

Abstract
Motivation: The growing availability of genome-scale datasets has attracted increasing attention to the development of computational methods for automated inference of functional similarities among genes and their products. One class of such methods measures the functional similarity of genes based on their distance in the Gene Ontology (GO). To measure the functional relatedness of a gene set, these measures consider every pair of genes in the set, and the average of all pairwise distances is calculated. However, as more data becomes available and gene sets used for analysis become larger, such pair-based calculation becomes prohibitive.
Results: In this article, we propose GS2 (GO-based similarity of gene sets), a novel GO-based measure of gene set similarity that is computable in linear time in the size of the gene set. The measure quantifies the similarity of the GO annotations among a set of genes by averaging the contribution of each gene's GO terms and their ancestor terms with respect to the GO vocabulary graph. To study the performance of our method, we compared our measure with an established pair-based measure when run on gene sets with varying degrees of functional similarities. In addition to a significant speed improvement, our method produced comparable similarity scores to the established method. Our method is available as a web-based tool and an open-source Python library.
Availability: The web-based tools and Python code are available at: http://bioserver.cs.rice.edu/gs2.
Contact: troy.ruths@rice.edu
1 INTRODUCTION
Genomic analysis based on multiple species' genomes and gene interaction networks generated by high-throughput technologies is making very large gene set analysis commonplace. Given such large datasets, a major point of investigation among researchers is functional similarity and divergence among groups of genes within and across species, biological processes and cell types (Lamb et al., 2006; Lein et al., 2007; Su et al., 2002). Such research is aided by the use of ontologies which provide unified vocabularies to describe genes and their classifications (e.g. Ashburner et al., 2000; Kanehisa, 1997; Kanehisa and Goto, 2000; Khatri et al., 2005). Here, we focus on the Gene Ontology (GO) (Ashburner et al., 2000) which was introduced to provide a vocabulary that encodes various functional characteristics of genes and has been widely adopted within the biology community. The GO classification system classifies a gene according to how its products (i.e. RNA and proteins) behave. This behavior is characterized in three orthogonal categories: the cellular components it belongs to, the biological processes it is involved in and the molecular functions it performs. These three aspects of gene activity provide a way of characterizing and quantifying similar functions among genes.
There are three main categories of tools used for assessing functional similarity among genes based on their GO identifiers: GO browsers, gene list annotation and statistical tools, and computational similarity measures (a complete list of available tools can be found at http://geneontology.org/GO.tools.shtml).
GO browsers, such as AmiGO (http://amigo.geneontology.org/) and QuickGO (http://www.ebi.ac.uk/ego/), provide information retrieval capabilities, allowing for manual comparisons of genes and their annotations. These tools produce output through visualizations and textual data that the scientist can use to manually gauge similarities among genes. The actual assessment of similarity is left up to the interpretation of the researcher. Though this can allow the biologist to judge similarity most precisely, the manual nature of such approaches makes using them to analyze large gene sets infeasible.
One approach to addressing this scalability problem is the use of statistics to summarize the distribution of GO annotations within a gene set: gene functional similarities can be judged from the probability and density of occurrences. Popular tools, such as eGOn (Beisvag et al., 2006) and DAVID (Huang et al., 2007) provide web-based tools for this kind of analysis. These packages offer several similarity heuristics for gene lists that visualize and quantify the distribution of the gene list on the entire GO data structure. eGOn additionally allows for hypothesis testing of GO category representation. Bingo (Maere et al., 2005) and Ease (Huang et al., 2007) accept a gene set and calculate the GO term enrichment: the functional GO ‘themes’ in the gene list, or overrepresented parent terms in the GO hierarchy. GOTM (Zhang et al., 2004) also supports identifying enriched biological themes, in addition to providing a visual explorer of the GO tree created by a gene list. The proliferation of these tools and the number of reported citations they receive (over 800 for DAVID as of November 2008) underscore the usefulness of analyzing large-scale gene sets using GO. However, while all these tools let the researcher identify common GO terms and statistics for a gene set, none provides a formal similarity measure that allows for automated, comparable analysis of gene sets or clusters produced by microarrays, etc. Thus, while statistical tools allow scientists to explore trends in large data sets, they still do not permit automated inference of functional similarity among large sets of genes.
