

A method to modify proteins with glutaraldehyde under reducing conditions is presented. Treatment with glutaraldehyde and dimethylaminoborane was found to result in cyclic pentylation of free amines and facilitated the structural determination of a protein previously recalcitrant to the formation of diffraction quality crystals.
Keywords: pentapeptide-repeat proteins, chemical modification, glutaraldehyde, reductive cyclic pentylation
Abstract
The pentapeptide-repeat protein EfsQnr from Enterococcus faecalis protects DNA gyrase from inhibition by fluoroquinolones. EfsQnr was cloned and purified to homogeneity, but failed to produce diffraction-quality crystals in initial crystallization screens. Treatment of EfsQnr with glutaraldehyde and the strong reducing agent borane–dimethylamine resulted in a derivatized protein which produced crystals that diffracted to 1.6 Å resolution; their structure was subsequently determined by single-wavelength anomalous dispersion. Analysis of the derivatized protein using Fourier transform ion cyclotron resonance mass spectrometry indicated a mass increase of 68 Da per free amino group. Electron-density maps about a limited number of structurally ordered lysines indicated that the modification was a cyclic pentylation of free amines, producing piperidine groups.
1. Introduction
Crystallization remains a major bottleneck in the determination of the structure of proteins by X-ray methods. A number of methods are increasingly being utilized to increase the probability that a given protein will form diffraction-quality crystals (see Derewenda, 2004b ▶, and references therein). Surface-exposed residues play an inordinately large role in the formation of a stable crystal lattice, as these are the residues which are most accessible for the formation of intermolecular contacts (McElroy et al., 1992 ▶; D’Arcy et al., 1999 ▶). Since surface residues are typically the least conserved residues amongst orthologous proteins, a highly successful first approach is to attempt the crystallization of species variants of the protein of interest (Kendrew et al., 1954 ▶; Campbell et al., 1972 ▶). This essentially shuffles the properties of the entire surface, resulting in a fresh screening of crystallization space without altering the general information held in the core structures of the protein. Secondly, the molecular constructs can be altered by, for example, changing the type or position of affinity tags or removing flexible regions of the structure (Gao et al., 2005 ▶; Carson et al., 2007 ▶; Graslund et al., 2008 ▶). However, even then proteins may remain recalcitrant to crystallization and/or one may wish not to change species or constructs if a large body of biophysical work has already been amassed on a particular protein construct. The mutation of surface residues with high conformational entropy (Lys, Glu, Asn) to alanines has been found to increase the likelihood of crystallization (Longenecker et al., 2001 ▶; Czepas et al., 2004 ▶; Derewenda, 2004a ▶; Derewenda & Vekilov, 2006 ▶). Although these mutations can have numerous biophysical effects (e.g. altering solubility and surface charge), it is often subsequently found that these residues participate directly or indirectly in crystal contacts. For proteins with an unknown structure, the surface-entropy reduction prediction (SERp) server can be utilized to suggest high-entropy residues which are in close proximity in the sequence and are likely to be on the surface (Goldschmidt et al., 2007 ▶). Finally, one may want a quicker and more direct method of altering the surface properties of a particular protein construct or several variants produced by previously mentioned methods. The reductive dimethylation of free amino groups (lysines, NH2-termini) with formaldehyde and strong reducing agents (sodium borohydride or borane–dimethylamine) has been successfully used to crystallize recalcitrant proteins (Rypniewski et al., 1993 ▶; Kobayashi et al., 1999 ▶; Kurinov & Uckun, 2003 ▶; Schubot & Waugh, 2004 ▶; Walter et al., 2006 ▶; Rauert et al., 2007 ▶; Shaw et al., 2007 ▶; Kim et al., 2008 ▶). Through the modification of the high surface-entropy residue lysine, reductive dimethylation of a protein can significantly alter the surface properties at several sites, which can result in novel crystal contacts and/or change the crystallization kinetics.
