stem-loop RNA may not form strong hydrogen bonds with the CTD as suggested by the dsRNA in the KsgA-RNA-SAH structure (Figure 4C), the crucial role of motif VIII in catalysis is clearly implicated (Figure 6A). Ec-KsgA can only methylate 30S subunits in a translationally inactive form (Desai and Rife, 2006). Likely, this is also true for Aa-KsgA. Thus, the enzyme may be embedded in the 30S and interact with RNA extensively, which should be sufficient to position the A1518-A1519 half of the tetraloop in contact with motif VIII.
Figure 6. The putative catalytic center of KsgA.
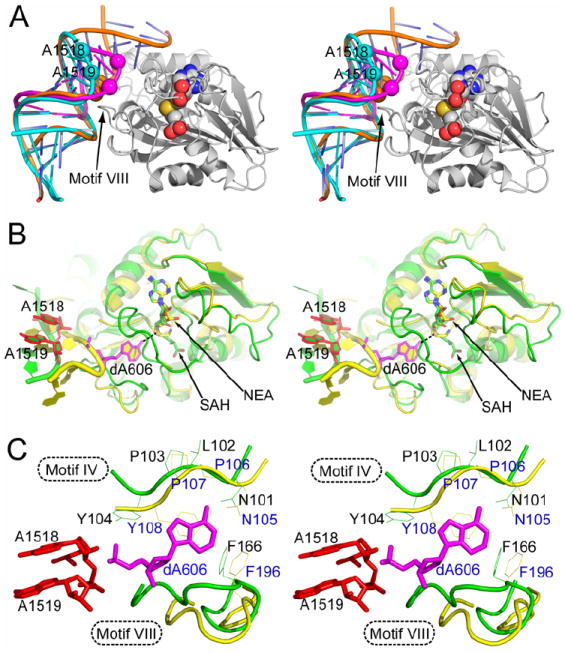
(A) Stereoview showing the superposition of tetraloop structures with the KsgA-RNA-SAH complex. SAH is shown as spheres (carbon in grey, nitrogen in blue, oxygen in red, and sulfur in yellow). KsgA is shown as a ribbon diagram in grey. RNAs are illustrated as tube-and-stick models with the positions of A1518 and A1519 indicated by spheres. RNA in KsgA-RNA-SAH is shown in orange, a structure of product RNA with dimethylated A1518 and A1519 (PDB entry 2AW7) in cyan, and a model for substrate RNA with unmethylated adenosines (PDB entry 1AFX) in magenta.
(B) Stereoview showing the superposition of the putative catalytic center of KsgA as observed in the ternary KsgA-RNA-SAH complex (green) with the catalytic center assembly of M·TaqI as observed in the M·TaqI-DNA-NEA structure (PDB entry 1G38, yellow). The alignment was based on the cofactor SAH (stick model with carbon in green, nitrogen in blue, oxygen in red and sulfur in yellow) and the cofactor analog NEA (stick model with carbon in yellow). The target adenine dA606 for M·TaqI is shown as a stick model in magenta, whereas the A1518 and A1519 for KsgA are shown as stick models in red. The contact between atom N6 of dA606 in the M·TaqI complex and atom S of SAH in the KsgA complex is indicated with a dashed line in black.
(C) A close-up stereoview showing the details of the adenine-binding site. The amino acid side chains in motif IV (N105, P106, P107 and Y108) and motif VIII (F196) in M·TaqI are shown as line models in yellow. The corresponding side chains in KsgA (N101, L102, P103, Y104 and F166) are shown in green. The adenine substrate is shown in magenta.
In the KsgA-RNA-SAH structure, the distance between the acceptor (atom N6 of adenine) and the donor (atom S of SAH) of the methyl group transfer reaction is ~20 Å (Figure 5B). In the KsgA-substrate model, the acceptor-donor distance is also ~20 Å (Figure 6A). For a direct methyl group transfer mechanism, this distance needs to be ~5 Å (Goedecke et al., 2001; Klimasauskas et al., 1994). The DNA double helix in the M·TaqI-DNA-NEA structure (PDB entry 1G38) is located at a distance similar to that for the RNA duplex in KsgA-RNA-SAH, suggesting that a base flipping mechanism may bring the acceptor in proximity to the donor of the reaction (Figure 6B). Thus, the catalytic center assembly of KsgA may be modeled on the basis of KsgA-RNA-SAH and M·TaqI-DNA-NEA (PDB entry 1G38).