In order to address this need for automated tools, methods have emerged that compute pairwise similarity of genes based on their GO annotations. The first methods used for comparison were developed for other semantic taxonomies, mostly lexical taxonomies (Jiang and Conrath, 1997; Lin, 1998; Resnik, 1999). These methods determine the similarity of two genes based on their distance to the closest common ancestor and the annotation statistics of their common ancestor terms. In a recent study addressing the applicability of these measures to the GO (Sevilla et al., 2005), the method of Resnik (1999) was found to be the most accurate; however, they did not provide any direct biological evidence of the functional similarity. This gap was bridged by the semantic similarity measure of Wang et al. (2007), which used weights to quantify the different types of relationships encoded in the GO data structure. Their method targeted the drawbacks of the lexical measures with respect to shallow annotations, requiring annotation statistics, and addressing the semantic relationships expressed through edges of the GO data structure. Using several biological case studies, Wang et al. (2007) demonstrated better performance than the method by Resnik (1999) in comparison with the ground truth estimated manually by biologists. However, while this method provides accurate estimates of functional similarity based on GO annotation similarity, it is pairwise and does not scale well to large gene sets. As we show in our results section, even computing annotation similarity on sets of 300 genes results in extremely long compute times.
Genomic analysis based on multiple species' genomes and gene interaction networks generated by high-throughput technologies is making very large gene set analysis commonplace. As a result, it is important to have efficient tools for estimating the functional relatedness for these large sets.
In this article, we propose GS2 (GO-based similarity of gene sets), an efficient GO-based measure of functional similarity of a gene set. The method operates in linear time in the size of the gene set under study. We compared our method with the leading GO pairwise measure of Wang et al. (2007) (extended appropriately to yield set-based values). Our method provides similar accuracy to that of Wang et al. (2007), yet much faster. This makes our method very appropriate for large-scale studies of gene sets and their GO annotations.
2 METHODS
In this section, we describe GS2, our measure of similarity of a gene set based on the GO terms used to annotate these genes. To compute the similarity of a gene set using the established similarity measures, all of which are pairwise measures, one has to compute all pairwise distances of the set and average their sum by the number of pairs. On the other hand, our measure is inherently set-wise, and provides significant gains in computational efficiency over the standard pairwise measures applied to a set.
2.1 GO vocabulary structure
GO provides a functional vocabulary for genes in terms of biological process, cellular component and molecular function. Each gene has a set of GO annotations that convey functionality through these three inter-related ontologies. The GO tree, as it is referred to in literature, is encoded as a collection of three directed acyclic graphs (DAGs), each representing a different ontology. While largely disconnected, these ontologies can be connected by edges representing regulation relationships. A term in the GO tree represents an annotatable concept, and is related to other terms in the tree largely through is-a semantics; however, other relationship types occur in the GO including part-of, regulates, positively regulates and negatively regulates. All these relationships manifest as directed edges in the graph, and each term in the tree must follow the true path rule: ‘the pathway from a child term all the way up to its top-level parent(s) must always be true’ (http://www.geneontology.org).
For example, Figure 1 displays the relationships of GO terms on the path from cellular structure morphogenesis (GO:0032989) to the biological process root. This is a subgraph of the biological process ontology, in that there exist other terms that point towards developmental process (GO:0032502) or cellular process (GO:0009987) but they are not part of the inheritance of cellular structure morphogenesis. While most relationships are is-a, there is one part-of edge connecting anatomical structure morphogenesis to anatomical structure development. Figure 2, the GO subgraph induced by the paths connecting regulation of transcription, DNA-dependent (GO:0006355) to the GO root, is far more complex and demonstrates the need for computational analysis methods.
Fig. 1.

An example of the GO DAG structure between the biological process root and GO:0032989 (cellular structure morphogenesis).
Fig. 2.

The inheritance graph of GO : 0006355 (regulation of transcription, DNA-dependent). The number of inheritance terms can grow rapidly; in this case there are 35 members in the graph.
2.2 Gene set similarity
Our method calculates the similarity of a set of genes based on their GO annotations. Throughout this article, we use gi to denote a gene and Gi to denote the set of GO terms that annotate it. Given a universal set 𝒰 of genes, and a set 𝒢={g1, g2,…, gk} of k genes, 𝒢⊆𝒰, the goal is to derive a measure m: 2𝒰 → [0, 1] such that m(𝒢) is the functional similarity of the gene set 𝒢.
