

Abstract
We measured the folding and unfolding kinetics of mutants for a simple protein folding reaction to characterize the structure of the transition state. Fluorescently labeled S-peptide analogues combine with S-protein to form ribonuclease S analogues: initially, S-peptide is disordered whereas S-protein is folded. The fluorescent probe provides a convenient spectroscopic probe for the reaction. The association rate constant, kon, and the dissociation rate constant, koff, were both determined for two sets of mutants. The dissociation rate constant is measured by adding an excess of unlabeled S-peptide analogue to a labeled complex (RNaseS*). This strategy allows kon and koff to be measured under identical conditions so that microscopic reversibility applies and the transition state is the same for unfolding and refolding. The first set of mutants tests the role of the α-helix in the transition state. Solvent-exposed residues Ala-6 and Gln-11 in the α-helix of native RNaseS were replaced by the helix destabilizing residues glycine or proline. A plot of log kon vs. log Kd for this series of mutants is linear over a very wide range, with a slope of −0.3, indicating that almost all of the molecules fold via a transition state involving the helix. A second set of mutants tests the role of side chains in the transition state. Three side chains were investigated: Phe-8, His-12, and Met-13, which are known to be important for binding S-peptide to S-protein and which also contribute strongly to the stability of RNaseS*. Only the side chain of Phe-8 contributes significantly, however, to the stability of the transition state. The results provide a remarkably clear description of a folding transition state.
The single-exponential folding kinetics often observed for small proteins (1–3) show that a dominant free energy barrier of at least 4 kcal⋅mol−1 is present in the protein folding process (4–7). Simulations suggest that an ensemble of conformations is present at this barrier (reviewed in refs. 8 and 9), but for simplicity we refer to this ensemble as the transition state for folding. We study the bimolecular RNaseS* system, in which fluorescently labeled analogues of unfolded S-peptide are allowed to combine with folded S-protein. Our aim is to characterize the transition state of the simplest two-state folding reaction studied to date. The smallest monomeric polypeptide that folds via a two-state mechanism is 36 residues (10) whereas typical small proteins studied are ≈60 residues in length. Only a single α-helix becomes ordered as S-peptide folds via combination with folded S-protein. In most folding reactions of monomeric proteins, the unfolding reaction cannot be measured directly under native conditions, and so the transition state may change between conditions for studying unfolding and refolding. By contrast, the unfolding–dissociation rate constant for S-peptide in RNaseS* can be measured in native conditions by mixing the complex with an excess of unlabeled S-peptide analogue. Both the rate constants for association–folding (kon) and for unfolding–dissociation (koff) are obtained under identical strongly native conditions, and the principle of microscopic reversibility applies in analyzing the transition state. Moreover, the stability of the complex under native conditions is given by Kd = koff/kon because combination of S-peptide with S-protein is two-state (11).
The complex between S-protein and the wild-type S-peptide analogue (Table 1) is remarkably strong: ΔG for dissociation = 14.5 kcal⋅mol−1 at 10°C, pH 6.8, in 10 mM Mops (Kd = 6 pM) (11). The dependence of kon and koff on denaturant suggests that the transition state is 55% sequestered from solvent, compared with the total change in buried surface area for the reaction shown in Eq. 1:
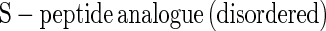 |
1 |
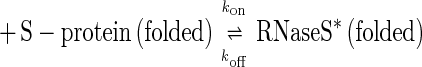 |
Similarly, the effect of temperature on kon and koff suggests that 66% of the change occurs in the transition state (12). In these respects, the behavior of this simple bimolecular reaction is remarkably similar to that of typical small monomeric proteins (see refs. 3, 13, and 14). The association–folding reaction is diffusion-controlled but not necessarily encounter-limited and displays an ionic strength dependence typical of protein association reactions (15).
Table 1.