The chromosomally encoded Enterococcus faecalis protein EfsQnr has been demonstrated to provide intrinsic resistance to fluoroquinolones (Arsene & Leclercq, 2007 ▶), presumably through its interaction with DNA gyrase. EfsQnr belongs to a family of proteins termed pentapeptide-repeat proteins (PRPs), the repeating signature sequence (STAV)1(D,N)2(L,F)3(STR)4(G)5 of which folds into a right-handed quadrilateral parallel β-helix (Hegde et al., 2005 ▶; Buchko et al., 2006 ▶; Vetting et al., 2006 ▶). Here, we demonstrate a novel modification of free protein amines by treatment with glutaraldehyde in a strongly reducing environment. This modification facilitated the crystallization and structure determination of EfsQnr and may be broadly applicable to other proteins that resist crystallization.
2. Methods
2.1. Cloning and purification
Details of the cloning and purification of EfsQnr will be described elsewhere.
Briefly, the gene for EfsQnr (NCBI GeneID 1199794) was PCR-amplified from genomic DNA and ligated into the overexpression vector pET28a using NdeI and XhoI restriction sites, yielding a protein with an N-terminal thrombin-cleavable hexahistidine tag (tag sequence MGSSHHHHHHSSGLVPRGSH-Met1). EfsQnr was expressed in Mueller–Hinton broth at 298 K and induced overnight with 0.5 mM IPTG after the cells reached an A 600 of 0.6. Recombinant EfsQnr was purified to homogeneity by Ni–NTA chromatography followed by size-exclusion chromatography on a Superdex S75 column and the N-terminal His tag was cleaved using thrombin, leaving the native protein with an additional three amino acids at the N-terminus (GSH-Met1). EfsQnr was concentrated by ultracentrifugation to 20–50 mg ml−1 in 20 mM Tris pH 8.0, 200 mM NaCl and stored as a 50%(v/v) glycerol stock at 253 K.
2.2. Treatment with glutaraldehyde
Reductive cyclic pentylation of free amines is based on the protocols for reductive dimethylation utilizing formaldehyde and a strong reducing agent (Rayment, 1997 ▶). EfsQnr was extensively dialyzed against 50 mM HEPES pH 7.5. All experiments were carried out with EfsQnr at a concentration of 10 mg ml−1 (3.2 mM free amine) and were incubated on a rocking platform shaker at 277 K in the dark. Stock solutions of 1 M borane–dimethylamine (DMAB; Sigma 180238) and 0.5 M glutaraldehyde [diluted from 25%(w/v) stock; Sigma G-5882] were freshly prepared. 20 µl DMAB was added per millilitre of protein solution followed by addition of 40 µl glutaraldehyde per millilitre of protein solution. This was incubated for 2 h prior to the addition of another aliquot of DMAB (20 µl per millilitre) and glutaraldehyde (40 µl per millilitre). After an additional 2 h incubation period, a final aliquot of DMAB (10 µl per millilitre) was added prior to incubation overnight (∼16 h). The final concentrations of DMAB and glutaraldehyde were 50 and 40 mM, respectively. The reductive cyclic pentylation of lysozyme was performed in a similar manner using lysozyme (Sigma L6876) at 10 mg ml−1 (5 mM free amine).
Treated samples were diluted 1:10 in buffer A (100 mM HEPES, 1 M ammonium sulfate, 1 mM DTT, 1 mM EDTA pH 7.5), applied onto a Phenyl-Sepharose CL-4B (1.5 × 15 cm; GE Healthcare) column equilibrated with buffer A and eluted with a linear gradient from buffer A to buffer B (100 mM HEPES, 1 mM DTT, 1 mM EDTA pH 7.5) over ten column volumes. Peak fractions were pooled and dialyzed for 12 h against buffer C (15 mM HEPES, 1 mM DTT, 1 mM EDTA pH 7.5). Samples were concentrated to 10–20 mg ml−1 by Amicon filtration and snap-frozen in liquid nitrogen for storage at 193 K.