A straightforward way to obtain such a measure is to use any of the pairwise similarity measures discussed in the introduction on the set. More formally, let m′ : 𝒰 × 𝒰 → [0, 1] be any pairwise measure, e.g. the measure of Wang et al. (2007). We define m(𝒢) as
| (1) |
This pair-based calculation of the similarity of gene set 𝒢 takes time O(|𝒢|2 · ℓ), where ℓ is the time it takes to compute m′ on a pair of genes.
As we discussed above, inferring the functional similarity of large gene sets using this calculation may become prohibitive. We now describe our measure GS2 that is computable in linear time in the size of the gene set, thus providing a linear speedup over the pair-based calculation.
2.2.1 Definitions
We now briefly describe the GO tree structure that we will use in defining our method. Given a GO tree (DAG) T = (V, E), where V is the set of the nodes and E is the set of directed edges, with set of roots R⊂V for each ontology, we define A{i}, the set of ancestors of term i, to be the set containing all terms that are on the path from i to a root, including term i itself.
This definition can be generalized to a set ℐ of GO terms in a straightforward manner.
Intuitively, the ancestors of a set ℐ of GO terms are all terms lying on the paths from terms in ℐ to a root in R. Note that since there are three roots in the GO tree, the ancestors of terms in different ontologies will terminate at different roots. In general such ancestor sets will not share terms, with the exception of regulates relationships which can link terms in different ontologies. Ancestors will be useful in calculating how many of the genes in 𝒢 share common functionality, which is expressed in terms of the shared inner terms of the GO tree. For each GO term i, we associate the set of genes in 𝒢 that are annotated by GO terms which are ancestors of i; we call the size of this set the rank of term i with respect to set 𝒢, and denote it by Rank𝒢(i). Formally,
| (2) |
As terms are chosen closer to the root in the GO tree, more genes will share the fundamental functionality. Consequently, this rank will be useful in describing the distribution of genes with respect to functionality. The maximum value of Rank𝒢(i) of a term i is |𝒢|, and the minimum size is 0 (i.e. no genes have that functionality).
2.2.2 The GS2 measure
Our measure averages the similarity contributed by each gene in 𝒢. Each gene is compared with the remaining set of genes by calculating how closely that gene follows the functionality distribution of the remaining genes. The functionality distribution is represented by the distribution of ancestor GO terms for each gene. We will use the rank set [Equation (2)] to quantify the distribution of ancestor terms.
| (3) |
where
| (4) |
Our GS2 similarity measure [Equation (3)] averages the comparison values for each gene against the rest of the set. This leaves most of the work in the Comp function, which compares the given gene, gi, to the remainder set of genes, that is 𝒢 − {gi}.
It is important to note that our method is defined for sets of genes with at least two members, so that we never compare a gene with an empty set of genes. With this said, we compute the pairwise distance of two genes by creating a gene set with only those two genes as members.
In comparing one gene (target) with a set of genes (source), ideally we want to return a value of 1 when the target and source genes share all the same GO annotations, and consequently all the same ancestor terms of those annotations. Having identical ancestor sets implies identical functionality in terms of the GO tree; therefore, a value of 1 implies identical functionality. This will only happen when all genes map to the same set of GO terms.
Our method employs a simple counting scheme to measure the comparison between target and source genes. We average the contribution by each annotated term of the target. For each annotated term, we calculate how similar its ancestor set is in comparison to the ancestors of the source genes. This is accomplished by counting the number of source genes that share each ancestor term of the annotation. We have defined this already as the rank, and normalize the value by the maximum possible rank, which is |𝒢|. This value is then averaged over all ancestor terms of the annotation. Since Comp returns a value between 0 and 1, the average of comparisons for each gene will yield a value between 0 and 1 as well. The value of GS2 shares the same intuition as Comp; if all genes have high comparison values (each gene is similar to all other genes), then the similarity of the set should be high. Likewise, low comparison values will yield a low set similarity value. It is important to note that our measure quantifies similarity based on graph connectivity rather than edge types, which means GS2 is robust to new relationships introduced into the GO.