Although the overall structures of the M·TaqI and KsgA complexes are different, the two N6-adenine MTases share a very similar cofactor-binding site (Figure 6B). Five residues of M·TaqI in motif IV (N105, P106, P107 and Y108) and motif VIII (F196) are involved in the binding and stabilization of the target adenosine (dA606). The counterparts of these five residues in KsgA are N101, L102, P103, Y104 and F166 (Figure 6C). The importance of these residues in catalysis has been demonstrated by mutational studies. Mutations of the Asn and Phe residues are poorly tolerated in DNA MTases and ErmC′ (Guyot et al., 1993; Kong and Smith, 1997; Maravic et al., 2003b; Pues et al., 1999; Roth et al., 1998; Willcock et al., 1994), while mutation of the Tyr residue totally abolishes the activity of both M·TaqI and ErmC′, both in vitro and in vivo (Pues et al., 1999). Four out of these five residues are either absolutely (N101 and P103) or highly (Y104 and F166) conserved in KsgA (Figure 1). Residue P106 in M·TaqI does not interact with the target adenine; its counterpart L102 in KsgA is not highly conserved either.
In addition to the conserved residues in M·TaqI which are involved in the stabilization of the target adenine, residues V21 from motif X and K199 from motif VIII are in contact with the substrate adenine. The counterparts of V21 and K199 in KsgA are residues H11 from motif X and P168 from motif VIII. Residue H11 may play multiple roles because (1) the amide of H11 is very close to the sulfur of SAH (Figure 5A) and (2) the side chain of H11 is hydrogen bonded with motif VIII residue P168 and in close contact with motif IV residue Y104 (Figure 5B). Thus, H11 seems to function as a hub in the active center of KsgA by interacting with both the cofactor and the target adenine and linking two catalytically very important motifs (IV and VIII). Residue P168 of KsgA may mimic the functional role of K199 in M·TaqI, which is part of DNA-binding loop and undergoes a conformational change upon DNA binding. Similarly, P168 of KsgA is in the middle of flexible motif VIII that also undergoes significant conformational changes upon RNA binding (Figure 4A).
Conclusions
Our ligand-free structures reveal two distinct conformations for both motif I and motif VIII (Figure 2). Our complex structures provide insights into the molecular details of ligand recognition by Aa-KsgA and the relationship between RNA binding and cofactor binding. We show how the RNA is bound, how the RNA-bound KsgA is changed for SAH binding, and how SAH is bound. The protein binds to the RNA duplex through motif VIII in the NTD and helix α8 in the CTD (Figures 3C and 4B). It binds SAH mainly through motifs I, II, III, IV and X (Figures 4B and 5A). Before RNA binding, motifs I and VIII are flexible (Figure 2B). Upon RNA binding, motif VIII is stabilized which in turn stabilizes motif I, allowing the cofactor-binding site to form. Motif X, along with residues 1-6 at the N-terminus, exhibits ordered structure in KsgA-RNA-SAH. A comparison of protein-RNA interactions between KsgA-RNA and KsgA-RNA-SAH indicates that 10 more residues of the protein in the ternary complex are involved in RNA binding (Figures 3D and 4C). For catalysis, the substrate adenine must become an integral part of the catalytic center assembly, for which we have proposed a base-flipping mechanism that has yet to be elucidated. Whether the dimethylation of the two adenosines is carried out by a single- or multi-binding event remains to be seen.