2.3. Crystallization
EfsQnr was crystallized by vapor diffusion under oil in 96-well round-bottom plates (Costar-3795). Equal volumes of EfsQnr (10 mg ml−1 in buffer C plus 400 mM ammonium sulfate) and precipitant [100 mM MES pH 6.0, 5–15%(w/v) PEG 6000, 1 M LiCl] were combined under 150 µl silicon oil (Fisher Scientific). Plates were stored at 277 K open to room humidity. Initial crystals, which appeared after significant incubation (1–2 months), were crushed and used as microseeds to streak-seed crystallization conditions identical to the initial screen (Stura & Wilson, 1991 ▶). Streak-seeded drops produced large thick plates that grew to maximum size (0.25 × 0.25 × 0.05 mm) in approximately 2–7 d. Crystals were incubated (30 s) in a solution of 100 mM MES pH 6.0, 15%(w/v) PEG 6000, 1.5 M LiCl prior to being mounted on 0.2 mm LithoLoops (Molecular Dimensions Ltd) and undergoing vitrification by plunging into liquid nitrogen. Diffraction data were collected at 100 K on a Rigaku RU-H3R Cu Kα rotating-anode generator (50 kV, 100 mA) equipped with an R-AXIS IV++ detector (Rigaku/MSC) and Osmic Blue confocal mirrors. Data were processed and scaled using MOSFLM (Leslie, 2006 ▶) and SCALA (Evans, 2006 ▶).
2.4. Structure determination
The structure of EfsQnr was determined by single-wavelength anomalous dispersion (SAD). A single crystal of EfsQnr was soaked for 120 s at 277 K in a cryosolution doped with 100 mM samarium acetate. Heavy-atom substructure determination and phasing (overall SAD figure of merit = 0.27) were performed using PHENIX (Adams et al., 2004 ▶). The automated fitting program ARP/wARP (Perrakis et al., 1997 ▶) was utilized to produce an initial model starting with the solvent-flattened SAD phases. This model was sufficient to place the structure of MfpA, a related pentapeptide-repeat protein, into the data, which guided further model building. The final model was built by iterative cycles of model building in Coot (Emsley & Cowtan, 2004 ▶) and positional and restrained individual B-factor refinement in REFMAC (Murshudov et al., 1997 ▶). The molecular-replacement program AMoRe (Navaza, 1994 ▶) and a partially built EfsQnr model were used to phase a high-resolution data set. Waters were added at sites exhibiting peaks in F o − F c (>3.5σ) and 2F o − F c (>1.0σ) maps if at least one hydrogen-bonding partner was present. There are two EfsQnr molecules per asymmetric unit, yielding a solvent content of 48.5%. Noncrystallographic symmetry restraints were not utilized during refinement owing to the high resolution of the data. Translation/libration/screw (TLS) refinement was used in the last steps of refinement (Winn et al., 2001 ▶). A total of four TLS groups were used per chain and the groups were determined by submission of the PDB file to the TLSMD server (Painter & Merritt, 2006 ▶). Model quality was checked using the MOLPROBITY web server (Davis et al., 2004 ▶). Data-collection and model statistics are shown in Table 1 ▶. OMIT maps were utilized to check for crystallographic evidence of cyclic pentylation. All lysines were exchanged for serines and all atoms underwent randomized deviations, yielding an r.m.s.d. of 0.4 Å for the entire model to the parent structure. The omitted and shaken model underwent positional and constrained B-factor refinement prior to synthesis of OMIT maps.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the outer shell.
| EfsQnr (modified) | Sm acetate | |
|---|---|---|
| Space group | P21 | P21 |
| Unit-cell parameters (Å, °) | a = 36.9, b = 63.1, c = 94.6, β = 96.1 | a = 36.6, b = 65.5, c = 94.1, β = 97.5 |
| Resolution (Å) | 20–1.6 (1.69–1.6) | 38–2.0 (2.11–2.0) |
| Completeness (%) | 94.7 (86.0) | 100 (100) |
| Redundancy | 2.9 (2.7) | 7.5 (3.9) |
| Mean (I)/σ(I) | 19.3 (3.1) | 20.3 (4.1) |
| Rmerge† | 0.043 (0.253) | 0.091 (0.322) |
| Wilson B factor (Å2) | 22.4 | 15.4 |
| Model and refinement data | ||
| Resolution (Å) | 20–1.6 (1.64–1.6) | |
| Unique reflections | 51172 (3093) | |
| Rcryst‡ (%) | 18.7 (36.0) | |
| Rfree‡ (5% of data) (%) | 22.1 (44.9) | |
| Contents of model | ||
| Residues (1–211 + tag) | A1–A8, A13–A211, B4–B211 | |
| Waters | 417 | |
| Other | 4 Cl | |
| Total atoms | 3910 | |
| Average B factor (Å2) | ||
| Protein | 13.6 | |
| Waters | 29.7 | |
| R.m.s.d. | ||
| Bond lengths (Å) | 0.014 | |
| Angles (°) | 1.59 | |
| MOLPROBITY statistics | ||
| Ramachandran favored/outliers (%) | 98.2/0.0 | |
| Rotamer outliers (%) | 1.1 | |
| Clashscore§ | 7.2 [85th percentile, N = 718, 1.35–1.85 Å] | |
| Overall score§ | 1.4 [92nd percentile, N = 7200, 1.35–1.85 Å] |
R
merge = 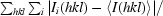
 , where I
i(hkl) is the intensity measurement for reflection i and 〈I(hkl)〉 is the mean intensity for multiply recorded reflections.