To illustrate our method, consider genes G1 = {GO:0016043}, G2 = {GO:0048869, GO:0009653}, G3 = {GO:0032989}, where 𝒢 = {g1, g2, g3 }. These GO terms correspond to the inheritance graph in Figure 1. In order to compute GS2(𝒢), we must first calculate the rank value for each term in the graph. This calculation is shown in Figure 3, where annotated terms for each gene are labeled and highlighted, and the number to the left of the GO accession number is the rank value for each term. Since our method is robust to relationship types, edge labels are elided. According to Equation (3), we average the contribution of each gene (g1, g2, g3). To compute this contribution, we calculate the rank set for each ancestor term per annotated term. Recall that Comp compares one gene to a set of genes, or in our example, to the two other genes. Instead of recalculating the rank values for each call to Comp, we can just subtract one from the rank calculation with all genes present. This emulates removing the target gene from the set in the Comp calculation. Also, we need to decrease the size of the source gene set by one as well. In order to calculate Comp for g1:
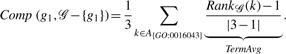 |
Table 1 shows the computation of the Comp value for each gene. As you can expect, the Comp value for g1 is the highest since it proportionally shares the most functionality with genes g2 and g3. Since we are dealing with shallow annotations, we can expect the fidelity of the method to decrease. The final computation of GS2 follows:
Fig. 3.

The counting phase of GS2 applied to three genes. The annotation term(s) for each gene are highlighted by a black box. In this case gene 2 has annotations: GO : 0048869 and GO : 0009653. The rank of each term with respect to the entire set is the number to the left of the GO identifier.
Table 1.
The legwork for the Comp calculation of an example gene set
| Gene | Term | Ancestors | Rank | Term avg. | Comp |
|---|---|---|---|---|---|
| g1 | GO:0016043 | GO:0016043 | 1 | 0.83 | 0.83 |
| GO:0009987 | 2 | ||||
| GO:0008150 | 2 | ||||
| g2 | GO:0048869 | GO:0048869 | 1 | 0.75 | 0.69 |
| GO:0009987 | 2 | ||||
| GO:0032502 | 1 | ||||
| GO:0008150 | 2 | ||||
| GO:0009653 | GO:0009653 | 1 | 0.63 | ||
| GO:0048856 | 1 | ||||
| GO:0032502 | 1 | ||||
| GO:0008150 | 2 | ||||
| g3 | GO:0032989 | GO:0032989 | 0 | 0.56 | 0.56 |
| GO:0016043 | 1 | ||||
| GO:0048869 | 1 | ||||
| GO:0009987 | 2 | ||||
| GO:0032502 | 1 | ||||
| GO:0009653 | 1 | ||||
| GO:0048856 | 1 | ||||
| GO:0008150 | 2 |
2.2.3 Complexity
As we now show, our method is computable in time O(md|𝒢|), where 𝒢 is the gene set, m is the maximum number of GO annotation terms per gene, and d is the maximum size of an ancestor set. For large gene sets, which is the emphasis in this work, we have m ≪ |𝒢| and d ≪ |𝒢|; therefore, for large gene sets, GS2 is computable in O(|𝒢|) time.
The time complexity of GS2 comes from two steps: computation of the ancestors and ranks, and computation of similarity.
In our implementation of GS2, we simultaneously precompute the rank [Equation (2)] and cache each ancestor set per GO term. In the worst case, no gene shares GO terms with any other gene and each GO term has a unique path to the root of the ontologies. This means we need to compute the rank set for each of these unique terms. Since each gene has O(m) terms with O(d) ancestors, there are O(md|𝒢|) rank values we need to precompute. The rank, however, can easily be computed with an O(1) operation while constructing the ancestor sets. Basically, we maintain a mapping for each GO term to the number of genes sharing that term. As we generate the ancestor sets for each gene in O(md|𝒢|) time, we increment the mapping. Ultimately, this mapping is the size of the rank, generated in O(md|𝒢|).
The computation of similarity uses two equations, one for the comparison [Equation (4)] and another for the similarity [Equation (3)]. Since we have precomputed both the ancestors set and rank values, Equation (4) requires O(md) time, since it loops over the terms of the gene and then the ancestors for each of those terms. Retrieving the ancestor set and the ranks costs O(1). Equation (3), then, is computable in O(md|𝒢|) time, since it computes the comparison value for each gene in the set.