S-peptide analogues used in this study†
| Name | Sequence | ΔΔASAnp, Å2‡ | Percent of helix§ |
|---|---|---|---|
| Pep-1F¶ | ac-YETAAAK′FERQHMDS-NH2 | 0 | 21∥ |
| Helix backbone series | |||
| A6G | ac-YETAAGK′FERQHMDS-NH2 | −28 | 4 |
| A6P | ac-YETAAPK′FERQHMDS-NH2 | 35 | 3 |
| Q11P | ac-YETAAAK′FERPHMDS-NH2 | 16 | 10 |
| Side chain series | |||
| F8A | ac-YETAAAK′AERQHMDS-NH2 | −166 | 42 |
| H12A | ac-YETAAAK′FERQAMDS-NH2 | −64 | 34 |
| M13A | ac-YETAAAK′FERQHADS-NH2 | −103 | 25 |
| H12A/M13MS⩵O‡‡ | ac-YETAAAK′FERQAMS⩵ODS-NH2 |
K′, 5-carboxyfluoresceinyl ɛN lysine.
The predicted effects of mutations on the change in accessible nonpolar surface area associated with forming RNaseS* from isolated S-peptide analogues and native S-protein (see Materials and Methods).
The fractional helicities for the mutant peptides calculated from the Lifson-Roig model with Pep-1F as a reference (see Materials and Methods), unless noted.
The suffix “1F,” which indicates the presence of 5-carboxyfluorescein, is dropped from the names of the mutants.
∥ From ref. 11.
MS⩵O is methionine sulfoxide.
We mutated to alanine, one by one, the three S-peptide residues (Table 1) that make the greatest contribution to the buried hydrophobic surface when S-peptide binds to S-protein (16) and studied a highly destabilizing double variant in which His-12 is replaced with alanine and Met-13 is oxidized to the sulfoxide level. We also changed residues Ala-6 and Gln-11 in the 3–13 α-helix to the helix-destabilizing residues glycine or proline to determine the role of the α-helix in the transition state. These sites are solvent-exposed in the helix of native RNaseS (Table 1), so that the mutations should affect helix stability without perturbing hydrophobic packing interactions.
MATERIALS AND METHODS
Materials.
S-protein and S-peptide analogues were prepared as described (11). Additional peptides were purchased from Research Genetics (Huntsville, AL) and were purified as described (11). Peptide structures were confirmed by mass spectrometry.
Peptide Design.
The sequences of the peptides used in this study are given in Table 1. The peptide with the sequence most similar to wild-type S-peptide is Pep-1F; it differs from wild-type S-peptide in several ways: The five C-terminal residues of S-peptide are disordered in crystal structures of RNaseS (17, 18) and relatively unimportant for binding (19, 20) and are not included. Lys-1 of S-peptide was changed to αN acetyl tyrosine to provide a spectroscopic probe for the unlabeled peptide and to render ɛN of Lys-7 the sole reactive amine for labeling with 5-carboxyfluorescein succinimidyl ester (Molecular Probes). Pep-1F is amidated at its C terminus in order not to introduce an unnatural carboxylate group into RNaseS. The largest change in Pep-1F relative to S-peptide is the attachment of fluorescein to ɛN of Lys-7, which provides a convenient spectroscopic probe for the association and dissociation of RNaseS* The complex of the labeled peptide and S-protein is stable and retains enzymatic activity (11).
Choice of Mutations.
To assess the role of side chains in the folding of RNaseS*, the side chains of Phe-8, His-12, and Met-13 were targeted for mutation (Table 1). These three side chains comprise 60% of the nonpolar interface between S-peptide side chains and S-protein (16), and their importance for the stability of RNaseS is well known (19, 20). Helix destabilizing residues were introduced at sites in the S-peptide 3–13 helix that are solvent-exposed in native RNaseS* (Table 1). The A6G and A6P mutations change an alanine residue three residues into the native helix that accepts an N-cap hydrogen bond from Thr-3. Glutamine 11, which was replaced with proline, is two residues before the C terminus of the helix in native RNaseS and lies between residues that make important side chain contacts with S-protein.
Solvent Accessibility Calculations.