, where I
i(hkl) is the intensity measurement for reflection i and 〈I(hkl)〉 is the mean intensity for multiply recorded reflections.
R
work, R
free = 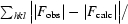
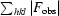 , where the working and free R factors are calculated using working and free reflections, respectively.
, where the working and free R factors are calculated using working and free reflections, respectively.
As calculated using the MOLPROBITY server; N is the number of sample structures in the resolution range.
2.5. Mass spectrometry
All mass spectra were acquired on a 9.4 T Fourier transform ion cyclotron resonance mass spectrometer (Ionspec, Lake Forest, California, USA). The protein was directly injected onto a C18 column at a rate of 10 µl min−1 in 50% acetonitrile containing 0.1% TFA.
3. Results
3.1. Crystallization of unmodified EfsQnr
The pentapeptide-repeat protein EfsQnr (211 amino acids) was purified to homogeneity utilizing Ni–NTA chromatography. EfsQnr was screened for crystallization utilizing approximately 600 solution conditions from various commercially available matrices (i.e. Hampton Research Crystal Screens I and II, PEG/Ion Screen etc.). Most of the conditions produced clear drops or precipitate, with only 2% of the conditions producing crystalline material. Over a period of 1–2 months, spherulite-shaped crystals appeared in drops containing medium-molecular-weight PEGs (2000–8000) in the pH range 5–7 (Fig. 1 ▶ a). These crystals could not be improved through seeding and were not large enough for determination of their space group or diffraction quality.
Figure 1.

Crystals of EfsQnr. (a) Crystals of unmodified EfsQnr. Crystals of reductively pentylated EfsQnr before (b) and after (c) crystal seeding.
3.2. Reductive pentylation
In an attempt to alter the crystallizability of EfsQnr, its lysine side chains were modified by chemical modification with glutaraldehyde under reducing conditions. EfsQnr was incrementally treated with glutaraldehyde in the presence of the strong reducing agent borane–dimethylamine (DMAB) over a period of 16 h. The reaction was quenched with 1 M ammonium sulfate and the protein was separated from free glutaraldehyde and DMAB by hydrophobic interaction chromatography (HIC). Hen egg-white lysozyme was treated using the same procedure in order to evaluate the universality of the technique. Neither EfsQnr nor lysozyme produced any precipitation at the protein concentrations used (10 mg ml−1) and protein recoveries were >80%, with no extraneous peaks in the HIC purification. SDS–PAGE of the proteins after modification suggested little or no intermolecular cross-linking (Fig. 2 ▶ a). Analysis by Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) was used to determine the molecular weights of treated and untreated samples (Figs. 2 ▶ b–2 ▶ e). Native EfsQnr was determined to have a molecular mass of 24 533.0 Da (expected 24 518.1 Da). The cause of the +15 mass difference is not known, but it does not affect the final analysis of untreated and treated EfsQnr. Glutaraldehyde/DMAB-treated EfsQnr exhibited a single peak in FTICR-MS with a mass of 25 077.5 Da, an increase of 544.5 mass units. EfsQnr has eight free amines (seven lysines and the N-terminus), giving an increase of 68.1 mass units per free amine. Similarly, the mass of lysozyme increased from 14 304.7 Da (expected 14 303.8 Da) to 14 782.1 Da after glutaraldehyde/DMAB treatment, an increase of 68.2 mass units per free amine (+477.4 Da; six lysines and the N-terminus). Based on the well documented reductive dimethylation of free amines, the FTICR-MS data suggest that a single glutaraldehyde molecule is reacting with each amine, as shown in Fig. 3 ▶. The first reaction is similar to the reductive dimethylation reaction: attack of a primary amine on an aldehyde followed by dehydration, resulting in a Schiff base which can then be reduced by a strong reducing agent. However, in the glutaraldehyde/DMAB reaction the next step is an intramolecular attack of the secondary amine on the remaining aldehyde moiety of glutaraldehyde followed by dehydration and reduction to produce a piperidine ring. The conversion of a ternary amine to a piperidine group would result in a mass difference of 68 Da (+C5H10, NH3→NH) as observed in the FTICR data. We are subsequently terming this reaction the cyclic pentylation of free amines.