Therefore, the time complexity of GS2 is O(md|𝒢|). For large gene sets, such as the ones we are interested in analyzing, we have m ≪ |𝒢| and d ≪ |𝒢|; therefore, for such gene sets, the time complexity of the method is dominated by the size of the gene set, which is O(|𝒢|).
Further, compared with the pairwise-based calculation of the functional similarity of a gene set, such as the one described in Equation (1), the GS2 method provides an O(|𝒢|) improvement.
3 RESULTS
To evaluate the performance of the GS2 measure, in terms of the similarity it measures and the efficiency of computing it, we conducted experiments on large sets of genes and compared the performance of our method with that of a pairwise-based similarity measure of gene sets. Equation (1) was used to compute set similarity from the pairwise measure.
Data: in our experiments, we used annotated genes from the human genome. Since GO annotations express functionality across many species, the human genome uses roughly 6000 unique identifiers out of about 27 000 terms in the entire GO tree. To verify that the human genome did not provide a significant sampling bias of the GO tree, we sampled at random 2000 unique GO terms and for each term measured the frequency of edge types on the path from that term to a root. The frequency of each edge type was averaged over 20 trials using multiple GO term distributions: Mus musculus, Danio rerio, Drosophila melanogaster and the entire GO tree. Figure 4 shows a doughnut plot of the relative edge type frequencies across the different organisms.
Fig. 4.

The relative frequencies of the different edge types across several GO term distributions: Homo sapien, M.musculus, D.rerio, D.melanogaster and the entire GO tree. Each ring in the doughnut plot represents a different distribution.
We downloaded the daily snapshot of the GO tree from the GO web site on September 28, 2008. We downloaded the human gene dataset and their GO annotations from the Ensembl BioMart (http://www.ensembl.org/biomart/martview) on September 1, 2008.
Implementation: we implemented the GS2 similarity measure in Python (http://www.python.org). Our decision to use Python rested on the language's strong scientific libraries, rapid prototyping capabilities and ease of transitioning code between computers. Transitioning code was important because we used Rice University's supercomputer to run many of the tests. Since Python is an interpreted language, it is more important to focus on the relative times between methods rather than the absolute running times.
For comparison purpose, we implemented the semantic pairwise similarity measure of Wang et al. (2007) in Python as well. We chose to use this method for comparison because it demonstrated higher accuracy than the method of Resnik (1999), which, in the study by Sevilla et al. (2005), outperformed all other similarity measures applicable to the GO structure. In short, the method of Wang et al. (2007) provides the highest accuracy of GO similarity and provided a clear benchmark against which to compare our method. While online tools for this method were created for both pairwise and set comparison, an accessible web API was not available. When considering the number of tests we wanted to run, and the need to compare runtimes, we opted for a local implementation. For all weighted tests we used the values recommended by Wang et al. (2007) of 0.8 and 0.6 for the is-a and part-of relationships in the GO tree; however, we also compared our method with the unweighted version, where the weights for is-a and part-of relationships were both set to 1. To obtain gene set similarity values, we used this pairwise measure as described in Equation (1). Since Wang et al. (2007) did not suggest tested values for regulates relationships, we disregarded them in the calculations; however, our method is robust to their inclusion.
Testing accuracy: we designed an experiment to test the accuracy of GS2 in comparison to the semantic pairwise method of Wang et al. (2007). This experiment also allowed us to analyze time efficiency with respect to the average number of terms per gene in the set. In this experiment, we started with sets of highly similar genes and gradually degraded the set similarity by introducing larger and larger percentages of random genes: if we start with a similar set of genes, replacing a percentage of those genes with randomly selected ones will cause the similarity of the set to decrease. On the other hand, if we start with a random set, replacement with random genes should have little effect on the similarity of the set. We chose our sets of similar genes by selecting a prototype gene around which to build the set. Then genes with exactly a certain number of GO terms in common with a prototype gene were selected. In the resulting set, each gene may have a varying total number of GO terms, but all share an exact number of GO terms with the prototype gene. In order to introduce dissimilarity, we replaced genes in this similar set with genes selected at random from the Ensembl human gene databank.