The effect of a mutation on the change in accessible nonpolar surface area accompanying folding is expressed as ΔΔASAnp = ΔASAU − ΔASAN, where ΔASAU is the difference in accessible nonpolar surface area between unfolded Pep-1F and unfolded mutant peptide, and ΔASAN is the difference in accessible nonpolar surface area between RNaseS* with Pep-1F or with mutant peptide. ΔASAU is estimated from the difference in nonpolar surface area between the stochastic standard states of the wild-type and mutant amino acids (21). The values of ΔASAN are the differences in nonpolar surface area between RNaseS [1rnu.pdb§ (18)] and the surface areas of RNaseS* complexes with mutant peptides and were calculated with naccess 2.1 (S. Hubbard and J. Thornton, University College, London) by using a probe radius of 1.4 Å and standard van der Waals radii (22). The coordinates for the mutant complexes were obtained by using the cara (23) module of look 3 (Molecular Applications Group, Palo Alto, CA) with the coordinates of neighboring residues fixed. Values of ΔΔASAnp also were calculated from models in which the coordinates of side chains within 4.5 Å of the mutated residues were allowed to vary; the values from these models are within 20% of those based on the models with fixed side chains. The value of ΔΔASAnp for the experimentally determined structure of M13A [1rbc.pdb (17)] is −121 Å2 vs. −104 Å2 for modeled M13A. The values from comparisons between experimental structures, which have different structures in disordered regions as well as at the mutation sites, and modeled structures are not expected to be identical (24).
Estimated Helicities of Isolated Mutant Peptides.
The fractional helicity of Pep-1F was determined from a CD measurement to be 21% (11). The fractional helicities of the mutated peptides were estimated (see Table 1) by using the literature values of helix propensities and helix–coil theory, including sidechain interactions, as described in refs. 25 and 26. Pep-1F is treated as a homopolymer except for Phe-8, His-12, and the site of the substitution. The stabilizing effect of the Phe-His pseudo H-bond interaction was included by using an equilibrium constant (p-value) of 1.65. The helix content of each peptide containing the parent residue at the site of the mutation was set to 21% by adjusting the average helix propagation parameter (w-value) for the host. The resulting host w-values ranged from 1.28 for Gln-11 to 1.55 for Met-13. (These are large w-values, compared with the measured helix propensities, because S-peptide contains a Glu-2–Arg-10 salt bridge.) The host N-cap parameter was set at 1. The fractional helicities for the mutant peptides then were estimated by inserting the helix propensities of the mutated residues. The p-value for the i, i + 4 Phe-His interaction was estimated to be 1.65 from published data (27) by a similar procedure, comparing host sequences in which His and Phe are spaced either four (interacting) or five (no interaction) residues apart.
Kinetic Measurements.
The visible absorbance and emission spectra of S-peptide analogues labeled with fluorescein at Lys-7 change on complex formation with S-protein, which provides a sensitive assay for complex formation (11). The association rate constant, kon (Eq. 1), was determined by following the time course of fluorescence emission after mixing a labeled S-peptide analogue with S-protein in a stopped flow spectrophotometer. The dissociation constant of RNaseS* (koff, Eq. 1) was measured after adding an excess of unlabeled S-peptide analogue. The excess was shown to be sufficient by increasing the concentration of the unlabeled analogue until the observed rate constant was concentration-independent. The excitation wavelength was 496 nm throughout, and the other details were described previously (11).
RESULTS
Association and Dissociation Time Courses.
Fluorescein-labeled S-peptide analogues bind to 6 μM S-protein after mixing in a stopped flow spectrophotometer at 10°C, pH 6.7–6.8, in 10 mM Mops, leading to a 13% decrease in the fluorescence emission (relative to free peptide) of Pep-1F, the host peptide similar in sequence to wild-type S-peptide (Fig. 1a). This decrease in fluorescence is caused by a 0.34-unit increase in the pKa of fluorescein in RNaseS* relative to free peptide (11). The fluorescence decreases are smaller for F8A, H12A/M13MS⩵O, and Q11P, which profoundly destabilize RNaseS* (Table 2), because these mutants show smaller extents of reaction with 6 μM S-protein and possibly also because of differences in the fluorescence quantum yield for F8A and Q11P. Fig. 1b shows dissociation time courses for Pep-1F, H12A/M13MS⩵O, and F8A, initiated by adding excess unlabeled Pep-1 under identical buffer and temperature conditions. All of the time courses, except for that of Pep-1F dissociation, are well described by a single exponential model of the form F(t) = −ΔF exp(−kobs t) + Ffinal. The early, small amplitude phase for Pep-1F is the result of contamination from ≈5% of M13MS⩵O that we are unable to remove. The binding of Pep-1F to 6 μM S-protein is complete in <0.1 second whereas dissociation of the resultant complex is not complete until after 36,000 seconds; this is the kinetic correlate of the impressive stability of the Pep-1F/S-protein complex (Table 2).