Figure 2.

SDS–PAGE and deconvoluted FTICR-MS of protein samples. (a) Coomassie-stained SDS–PAGE of proteins before and after modification. Lane 2, native lysozyme; lane 3, modified lysozyme; lane 4, modified lysozyme after HIC purification; lane 5, native EfsQnr; lane 6, modified EfsQnr; lane 7, modified EfsQnr after HIC purification. Lanes 1 and 8, 10–150 kDa molecular-weight markers. (b, c) FTICR-MS of EfsQnr before (b) and after (c) modification by glutaraldehyde. (d, e) FTICR-MS of lysozyme before (d) and after modification (e) by glutaraldehyde. The measured masses are indicated.
Figure 3.

Reductive cyclic pentylation reaction scheme. Proposed reaction of glutaraldehyde with lysine/N-termini in the presence of a strong reducing agent.
3.3. Structural evidence for cyclic pentylation
Glutaraldehyde/DMAB-treated EfsQnr was rescreened for crystallization. Similar to unmodified EfsQnr, modified EfsQnr mostly formed clear drops or precipitate. Over a period of 1–2 months, crystals with a paddle-wheel morphology (interjoined plates, 0.1 × 0.1 × 0.02 mm) formed in 10–15%(w/v) PEG 3350, 1 M LiCl, 0.1 M MES pH 6 (Fig. 1 ▶ b). The paddle wheels were broken apart and the individual plates diffracted to 2.0 Å resolution. Crystal and diffraction quality were improved by streak-seeding crushed paddle wheels through an identically produced PEG 3350/LiCl/MES screen. Individual plates with enhanced thickness (0.25 × 0.25 × 0.05 mm) grew over a period of a week (Fig. 1 ▶ c) and diffracted to 1.6 Å resolution. The structure of modified EfsQnr was determined by single anomalous dispersion (SAD) phasing using data collected on a Cu Kα home source from a samarium acetate-soaked crystal.
Structural analysis will be limited to the modified lysine residues; the full details of the EfsQnr structure will be presented elsewhere. Briefly, EfsQnr folds as a right-handed quadrilateral β-helix similar to that observed for other members of the pentapeptide-repeat family (PRPs) of proteins (Hegde et al., 2005 ▶; Buchko et al., 2006 ▶; Vetting et al., 2006 ▶). There is a molecular dimer in the asymmetric unit, with a C-terminal dimer interface similar to that observed for the PRP protein MfpA (Hegde et al., 2005 ▶). Three N-terminal residues remain from the thrombin-cleavable hexahistidine tag (Gly-Ser-His-Met1) used for purification. As such, the modified N-terminal residue is a cloning-artifact glycine and not the native methionine. The N-terminal residues are not observed in the electron-density maps, with the first modeled residues being Met1A and Thr4B (subunit designations are in given in italics). The lack of electron density for the N-termini suggests that they are solvent-accessible and available for modification. Apart from the N-terminal residues, all 211 residues were built into electron-density maps except for residues Pro9–Pro12 of subunit A, which are part of a flexible N-terminal fragment (see below). All seven lysines (Lys2, Lys110, Lys118, Lys150, Lys151, Lys170 and Lys193) are solvent-exposed with excellent electron density for the backbone atoms but weak electron density for most of their side chains, indicative of high mobility. This is consistent with all seven lysines being available for modification by glutaraldehyde as indicated by the FTICR-MS data. A select number of lysines are more ordered and exhibit electron density consistent with the proposed modification (Fig. 4 ▶). The electron density for Lys2A is consistent with a piperidine ring in the chair conformation and sp 3 hybridization of the amine group, confirming successful reduction of the final Schiff base in the reaction. The nonpolar edge of the piperidine group lies in a hydrophobic pocket bounded by the peptide planes of two rungs of the PRP β-helix (Asp111–Leu113, Arg91–Thr93), the outward projecting side chains from those rungs and the side chain of Leu7 (Fig. 5 ▶ a). Based on the pK a values of substituted piperidines (N-methylpiperidine, pK a 10.1; N-ethylpiperidine, pK a 10.4; Hall, 1957 ▶) the amine would be protonated under the experimental conditions (pH 6.0) and be positively charged. In the case of Lys2A the tertiary amine of the piperidine group forms a charged pair (d = 2.8 Å) with the side chain of Asp111.