Experimental Procedures
Cloning, protein expression and purification
The ORF of Aa-KsgA was amplified from genomic DNA by PCR using the following oligonucleotide primers: 5’-GAG AAC CTG TAC TTC CAG TCT ATG GTA AGA CTG AAA AAA TCC TTC-3’ and 5’-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT ATT ACT CTC CAG AAT CCT CAA TTA ATC-3’ (primer R). The PCR amplicon was subsequently used as template for a second PCR with the following primers: 5’-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC GGA GAA CCT GTA CTT CCA G-3’ and primer R. The amplicon from the second PCR was inserted by recombinational cloning into the entry vector pDONR221 (Invitrogen, Carlsbad, CA) and the nucleotide sequence confirmed experimentally. The open reading frame encoding the KsgA gene now with a recognition site for tobacco etch virus (TEV) protease (ENLYFQ/S) fused in-frame to its N-terminus, was moved by recombinational cloning into the destination vector pDEST-HisMBP (Nallamsetty and Waugh, 2006) to produce pBA1939. The pBA1939 directs the expression of Aa-KsgA as a fusion to the C-terminus of E.coli maltose binding protein (MBP) with an intervening TEV protease recognition site. The MBP contains an N-terminal hexahistidine tag for affinity purification by immobilized metal affinity chromatography. The fusion protein was expressed in the E.coli strain BL21-CodonPlus®(DE3)-RIL (Stratagene, La Jolla, CA). Cells were grown to mid-log phase (OD600 ~ 0.5) at 37°C in Luria broth containing 100 μg·ml-1 ampicillin, 30 μg·ml-1 chloramphenicol and 0.2% glucose. Overproduction of fusion protein was induced with isopropyl-β-D-thiogalactopyranoside at a final concentration of 1 mM for 4h at 30°C. The cells were pelleted by centrifugation and stored at -80°C.
All procedures were performed at 4-8°C. Ten grams of E.coli cell paste was suspended in 150 ml ice-cold 50 mM sodium phosphate (pH 7.5), 200 mM NaCl, 25 mM imidazole buffer (buffer A) containing 1 mM benzamidine HCl (Sigma Chemical Company, St. Louis, MO) and complete EDTA-free protease inhibitor cocktail tablets (Roche Molecular Biochemicals, Indianapolis, IN). The cells were lysed with an APV-1000 homogenizer (Invensys, Roholmsvej, Denmark) at 10,000 psi, and centrifuged at 30,000 g for 30 minutes. The supernatant was filtered through a 0.22 μm polyethersulfone membrane and applied to a HisPrep FF 16/10 column (GE Healthcare Bio-Sciences Corporation, Piscataway, NJ) equilibrated in buffer A. The column was washed to baseline with buffer A and eluted with a linear gradient of imidazole to 500 mM. Fractions containing recombinant His6-MBP-KsgA were pooled, concentrated using an Amicon YM30 membrane (Millipore Corporation, Bedford, MA), diluted with 50 mM sodium phosphate (pH 7.5), 200 mM NaCl buffer (buffer B) to reduce the imidazole concentration to about 25 mM, and digested overnight at 4°C with His6-tagged TEV protease. The digest was applied to the HisPrep FF 16/10 column equilibrated in buffer A and the KsgA emerged in the column effluent. The column effluent was incubated with dithiothreitol (10 mM), concentrated using an Amicon YM10 membrane and applied to a HiPrep 26/60 Sephacryl S-100 HR column (GE Healthcare Bio-Sciences Corporation) equilibrated in buffer B. The peak fractions containing KsgA were pooled, heat-treated at 90°C for 90 min, and clarified by centrifugation and filtration. The sample was concentrated as above and applied to a HiPrep 26/60 Sephacryl S-100 HR column equilibrated in 25 mM Tris (pH 7.2), 150 mM NaCl, 2 mM tris(2-carboxyethyl) phosphine (TCEP) buffer. The peak fractions containing recombinant KsgA were pooled and concentrated to 30-40 mg·ml-1 (estimated at 280 nm using a molar extinction coefficient of 15930 M-1·cm-1). Aliquots were flash-frozen in liquid nitrogen and stored at -80°C. The final product was judged to be >90% pure by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The molecular weight was confirmed by electrospray ionization mass spectroscopy.
Crystallization and data collection
Protein concentrations were adjusted to ~27 mg/ml in 25 mM Tris (pH 7.2), 150 mM NaCl, 2 mM TCEP buffer for crystallization trials. RNA oligos were purchased from Integrated DNA Technologies (Coralville, IA) and used without further purification. The molar ratio of RNA to protein was kept at 1.2:1 for crystallization. Initially, the ratio was for single-stranded RNA vs. protein, and later changed to dsRNA vs. protein, but the concentration of RNA did not make any difference in the composition of the protein-RNA complexes. RNAs were heated at 75 °C for 5 min and cooled on ice for 10 min before being mixed with the protein solution. The protein-RNA mixtures were heated at 70 °C for 5 min and slowly cooled down to and kept at the room temperature for 45 min before crystallization. For the ternary complex, ~10 mM SAM was added and the mixture was kept at room temperature for another 30 min, but SAH was observed in the ternary complex. Crystallization trials of the protein with either SAH alone or both SAH and RNA did not yield any crystals. A different RNA containing 28 nucleotide residues was present in the crystallization drops of KsgA1 and KsgA2, but only the protein crystallized.