Figure 1.

(a) Time course of the fluorescence emission decrease (λex = 496 nm) for forming RNaseS* from S-peptide analogues and 6 μM native S-protein at 10°C, pH 6.75 ± 0.05, in 10 mM Mops. The data were obtained under pseudo-first-order conditions with S-protein in large excess and are shown with single exponential fits. One-hundred percent fluorescence is defined as that of free peptide. S-peptide analogue concentrations are 0.2 μM (Pep-1F and H12A/M13MS⩵O) or 0.2 μM (Q11P and F8A). ●, Pep-1F; ⊞, H12A/M13MS⩵O; ■, F8A; ▴, Q11P. (b) Time course of the fluorescence emission increase for dissociation of RNaseS* in competition experiments. The conditions and symbols are identical to those for the association experiments. The concentration of RNaseS* was 0.8 or 1 μM, and labeled peptide was competed off of RNaseS* by 50 or 200 μM of (unlabeled) Pep-1. Single exponential fits to the data are shown.
Table 2.
Parameters describing the interaction between S-peptide analogues and native S-protein*
| kon, 107 M−1⋅s−1 | koff, s−1 | Kd, nM† | Kd, nM‡ | |
|---|---|---|---|---|
| Pep-1F | 1.8 ± 0.08 | 1.2 ± 0.08 × 10−4 | 6.5 ± 0.5 × 10−3 | |
| A6G | 1.2 ± 0.8 | 1.9 ± 0.08 × 10−4 | 16 ± 1 × 10−3 | |
| A6P | 0.32 ± 0.01 | 8.8 ± 0.4 × 10−3 | 2.8 ± 0.8 | |
| Q11P | 0.022 ± 0.004 | 1.9 ± 0.3 | 8600 ± 2100 | 4700 ± 60 |
| F8A | 0.12 ± 0.06 | 1.9 ± 0.1 | 1500 ± 200 | 1400 ± 700 |
| H12A | 3.9 ± 0.1 | 0.028 ± 0.003 | 0.72 ± 0.05 | |
| M13A | 1.65 ± 0.08 | 0.080 ± 0.006 | 4.9 ± 0.04 | |
| H12A/M13MS⩵O§ | 1.32 ± 0.08 | 78 ± 6 | 5900 ± 600 | 7700 ± 2000 |
pH 6.7 ± 0.1, 10 mM Mops, 9.9 ± 0.1°C.
Value from koff/kon.
Value from kinetic amplitude changes.
MS⩵O is methionine sulfoxide.
The H12A/M13MS⩵O, F8A, and Q11P variants all profoundly destabilize RNaseS* (Table 2), but H12A/M13MS⩵O binds to S-protein at about the same rate as does Pep-1F whereas F8A and Q11P bind 12- and 87-fold more slowly, respectively. These differences in kobs show that the α-helix and an interaction between Phe-8 and S-protein are at least partially formed in the transition state whereas the side chains of H12 and M13 are unfolded, as discussed below.
The Association and Dissociation Rate Constants.
For a reaction of the type shown in equation 1, under pseudo-first order conditions ([S-protein] ≥ 5 × [S-peptide analogue]), kobs = kon [S-protein] + koff. The slope of a plot of kobs vs. [S-protein] yields kon, the bimolecular rate constant for association–folding. Plots of kobs vs. [S-protein] are shown in Fig. 2 for the mutants described in Table 1, and the relative values of kon for the mutants may be readily discerned. The values of kon, koff, and Kd are given in Table 2. When the value of koff is significantly greater than the error in the extrapolated value of kobs at zero concentration of S-protein, as for H12A/M13MS⩵O, F8A, and Q11P, this rate constant may be obtained from the y intercept. koff also was measured directly for all analogues except Q11P, and the values are the same, within experimental error, as the intercept values. For certain analogues, H12A, A6G, and A6P, there is downward curvature in plots of kobs vs. [S-protein]. Here, we use the model kobs = kmax/(1 + KM/[S-protein]); kon is the limiting slope as [S-protein] approaches zero, or kmax/KM. This equation is identical to that describing the Michaelis-Menten model for the reaction of a substrate and enzyme. The downward curvature for these variants suggests the presence of a transient folding intermediate that increases in population at higher concentrations of S-protein.