In our experiment, we measured the similarity of gene sets with seven, eight and nine shared GO terms. For each of these, we introduced an increasing amount of random genes from 10% to 100% of the set size in increments of 10%, maintaining a set size of 100 genes. In this manner we started with similar sets but ended with completely random sets. We repeated this process 100 times. We kept track of the time for each method to calculate the similarity, the similarity reported, the average number of GO terms per gene and the average number of shared genes. Figure 5 plots the average similarity measured by the three different methods (pairwise with weights, pairwise without weights and GS2). Figure 6 plots the similarity measured by the pairwise semantic method with the similarity measured by our set method, and Figure 7 shows the speed boost of our set method over growing number of GO terms per gene. Note that these figures present the experimental results of gene sets with seven shared GO terms. Datasets with eight and nine shared GO terms yielded similar trends to those reported here.
Fig. 5.

The similarity measured by the three methods, on sets of 100 genes, with varying degrees of relatedness among the genes in a set. The x-axis shows the percentage of the gene set replaced by random genes. ‘Pairwise-based’ refers to the method by Wang et al. (2007) when plugged into the set similarity calculation given in Equation (1).
Fig. 6.

A comparison of the similarity computed by GS2 to that measured by the pairwise method of Wang et al. (2007). The dotted line is the 45○ line.
Fig. 7.

The time speedup of GS2 versus the pairwise method of Wang et al. (2007) as the number of GO terms per gene in the set increases. Values are normalized for comparison purposes.
Testing efficiency: we also designed a simple experiment to measure the performance of our method over varying gene set sizes. We calculated the similarity of random gene sets of size 50 to 3000 and recorded the time efficiency of our method. For comparison purposes, we projected the time cost of the pairwise method based on our results to the previous experiment. See Figure 8 for the results of this experiment.
Fig. 8.

The time efficiency of GS2 over the pairwise method of Wang et al. (2007) as the size of gene sets increases. Outliers are due to garbage collection in Python.
4 DISCUSSION
We analyzed the quality of GS2 measurements in two ways. First, we compared our method with an established method in the area (Wang et al., 2007), reasoning that returning results similar to this measure would at least capture the operational definition of similarity. Even though there are several established methods for pairwise GO similarity, the size of the gene sets prohibited a comparison to each one; rather, we chose the best performing method in terms of biological similarity. Wang et al. (2007) specifically compared their method with the method of Resnik (1999), which was found to provide the highest accuracy measure by Sevilla et al. (2005). Second, we designed our experiments in such a way that the similarity of genes was guaranteed to decrease: though we cannot assert the correct similarity value for a set of genes, we can say that a set of genes that share many GO terms in common is very likely to be more functionally similar than a set of genes whose annotations hardly overlap at all. In our experiment, we employed this reasoning by starting with a set of genes that shared many GO terms in common. We then introduced increasing amounts of random genes into this set by replacement (removing a certain number of ‘similar’ genes and replacing them with the same number of randomly chosen genes). As can be seen in Figure 5, the established method returned decreasing similarity scores as more random genes were added, confirming that our experimental methodology achieved its intended purpose.
In terms of accuracy, we can see in Figure 5 that our method closely follows the trend of the pairwise semantic similarity method. All methods demonstrated expected similarity to the introduction of random genes. However, note that the similarity values computed by GS2 mirrored the descent of the weighted pairwise method rather than the unweighted calculation. In Wang et al. (2007), sensitivity to semantic relationships (is-a and part-of) was identified as being important to correct estimation of functional similarity. It is notable that our method does not explicitly weight these semantic relationships, but nonetheless, performs similarly to methods that use them. To investigate this further, we plotted the weighted pairwise measure against the set similarity measure. We discovered that the covariance of the these distributions was close to zero (0.038), and the measurements were closer at low similarity values and farther at higher similarity ones. Figure 6 displays this high correlation. Therefore, in terms of accuracy, our method not only performs on par with established methods, but also demonstrates sensitivity to semantic relationships without explicitly using them. This latter point suggests that at the scale of large gene sets (in this case 100 genes), semantic relationships are either less important to consider than previously thought or somehow encoded in the structure of the tree. We suspect that the former condition holds for the GO tree.