Crystals were grown at 19 ± 1 °C. A Hydra II Plus One (Matrix Technologies Corporation) crystallization robot system was employed. The sitting drops of KsgA1 and KsgA2 contained 0.2 μl of protein solution and 0.2 μl reservoir solution. For KsgA1, the reservoir solution contained 20% PEG MME 5000 and 0.1M Bis-Tris, pH 6.5. For KsgA2, the reservoir contained 25% PEG 3000 and 0.1 M MES, pH 6.5. Crystals for both apo-forms did not appear till 12 months later and reached the final size of 0.20 mm×0.15 mm×0.05 mm. The drops of KsgA-RNA crystals contained 0.3 μl protein-RNA solution and 0.3 μl reservoir solution containing 0.2 M NH4NO3 and 40% MPD, and the crystal reached the size of 0.15 mm×0.05 mm×0.01 mm in 10 days. The drops of KsgA-RNA-SAH contained 0.4 μl protein-RNA-cofactor solution and 0.2 μl reservoir solution containing 0.2 M KF and 40% MPD, and the crystals reached the final size of 0.4 mm×0.05 mm×0.01 mm in 7-10 days.
The crystals were flash frozen in liquid nitrogen before data collection. The X-ray data of KsgA1, KsgA2 and KsgA-RNA-SAH were collected at beamline 22-BM, whereas the data of KsgA-RNA were collected at 22-ID, of the Southeast Regional Collaborative Access Team (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory. Data processing was carried out with the HKL2000 program suite (Otwinowski and Minor, 1997). Crystal data and processing statistics are summarized in Table 1.
Structure solution and refinement
The structures were solved by molecular replacement (MR) using program PHASER (McCoy, 2007). The search model for KsgA2 was chain A of the Ec-KsgA structure (PDB entry 1QYR) while the search model for KsgA1 was the refined KsgA2 structure. The two ligand-free structures were refined using PHENIX (Adams et al., 2002) and REFMAC (Murshudov et al., 1997). To solve the two complex structures, the refined KsgA1 structure and the helix 45 structure (residues 1507-1528) of the 16S rRNA structure (PDB entry 1J5E) (Wimberly et al., 2000) were used as search models. The MR solution contained one protein and two RNA molecules. The electron density revealed that the two RNA molecules formed a continued double helix. The two complex structures were refined using CNS (Brünger et al., 1998) and REFMAC (Murshudov et al., 1997).
Bulk solvent correction was employed in all of the refinement. The 2Fo − Fc and Fo − Fc electron density maps were used to inspect and improve the models during the refinement. The SAH molecule was built using the REFMAC library (Vagin et al., 2004). Solvent molecules, as peaks ≥3 σ on the Fo − Fc electron density map with reasonable hydrogen bond networks, were included as water molecules at the later stage of the refinement and verified with omit maps. All graphics work was carrying out using O (Jones et al., 1991) and COOT (Emsley and Cowtan, 2004). The refined structures were assessed using PROCHECK (Laskowski et al., 1993). Illustrations were prepared with PyMOL (DeLano Scientific LLC.).