Figure 4.

Electron density for the typical (Lys193A) and best ordered (Lys2A, Lys110A, Lys150A) lysine residues of EfsQnr. The final 2F o − F c electron density contoured at 1σ is shown in gray. The OMIT F o − F c electron density contoured at 2.5σ is shown in green. The boxed figure shows the electron density of Lys2A looking parallel to the piperidine ring. See §2 for the OMIT procedure.
Figure 5.

Environment around the N-terminal residues. (a) Stereoview around Lys2A (brown C atoms) and its interaction with the surface of the β-helix. Atoms are colored by atom type. The hydrogen bond from Asp111 to Lys2 is shown as dotted line. (b) Alternate conformation for the N-terminus in the A (black trace) and B (cyan trace) subunits. The position of Lys2A is marked by an asterisk. (c, d) Interactions of the two N-termini with symmetry-related subunits.
3.4. Change in crystallizability
Analysis of the position of the modified residues does not readily suggest a mechanism for the altered crystallization. While none of the modified residues directly participate in crystal contacts, Lys2A appears to be a residue which may influence the dynamics of a flexible N-terminus. The N-terminus (residues 1–16) runs down one of the faces of the β-helix prior to forming a cap on the N-terminal end of the β-helix (residues 17–26) and finally initiating the first coil (residues 27–47). The N-terminus takes alternative conformations in the two subunits of the dimer (Fig. 5 ▶ b), with each making numerous and non-equivalant polar interactions with the face of the β-helix. The piperidine group of Lys2A forms intimate interactions with the face of the β-helix, while Lys2B is not visible in electron-density maps. Interestingly, each of the N-termini is involved in extensive but non-equivalent crystal contacts. The A-subunit N-terminus interacts with one crystallographically related subunit, while the B-subunit N-terminus interacts with two crystallographically related subunits (Figs. 5 ▶ c and 5 ▶ d). It is possible that the dynamics of the N-terminus is important in the formation of a stable crystal lattice and that cyclic pentylation of Lys2 would affect crystal lattice formation. Alternatively, the cyclic pentylation of lysines in EfsQnr may be acting in a negative fashion to disrupt non-ordered aggregation under those conditions which yielded crystals.
4. Discussion
In order for a covalent modification to be a useful technique for positively altering protein crystallization, the modification must be homogeneous. At first glance, treatment with glutaraldehyde would seem to be an unlikely choice to produce a homogeneous population, as glutaraldehyde has been used for decades as a protein cross-linking agent. However, in the limited tests performed here on two biophysically different proteins (EfsQnr and lysozyme) the method appears to be robust in preparing homogenously modified protein with no cross-linking when used under reducing conditions. This surprising result most likely originates from several factors. Firstly, glutaraldehyde is in molar excess (10–40-fold) over free amine and once the first reaction takes place access to that amine by free glutaraldehyde is reduced. Secondly, the effective concentration of the bound aldehyde is vastly increased over free aldehyde, ensuring that the second reaction is intramolecular rather than intermolecular. Finally, in the modification of free amines by formaldehyde the monomethylated amine is more reactive to a second aldehyde (Means & Feeney, 1968 ▶; Rayment, 1997 ▶); if this holds true for modification by alternate aldehydes then the intramolecular reaction by a dialdehyde should be rapid.