Since the annotations of the human genome constitute a fraction of the total GO topology, our similarity calculations could be effected by the bias of the human GO subgraph. Since our measure does not specifically weight different semantic relationships, if human genes are annotated with terms that use is-a relationships more frequently than the rest of the GO tree, then our similarity results would not extend to other organisms that carry a different bias. To test this, we sampled the frequency of GO edge types from several GO term distributions. As shown in Figure 4, all distributions had nearly identical frequencies across all edge types. As expected, is-a occurs with the highest frequency (roughly 85%) across all distributions with part-of a distant second. This highlights two important points. First, the distribution of edge types is similar for the selected organisms as well as the entire GO tree, and consequently our method will extend to different GO distributions. Second, since the frequency of is-a relationships for large gene sets eclipses other edge types, similarity is a derivative of topology rather than edge semantics. This explains why our method parallels the performance of semantic methods without explicitly weighting edge types.
Our method also proved to be very efficient not only with respect to set size, but also in terms of the number of GO terms. As shown in Figure 7, as the number of GO terms increases, so does the efficiency boost provided by GS2. We also observe the effectiveness of our method over large sets. In Figure 8, our method takes 0.3 seconds to calculate the similarity for a set of 3000 genes. The outliers in the plot resulted from garbage collection in Python. In comparison, in the same amount of time, the pairwise method can calculate the similarity for a set with eight genes only.
It is important to note that the performance increase of our method over the pairwise method of Wang et al. (2007) would hold over all set similarity measures that are inherently pairwise-based computation. As the size of the set increases, the calculation time increases quadratically. This results in millions of calculations rather than the thousands we manage to compute while still preserving high-quality similarity measurements.
5 CONCLUSIONS
In this article, we provide an efficient and accurate gene set similarity measure, GS2. In addition to measuring similarity at remarkably fast speeds, our method performed on par with semantic methods without explicit modeling of semantics, such as in weighting GO-term relationships. A web-based implementation of GS2 is available at http://bioserver.cs.rice.edu/gs2.
ACKNOWLEDGEMENTS
The authors would like to thank the two anonymous reviewers for comments that helped improve the quality of this manuscript. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the Department of Energy (DOE), National Science Foundation, National Library of Medicine or the National Institutes of Health.
Funding: Department of Energy (DE-FG02-06ER25734); National Science Foundation (CCF-0622037, CCF-0829276); National Library of Medicine (R01LM009494); Rice Computational Research Cluster funded by National Science Foundation (CNS-0421109); partnership between Rice University, Advanced Micro Devices and Cray.
Conflict of Interest: none declared.
REFERENCES
- Ashburner M, et al. Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisvag V, et al. Genetools—application for functional annotation and statistical hypothesis testing. BMC Bioinformatics. 2006;7:470. doi: 10.1186/1471-2105-7-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, et al. David bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Conrath D. Proceedings of International Conference Research on Computational Linguistics (ROCLING X). Taiwan: 1997. Semantic similarity based on corpus statistics and lexical taxonomy. [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. A database for post-genome analysis. Trends Genet. 1997;13:375–376. doi: 10.1016/s0168-9525(97)01223-7. [DOI] [PubMed] [Google Scholar]
- Khatri P, et al. A semantic analysis of the annotations of the human genome. Bioinformatics. 2005;21:3416–3421. doi: 10.1093/bioinformatics/bti538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Lein E, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lin D. Fifteenth International Conference on Machine Learning. San Francisco, CA: Morgan Kaufmann; 1998. An information-theoretic definition of similarity, semantic similarity based on corpus statistics and lexical taxonomy; pp. 296–304. [Google Scholar]
- Maere S, et al. Bingo: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Resnik P. Semantic similarity in a taxonomy: an information-based measure and its application to problems of ambiguity in natural language. J. Artif. Intell. Res. 1999;11:93–130. [Google Scholar]
- Sevilla J, et al. Correlation between gene expression and go semantic similarity. IEEE/ACM Trans. Comput. Biol. Bioinform. 2005;2:330–338. doi: 10.1109/TCBB.2005.50. [DOI] [PubMed] [Google Scholar]
- Su A, et al. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl Acad. Sci. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. A new method to measure the semantic similarity of go terms. Bioinformatics. 2007;23:1274–1281. doi: 10.1093/bioinformatics/btm087. [DOI] [PubMed] [Google Scholar]
- Zhang B, et al. GOTree Machine (GOTM): a web-based platform for interpreting sets of interesting genes using gene ontology hierarchies. BMC Bioinformatics. 2004;5:16. doi: 10.1186/1471-2105-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]