Acknowledgments
X-ray diffraction data were collected at the 22-ID and 22-BM beamlines of SER-CAT, Advanced Photon Source, Argonne National Laboratory. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Coordinates and Structure Factors The coordinates and structure factors will be deposited with the Protein Data Bank under accession code 1111 for KsgA1, 2222 for KsgA2, 3333 for KsgA-RNA and 4444 for KsgA-RNA-SAH, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr, D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr, D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Bussiere DE, Muchmore SW, Dealwis CG, Schluckebier G, Nienaber VL, Edalji RP, Walter KA, Ladror US, Holzman TF, Abad-Zapatero C. Crystal structure of ErmC’, an rRNA methyltransferase which mediates antibiotic resistance in bacteria. Biochemistry. 1998;37:7103–7112. doi: 10.1021/bi973113c. [DOI] [PubMed] [Google Scholar]
- Butcher SE, Dieckmann T, Feigon J. Solution structure of the conserved 16 S-like ribosomal RNA UGAA tetraloop. J Mol Biol. 1997;268:348–358. doi: 10.1006/jmbi.1997.0964. [DOI] [PubMed] [Google Scholar]
- Cotney J, Shadel GS. Evidence for an early gene duplication event in the evolution of the mitochondrial transcription factor B family and maintenance of rRNA methyltransferase activity in human mtTFB1 and mtTFB2. J Mol Evol. 2006;63:707–717. doi: 10.1007/s00239-006-0075-1. [DOI] [PubMed] [Google Scholar]
- Desai PM, Rife JP. The adenosine dimethyltransferase KsgA recognizes a specific conformational state of the 30S ribosomal subunit. Arch Biochem Biophys. 2006;449:57–63. doi: 10.1016/j.abb.2006.02.028. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Goedecke K, Pignot M, Goody RS, Scheidig AJ, Weinhold E. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat Struct Biol. 2001;8:121–125. doi: 10.1038/84104. [DOI] [PubMed] [Google Scholar]
- Guyot JB, Grassi J, Hahn U, Guschlbauer W. The role of the preserved sequences of Dam methylase. Nucleic Acids Res. 1993;21:3183–3190. doi: 10.1093/nar/21.14.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helser TL, Davies JE, Dahlberg JE. Change in methylation of 16S ribosomal RNA associated with mutation to kasugamycin resistance in Escherichia coli. Nat New Biol. 1971;233:12–14. doi: 10.1038/newbio233012a0. [DOI] [PubMed] [Google Scholar]
- Helser TL, Davies JE, Dahlberg JE. Mechanism of kasugamycin resistance in Escherichia coli. Nat New Biol. 1972;235:6–9. doi: 10.1038/newbio235006a0. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- Klootwijk J, Klein I, Grivell LA. Minimal Post-Transcriptional Modification of Yeast Mitochondrial Ribosomal-Rna. J Mol Biol. 1975;97:337–&. doi: 10.1016/s0022-2836(75)80044-1. [DOI] [PubMed] [Google Scholar]
- Kong H, Smith CL. Substrate DNA and cofactor regulate the activities of a multi-functional restriction-modification enzyme, BcgI. Nucleic Acids Res. 1997;25:3687–3692. doi: 10.1093/nar/25.18.3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labahn J, Granzin J, Schluckebier G, Robinson DP, Jack WE, Schildkraut I, Saenger W. Three-dimensional structure of the adenine-specific DNA methyltransferase M.Taq I in complex with the cofactor S-adenosylmethionine. Proc Natl Acad Sci USA. 1994;91:10957–10961. doi: 10.1073/pnas.91.23.10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine D, Delcour J, Glasser AL, Desgres J, Vandenhaute J. The Dim1 gene responsible for the conserved M(2)(6)Am(2)(6)a dimethylation in the 3’-terminal loop of 18-S ribosomal-RNA Is essential in Yeast. J Mol Biol. 1994;241:492–497. doi: 10.1006/jmbi.1994.1525. [DOI] [PubMed] [Google Scholar]
- Lafontaine D, Vandenhaute J, Tollervey D. The 18S ribosomal-RNA dimethylase Dim1p is required for pre-ribosomal-RNA processing in Yeast. Genes & Development. 1995;9:2470–2481. doi: 10.1101/gad.9.20.2470. [DOI] [PubMed] [Google Scholar]