Figure 2.

Concentration dependence of the kinetics for S-peptide analogues binding to S-protein and the concentration-independent value of the dissociation rate constant for selected variants. All experiments were conducted under conditions described in Fig. 1a. Association kinetics were measured under pseudo-first-order conditions ([S-protein] = 5 × [S-peptide analogue]), and dissociation kinetics were measured in the presence of a 50- to 500-fold excess over RNaseS* of (unlabeled) Pep-1. Data for the slower-reacting variants are expanded in the inset. The concentration of the analogue is 0.4 μM, except for Pep-1F and H12A/M13MS⩵O, for which it is 0.2 μM. ●, Pep-1F; □, M13A;  , H12A; ⊞, H12A/M13MS⩵O; ■, F8A; ▿, A6G; ▾, A6P; ▴, Q11P. The symbols ⧫ and ◊ plotted at [S-protein] = 0 indicate the dissociation rate constants for H12A/M13MS⩵O and F8A, respectively, where the labeled analogues are competed-off RNaseS* by excess (unlabeled) Pep-1 (Fig. 1b). The data are fitted with a straight line or the Michaelis-Menten model (see text).
, H12A; ⊞, H12A/M13MS⩵O; ■, F8A; ▿, A6G; ▾, A6P; ▴, Q11P. The symbols ⧫ and ◊ plotted at [S-protein] = 0 indicate the dissociation rate constants for H12A/M13MS⩵O and F8A, respectively, where the labeled analogues are competed-off RNaseS* by excess (unlabeled) Pep-1 (Fig. 1b). The data are fitted with a straight line or the Michaelis-Menten model (see text).
Amplitude of the Folding Reaction.
The S-protein concentration dependences of the amplitudes of reactions with H12A/M13MS⩵O, F8A, and Q11P are well described by a standard binding titration model: F(N) = ΔF{( Kd + S + N) − sqrt[(Kd + S + N)2 − 4SN]}/2S, where F(N) is the observed fluorescence (minus an offset factor), ΔF is the total fluorescence change, Kd is the dissociation constant, S is the concentration of the S-peptide analogue, and N is the concentration of S-protein. For all three variants, the values of Kd are similar to that calculated from koff/kon (Table 2), indicating that these reactions are described by the two-state approximation.
DISCUSSION
The 3–13 α-Helix and Side Chains of Phe-8, His-12, and Met-13 of S-Peptide Greatly Stabilize RNaseS*.
In Fig. 3 the logarithm of the rate constant for refolding of the S-peptide analogues, kon, is plotted against the logarithm of the dissociation constant for RNaseS*, Kd. The extremely large displacements toward larger values of Kd reflect the key roles played by the 3–13 α-helix and the side chains of Phe-8, His-12, and Met-13 in stabilizing native RNaseS*. Below we discuss the extent to which the helix and the side chain interactions stabilize the transition state relative to the unfolded protein and thereby accelerate the folding process.
Figure 3.

Brønsted plot comparing the logarithm of the association rate constant (kon) to the logarithm of the dissociation constant (Kd) for S-peptide analogues binding to native S-protein under the conditions described in Fig. 1a. The symbols are defined in Fig. 2, and the data are fitted by straight lines.
The Transition State for RNaseS* Folding Contains α-Helical Structure.