The modification of free amines by cyclic pentylation is most likely to be sufficiently dissimilar to reductive dimethylation that the technique will generate a unique set of crystallization hits for recalcitrant proteins. The dimethylation of lysines produces a side chain with polarized methyl groups owing to the electron-withdrawing effect of the nitrogen. The dimethylated lysine is therefore able to make ionic interactions between the methyl groups and carbonyl/carboxyl O atoms of neighboring residues, an interaction that has recently been observed in crystal structures of dimethylated proteins (Shaw et al., 2007 ▶; Kim et al., 2008 ▶). In contrast, the cyclic pentylation of lysines produces a piperidine ring which, while retaining the charge of the lysine, buries that charge behind a saturated hydrocarbon ring. Therefore, a protein that has been treated with glutaraldehyde/DMAB should have a more hydrophobic surface than a protein treated with formaldehyde/DMAB and may form unique crystal contacts. Interestingly, glutaraldehyde/DMAB-treated lysozyme did not form crystals (data not shown), suggesting that treatment with glutaraldehyde can also have a negative effect on crystallization. However, it should be noted that the dimethylation of lysozyme also led to a decrease in crystallizability, with usable crystals requiring macroseeding techniques (Rypniewski et al., 1993 ▶). A recent study of a large number of proteins resistant to crystallization indicated that approximately 7% could be rescued (lead to finished structures; Kim et al., 2008 ▶). Inclusion of additional modification procedures such as cyclic pentylation should increase this success rate.
Supplementary Material
PDB reference: EfsQnr, 2w7z, r2w7zsf
Acknowledgments
The authors would like to thank Dr Steven Roderick for thoughtful comments on the manuscript. Mass analysis was performed using the resources of the Laboratory for Macromolecular Analysis and Proteomics (LMAP) at the Albert Einstein College of Medicine. Dr Hui Xiao performed the FTICR-MS mass analysis. This work was supported by NIH grant AI33696 (to JSB).
References
- Adams, P. D., Gopal, K., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Pai, R. K., Read, R. J., Romo, T. D., Sacchettini, J. C., Sauter, N. K., Storoni, L. C. & Terwilliger, T. C. (2004). J. Synchrotron Rad.11, 53–55. [DOI] [PubMed]
- Arsene, S. & Leclercq, R. (2007). Antimicrob. Agents Chemother.51, 3254–3258. [DOI] [PMC free article] [PubMed]
- Buchko, G. W., Ni, S., Robinson, H., Welsh, E. A., Pakrasi, H. B. & Kennedy, M. A. (2006). Protein Sci.15, 2579–2595. [DOI] [PMC free article] [PubMed]
- Campbell, J. W., Duee, E., Hodgson, G., Mercer, W. D., Stammers, D. K., Wendell, P. L., Muirhead, H. & Watson, H. C. (1972). Cold Spring Harb. Symp. Quant. Biol.36, 165–170. [DOI] [PubMed]
- Carson, M., Johnson, D. H., McDonald, H., Brouillette, C. & DeLucas, L. J. (2007). Acta Cryst. D63, 295–301. [DOI] [PubMed]
- Czepas, J., Devedjiev, Y., Krowarsch, D., Derewenda, U., Otlewski, J. & Derewenda, Z. S. (2004). Acta Cryst. D60, 275–280. [DOI] [PubMed]
- D’Arcy, A., Stihle, M., Kostrewa, D. & Dale, G. (1999). Acta Cryst. D55, 1623–1625. [DOI] [PubMed]
- Davis, I. W., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2004). Nucleic Acids Res.32, W615–W619. [DOI] [PMC free article] [PubMed]
- Derewenda, Z. S. (2004a). Structure, 12, 529–535. [DOI] [PubMed]
- Derewenda, Z. S. (2004b). Methods, 34, 354–363. [DOI] [PubMed]
- Derewenda, Z. S. & Vekilov, P. G. (2006). Acta Cryst. D62, 116–124. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gao, X., Bain, K., Bonanno, J. B., Buchanan, M., Henderson, D., Lorimer, D., Marsh, C., Reynes, J. A., Sauder, J. M., Schwinn, K., Thai, C. & Burley, S. K. (2005). J. Struct. Funct. Genomics, 6, 129–134. [DOI] [PubMed]
- Goldschmidt, L., Cooper, D. R., Derewenda, Z. S. & Eisenberg, D. (2007). Protein Sci.16, 1569–1576. [DOI] [PMC free article] [PubMed]
- Graslund, S., Sagemark, J., Berglund, H., Dahlgren, L. G., Flores, A., Hammarstrom, M., Johansson, I., Kotenyova, T., Nilsson, M., Nordlund, P. & Weigelt, J. (2008). Protein Expr. Purif.58, 210–221. [DOI] [PubMed]
- Hall, H. K. J. (1957). J. Am. Chem. Soc.79, 5441–5444.