- Lafontaine DLJ, Preiss T, Tollervey D. Yeast 18S rRNA dimethylase Dim1p: a quality control mechanism in ribosome synthesis? Mol Cell Biol. 1998;18:2360–2370. doi: 10.1128/mcb.18.4.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Law SF, Zhang YZ, Klein-Szanto AJ, Golemis EA. Cell cycle-regulated processing of HEF1 to multiple protein forms differentially targeted to multiple subcellular compartments. Mol Cell Biol. 1998;18:3540–3551. doi: 10.1128/mcb.18.6.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone T, Blumenthal RM, Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J Mol Biol. 1995;253:618–632. doi: 10.1006/jmbi.1995.0577. [DOI] [PubMed] [Google Scholar]
- Maravic G, Bujnicki JM, Feder M, Pongor S, Flogel M. Alanine-scanning mutagenesis of the predicted rRNA-binding domain of ErmC’ redefines the substrate-binding site and suggests a model for protein-RNA interactions. Nucleic Acids Res. 2003a;31:4941–4949. doi: 10.1093/nar/gkg666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravic G, Feder M, Pongor S, Flogel M, Bujnicki JM. Mutational analysis defines the roles of conserved amino acid residues in the predicted catalytic pocket of the rRNA:m6A methyltransferase ErmC’. J Mol Biol. 2003b;332:99–109. doi: 10.1016/s0022-2836(03)00863-5. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch V, Seidel-Rogol BL, Shadel GS. A human mitochondrial transcription factor is related to RNA adenine methyltransferases and binds S-adenosylmethionine. Mol Cell Biol. 2002;22:1116–1125. doi: 10.1128/MCB.22.4.1116-1125.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P, Osswald M, Brimacombe R. Identification of intermolecular RNA cross-links at the subunit interface of the Escherichia coli ribosome. Biochemistry. 1992;31:3004–3011. doi: 10.1021/bi00126a023. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nallamsetty S, Waugh DS. Solubility-enhancing proteins MBP and NusA play a passive role in the folding of their fusion partners. Protein Expr Purif. 2006;45:175–182. doi: 10.1016/j.pep.2005.06.012. [DOI] [PubMed] [Google Scholar]
- O’Farrell HC, Pulicherla N, Desai PM, Rife JP. Recognition of a complex substrate by the KsgA/Dim1 family of enzymes has been conserved throughout evolution. RNA. 2006;12:725–733. doi: 10.1261/rna.2310406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell HC, Scarsdale JN, Rife JP. Crystal structure of KsgA, a universally conserved rRNA adenine dimethyltransferase in Escherichia coli. J Mol Biol. 2004;339:337–353. doi: 10.1016/j.jmb.2004.02.068. [DOI] [PubMed] [Google Scholar]
- Oh JH, Yang JO, Hahn Y, Kim MR, Byun SS, Jeon YJ, Kim JM, Song KS, Noh SM, Kim S, et al. Transcriptome analysis of human gastric cancer. Mamm Genome. 2005;16:942–954. doi: 10.1007/s00335-005-0075-2. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Poldermans B, Bakker H, Van Knippenberg PH. Studies on the function of two adjacent N6,N6-dimethyladenosines near the 3’ end of 16S ribosomal RNA of Escherichia coli. IV. The effect of the methylgroups on ribosomal subunit interaction. Nucleic Acids Res. 1980;8:143–151. doi: 10.1093/nar/8.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poldermans B, Roza L, Van Knippenberg PH. Studies on the function of two adjacent N6,N6-dimethyladenosines near the 3’ end of 16 S ribosomal RNA of Escherichia coli. III. Purification and properties of the methylating enzyme and methylase-30 S interactions. J Biol Chem. 1979a;254:9094–9100. [PubMed] [Google Scholar]
- Poldermans B, Van Buul CP, Van Knippenberg PH. Studies on the function of two adjacent N6,N6-dimethyladenosines near the 3’ end of 16 S ribosomal RNA of Escherichia coli. II. The effect of the absence of the methyl groups on initiation of protein biosynthesis. J Biol Chem. 1979b;254:9090–9093. [PubMed] [Google Scholar]
- Pues H, Bleimling N, Holz B, Wolcke J, Weinhold E. Functional roles of the conserved aromatic amino acid residues at position 108 (motif IV) and position 196 (motif VIII) in base flipping and catalysis by the N6-adenine DNA methyltransferase from Thermus aquaticus. Biochemistry. 1999;38:1426–1434. doi: 10.1021/bi9818016. [DOI] [PubMed] [Google Scholar]
- Rife JP, Moore PB. The structure of a methylated tetraloop in 16S ribosomal RNA. Structure. 1998;6:747–756. doi: 10.1016/s0969-2126(98)00076-8. [DOI] [PubMed] [Google Scholar]