Assuming that the transition state approximation is valid in protein folding, the rate constant for a folding reaction may be expressed as kforward ≈ K‡k‡, where K‡ is the equilibrium constant between the unfolded and the activated states, and k‡ is the currently unknown rate constant of the process that converts the transition state to the final product. We assume that mutations affect K‡ and not k‡, so that changes in kforward (kon in this case) reflect changes in −RT ln K‡, the size of free-energy barrier for refolding. The ratio Δlog kforward/Δlog Keq equals the φ-value for a mutant (28, 29) when the wild-type values of kforward and Keq are used as the reference. A relatively large decrease in log kon, or a large φ-value, means that the mutated structure stabilizes the transition state of the wild type whereas a relatively small change in log kon, or a small φ-value, means that the affected structure is unimportant until later in the folding process. Therefore, the large decreases in log kon caused by the potent helix-destabilizing mutations at exposed residues Ala-6 and Gln-11 (Fig. 3) indicate the presence of α-helical structure in the transition state for RNaseS* refolding. In fact, the Q11P mutation causes a larger decrease in rate than does removal of the Phe-8 benzyl group, indicating that the presence of helical structure is essential, and does more than merely present the side chain of Phe-8 in the correct orientation. The greater impact on kon of Q11P than A6P shows that the C terminus of the helix is more important than the N terminus and, because His-12 and Met-13 are unfolded in the transition state (see below), suggests a role for the polar backbone interactions between S-peptide and S-protein that are observed in crystal structures of RNaseS (18, 30). This possible role may be tested in competition experiments with nonspecific unlabeled peptides with high helical content.
A Folding Pathway, Not a Funnel.
Multiple pathway models, such as funnel models, for protein folding predict that strong helix-destabilizing mutations will not slow down folding in the limit in which alternative, nonhelical pathways dominate the process (31). This means that a plot of log kon vs. log Kd should level off for sufficiently helix-destabilizing mutations. There is no evidence for such leveling-off in Fig. 3, indicating that the major folding pathway (that used by most of the molecules) is strongly dominant and involves helix formation in the transition state. A previous study of CI2 reached a similar conclusion tentatively (31), but curvature could have been masked by scatter in the data over the free energy range investigated (32); the linearity observed here in the plot of log kon vs. log Kd over a range of 8 kcal⋅mol−1 (Fig. 3) removes this uncertainty. Funnel-like processes may occur during other stages of refolding, but formation of the transition state is largely restricted to helical conformations.
The φ-value is usually taken as a measure of the extent to which the structure formed by a residue in native protein stabilizes the transition state and is expected to vary from 0 to 1 as the structure of this residue in the transition state varies from being disrupted to becoming more like that of the native structure (28, 29). Fractional φ-values may occur if an interaction is formed partially in a dominant transition state or if it is fully formed in a fraction of the transition states on a funnel-like landscape (31, 32). The φ-values observed here for the helix-destabilizing mutations are ≈0.3 (Fig. 3). The consistency of this value over several orders of magnitude (that is, the linearity in the plot of log kon vs. log Kd for the entire set of helix-destabilizing mutations) indicates that the fractional φ-values in this case stem from a structure that is less stable in a dominant transition state than in the native protein.
The Transition State Involves a Specific Side Chain Interaction Between S-Protein and Phe-8 of S-Peptide.
The mutation to alanine of Phe-8 reduces the refolding rate of RNaseS*, but similar mutations of His-12 and Met-13 have almost no effect on the rate of folding (Fig. 3), although all three mutations strongly affect Keq. Even the H12A/M13MS⩵O variant, which profoundly destabilizes RNaseS* by removing an imidazole group and burying a polar group in the Met-13 binding site, has little effect on kon. This means that the side chain of Phe-8 stabilizes the transition state but those of Met-13 and His-12 do not. This specificity is surprising, given that His-12 and Met-13 together contribute more nonpolar surface area to the interface between S-peptide and S-protein than does Phe-8 (16) and that together they stabilize native RNaseS* by as much as does Phe-8 (Table 2 and Fig. 3). The small increase in kon for the H12A mutation probably stems from the increase in negative charge that this mutation confers on S-peptide at neutral pH, enhancing the electrostatic attraction between the S-peptide analogue and S-protein that is evidenced by the effect of ionic strength on kon (11).
The rate constants for the refolding and unfolding of RNaseS* are inversely proportional to the solvent viscosity (12), indicating that the reaction is fully diffusion-controlled. Mutations that affect the packing between S-peptide and S-protein are not predicted to change the rate constant of a purely encounter-limited reaction (33), for which every collision of the reactants in the proper orientation and with appropriate secondary structure leads to product. The effect on kon of the F8A mutation therefore suggests that the rate-determining step occurs after the encounter complex is formed and that the viscosity sensitivity is conferred by a diffusion-limited step within this complex.