- Hegde, S. S., Vetting, M. W., Roderick, S. L., Mitchenall, L. A., Maxwell, A., Takiff, H. E. & Blanchard, J. S. (2005). Science, 308, 1480–1483. [DOI] [PubMed]
- Kendrew, J. C., Parrish, R. G., Marrack, J. R. & Orlans, E. S. (1954). Nature (London), 174, 946–949. [DOI] [PubMed]
- Kim, Y. et al. (2008). Nature Methods, 5, 853–854. [DOI] [PMC free article] [PubMed]
- Kobayashi, M., Kubota, M. & Matsuura, Y. (1999). Acta Cryst. D55, 931–933. [DOI] [PubMed]
- Kurinov, I. V. & Uckun, F. M. (2003). Biochem. Pharm.65, 1709–1717. [DOI] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Longenecker, K. L., Garrard, S. M., Sheffield, P. J. & Derewenda, Z. S. (2001). Acta Cryst. D57, 679–688. [DOI] [PubMed]
- McElroy, H. E., Sisson, G. W., Schoettlin, W. E., Aust, R. M. & Villafranca, J. E. (1992). J. Cryst. Growth, 122, 265–272.
- Means, G. E. & Feeney, R. E. (1968). Biochemistry, 7, 2192–2201. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- Painter, J. & Merritt, E. A. (2006). J. Appl. Cryst.39, 109–111.
- Perrakis, A., Sixma, T. K., Wilson, K. S. & Lamzin, V. S. (1997). Acta Cryst. D53, 448–455. [DOI] [PubMed]
- Rauert, W., Eddine, A. N., Kaufmann, S. H. E., Weiss, M. S. & Janowski, R. (2007). Acta Cryst. F63, 507–511. [DOI] [PMC free article] [PubMed]
- Rayment, I. (1997). Methods Enzymol.276, 171–179. [PubMed]
- Rypniewski, W. R., Holden, H. M. & Rayment, I. (1993). Biochemistry, 32, 9851–9858. [DOI] [PubMed]
- Schubot, F. D. & Waugh, D. S. (2004). Acta Cryst. D60, 1981–1986. [DOI] [PubMed]
- Shaw, N., Cheng, C., Tempel, W., Chang, J., Ng, J., Wang, X. Y., Perrett, S., Rose, J., Rao, Z., Wang, B.-C. & Liu, Z.-J. (2007). BMC Struct. Biol.7, 46. [DOI] [PMC free article] [PubMed]
- Stura, E. A. & Wilson, I. A. (1991). J. Cryst. Growth, 110, 270–282.
- Vetting, M. W., Hegde, S. S., Fajardo, J. E., Fiser, A., Roderick, S. L., Takiff, H. E. & Blanchard, J. S. (2006). Biochemistry, 45, 1–10. [DOI] [PMC free article] [PubMed]
- Walter, T. S., Meier, C., Assenberg, R., Au, K. F., Ren, J., Verma, A., Nettleship, J. E., Owens, R. J., Stuart, D. I. & Grimes, J. M. (2006). Structure, 14, 1617–1622. [DOI] [PMC free article] [PubMed]
- Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001). Acta Cryst. D57, 122–133. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: EfsQnr, 2w7z, r2w7zsf
PDB reference: EfsQnr, 2w7z, r2w7zsf