- Roth M, Helm-Kruse S, Friedrich T, Jeltsch A. Functional roles of conserved amino acid residues in DNA methyltransferases investigated by site-directed mutagenesis of the EcoRV adenine-N6-methyltransferase. J Biol Chem. 1998;273:17333–17342. doi: 10.1074/jbc.273.28.17333. [DOI] [PubMed] [Google Scholar]
- Schluckebier G, Kozak M, Bleimling N, Weinhold E, Saenger W. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and Sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J Mol Biol. 1997;265:56–67. doi: 10.1006/jmbi.1996.0711. [DOI] [PubMed] [Google Scholar]
- Schluckebier G, Zhong P, Stewart KD, Kavanaugh TJ, Abad-Zapatero C. The 2.2 Å structure of the rRNA methyltransferase ErmC’ and its complexes with cofactor and cofactor analogs: implications for the reaction mechanism. J Mol Biol. 1999;289:277–291. doi: 10.1006/jmbi.1999.2788. [DOI] [PubMed] [Google Scholar]
- Schubot FD, Chen CJ, Rose JP, Dailey TA, Dailey HA, Wang BC. Crystal structure of the transcription factor sc-mtTFB offers insights into mitochondrial transcription. Protein Sci. 2001;10:1980–1988. doi: 10.1110/ps.11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- Seidel-Rogol BL, McCulloch V, Shadel GS. Human mitochondrial transcription factor B1 methylates ribosomal RNA at a conserved stem-loop. Nat Genet. 2003;33:23–24. doi: 10.1038/ng1064. [DOI] [PubMed] [Google Scholar]
- Skinner R, Cundliffe E, Schmidt FJ. Site of action of a ribosomal RNA methylase responsible for resistance to erythromycin and other antibiotics. J Biol Chem. 1983;258:12702–12706. [PubMed] [Google Scholar]
- Thammana P, Held WA. Methylation of 16S RNA during ribosome assembly in vitro. Nature. 1974;251:682–686. doi: 10.1038/251682a0. [DOI] [PubMed] [Google Scholar]
- Tokuhisa JG, Vijayan P, Feldmann KA, Browse JA. Chloroplast development at low temperatures requires a homolog of DIM1, a yeast gene encoding the 18S rRNA dimethylase. Plant Cell. 1998;10:699–711. doi: 10.1105/tpc.10.5.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr, D. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- Van Buul CP, Damm JB, Van Knippenberg PH. Kasugamycin resistant mutants of Bacillus stearothermophilus lacking the enzyme for the methylation of two adjacent adenosines in 16S ribosomal RNA. Mol Gen Genet. 1983;189:475–478. doi: 10.1007/BF00325912. [DOI] [PubMed] [Google Scholar]
- van Buul CP, van Knippenberg PH. Nucleotide sequence of the ksgA gene of Escherichia coli: comparison of methyltransferases effecting dimethylation of adenosine in ribosomal RNA. Gene. 1985;38:65–72. doi: 10.1016/0378-1119(85)90204-5. [DOI] [PubMed] [Google Scholar]
- van Gemen B, Twisk J, van Knippenberg PH. Autogenous regulation of the Escherichia coli ksgA gene at the level of translation. J Bacteriol. 1989;171:4002–4008. doi: 10.1128/jb.171.7.4002-4008.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Knippenberg PH, Van Kimmenade JM, Heus HA. Phylogeny of the conserved 3’ terminal structure of the RNA of small ribosomal subunits. Nucleic Acids Res. 1984;12:2595–2604. doi: 10.1093/nar/12.6.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisblum B. Erythromycin resistance by ribosome modification. Antimicrob Agents Chemother. 1995;39:577–585. doi: 10.1128/AAC.39.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcock DF, Dryden DT, Murray NE. A mutational analysis of the two motifs common to adenine methyltransferases. Embo J. 1994;13:3902–3908. doi: 10.1002/j.1460-2075.1994.tb06701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM, Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- Xu Z, O’Farrell HC, Rife JP, Culver GM. A conserved rRNA methyltransferase regulates ribosome biogenesis. Nat Struct Mol Biol. 2008;15:534–536. doi: 10.1038/nsmb.1408. [DOI] [PubMed] [Google Scholar]
- Yu L, Petros AM, Schnuchel A, Zhong P, Severin JM, Walter K, Holzman TF, Fesik SW. Solution structure of an rRNA methyltransferase (ErmAM) that confers macrolide-lincosamide-streptogramin antibiotic resistance. Nat Struct Biol. 1997;4:483–489. doi: 10.1038/nsb0697-483. [DOI] [PubMed] [Google Scholar]