An Emerging Picture of the Transition State.
The simplicity of the process in which S-peptide combines with folded S-protein and then refolds allows us to develop a remarkably clear concept of the ensemble of structures present at the free energy barrier for folding/unfolding (Fig. 4). The α-helix present in the native complex is at least partially formed in the transition state of the dominant folding pathway, and its C terminus is more important than its N terminus because substitution of a proline residue at position 11 destabilizes the transition state more than a proline residue at position 6 and both sites are solvent exposed. Phe-8, but not His-12 or Met-13, makes favorable interactions with S-protein in the transition state. The reason for this specificity is unknown, but it seems reasonable that a given amount of buried hydrophobic surface area in one patch (Phe-8) stabilizes the transition state, containing it in preference to the transition state containing a similar amount of buried nonpolar area spread over two sites (His-12 and Met-13). Phe-8 is located at the center of the 3–13 helix and is predicted to have a higher backbone conformation in helical form than His-12 or Met-13 because of end-fraying effects (34, 35).
Figure 4.

A model for the transition state for S-peptide binding to native S-protein. Fn, fluorescein.
Transient Intermediates.
The downward curvature observed in plots of kobs vs. [S-protein] for A6G, A6P, and H12A below 10 μM S-protein (Fig. 2) and for Pep-1F and M13A above 10 μM S-protein (11) hints at the presence of transient intermediates that are increasingly populated at higher concentrations of S-protein. The existence of possible transient intermediates needs to be addressed in future studies at high concentrations of S-protein and in competition experiments with unlabeled nonspecific peptides.
Is Folding Hierarchic or Nonhierarchic?
What happens first when unfolded S-peptide combines with folded S-protein to form RNaseS*? Does the helix form first and then dock with S-protein via Phe-8 (a hierarchic pathway)? Or does the interaction between Phe-8 and S-protein form first, creating a nucleus for further folding involving helix formation (a nonhierarchic pathway)? There is a long-standing interest in the possibility that folding is initiated by nonspecific collapse (reviewed in ref. 6), driven mainly by hydrophobic interactions (also a nonhierarchic pathway). The special role in the transition state played by Phe-8 argues against the nonspecific collapse model, which predicts that the equally disruptive H12A/M13MS⩵O mutation should have a similar effect on kon. Comparison of kon values for S-peptide variants cannot tell us whether the helix forms first or the interaction between Phe-8 and S-protein forms first. All that can be learned from studying the transition state of this two-state folding reaction is that both helical structure and the interaction involving Phe-8 are present in the transition state. Characterization of the transient folding intermediates postulated above might distinguish between hierarchic and nonhierarchic folding mechanisms for RNaseS*.
Other Studies of Transition States.
The nature of the transition state for protein folding is still an open question. An extensive study of CI2 (36) gives an approximately linear Brønsted plot for all residues of the protein, with an average φ-value of 0.3 in the folding direction. This result suggests that the transition state is formed cooperatively and involves the entire protein but that close packing is incomplete and all interactions are considerably less stable than in the native protein. On the other hand, recent studies of two SH3 domains, [one from src (37) and the other from α-spectrin (38)] show sharply polarized transition states with similar structures and with some φ-values >0.7 and others <0.2, in the case of src (37). Our results resemble the SH3 results in showing a single dominant transition state with a strongly polarized structure, but they resemble the CI2 results in showing low φ-values for folding, which indicate that the interactions present in the transition state are substantially less stable than in the native protein.
The technique of measuring φ-values for helix-destabilizing substitutions at solvent-exposed sites, to test whether a helix is an important structural element of a transition state, has been used only rarely, and our study is unusual in giving a clear positive result. For example, a study of the transition state of GCN4 (a dimeric coiled-coil) failed to reveal the presence of a unique helical nucleus (39). The difference between our result and theirs may reside in a single dominant transition state in our system versus multiple, nearly equivalent, transition states in their system.
Acknowledgments
We thank Drs. Carol Rohl, Michael Kay, Janet Smith, and Pat Jennings for valuable discussions and Drs. Simon Hubbard and Janet Thornton (Department of Biochemistry, and Molecular Biology, University College, London) for the naccess computer program. Matrix-assisted laser desorption ionization spectroscopy was carried out by the Protein and Nucleic Acid Facility, Stanford, CA. This work was supported by a grant from the National Institutes of Health (GM 19988). The Mass Spectrometry Facility at the University of California, San Francisco was supported by National Institutes of Health Grant RR 01614.
Footnotes
The A conformations of the side-chains with multiple conformations were used for the calculations.
References
- 1.Tan Y J, Oliveberg M, Fersht A R. J Mol Biol. 1996;264:377–389. doi: 10.1006/jmbi.1996.0647. [DOI] [PubMed] [Google Scholar]
- 2.Schönbrunner N, Pappenberger G, Scharf M, Engels J, Kiefhaber T. Biochemistry. 1997;36:9057–9065. doi: 10.1021/bi970594r. [DOI] [PubMed] [Google Scholar]
- 3.Scalley M L, Yi Q, Gu H, McCormack A, Yates J R, III, Baker D. Biochemistry. 1997;36:3373–3382. doi: 10.1021/bi9625758. [DOI] [PubMed] [Google Scholar]
- 4.Shastry M C, Roder H. Nat Struct Biol. 1998;5:385–392. doi: 10.1038/nsb0598-385. [DOI] [PubMed] [Google Scholar]
- 5.Laurents D V, Baldwin R L. Biophys J. 1998;75:428–434. doi: 10.1016/S0006-3495(98)77530-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan H S, Dill K A. Proteins Struct Funct Genet. 1998;30:2–33. doi: 10.1002/(sici)1097-0134(19980101)30:1<2::aid-prot2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 7.Doyle R, Simons K, Qian H, Baker D. Proteins Struct Funct Genet. 1997;29:282–291. doi: 10.1002/(sici)1097-0134(199711)29:3<282::aid-prot3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin R L. J Biomol NMR. 1995;5:103–109. doi: 10.1007/BF00208801. [DOI] [PubMed] [Google Scholar]
- 9.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 10.McKnight C J, Doering D S, Matsudaira P T, Kim P S. J Mol Biol. 1996;260:126–134. doi: 10.1006/jmbi.1996.0387. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg J M, Baldwin R L. Biochemistry. 1998;37:2546–2555. doi: 10.1021/bi972402y. [DOI] [PubMed] [Google Scholar]
- 12.Goldberg J M, Baldwin R L. Biochemistry. 1998;37:2556–2563. doi: 10.1021/bi972403q. [DOI] [PubMed] [Google Scholar]
- 13.Schindler T, Schmid F X. Biochemistry. 1996;35:16833–16842. doi: 10.1021/bi962090j. [DOI] [PubMed] [Google Scholar]
- 14.Oliveberg M, Tan Y J, Fersht A R. Proc Natl Acad Sci USA. 1995;92:8926–8929. doi: 10.1073/pnas.92.19.8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vijayakumar M, Wong K Y, Schreiber G, Fersht A R, Szabo A, Zhou H X. J Mol Biol. 1998;278:1015–1024. doi: 10.1006/jmbi.1998.1747. [DOI] [PubMed] [Google Scholar]
- 16.Richards F M, Wyckoff H W, Carlson W D, Allewell N M, Lee B, Mitsui Y. Cold Spring Harb Symp Quant Biol. 1972;36:35–43. doi: 10.1101/sqb.1972.036.01.008. [DOI] [PubMed] [Google Scholar]
- 17.Varadarajan R, Richards F M. Biochemistry. 1992;31:12315–12327. doi: 10.1021/bi00164a005. [DOI] [PubMed] [Google Scholar]
- 18.Kim E E, Varadarajan R, Wyckoff H W, Richards F M. Biochemistry. 1992;31:12304–12314. doi: 10.1021/bi00164a004. [DOI] [PubMed] [Google Scholar]
- 19.Richards F M, Wyckoff H W. Enzymes. 1971;4:647–806. [Google Scholar]
- 20.Blackburn P, Moore S. Enzymes. 1982;15:317–433. [Google Scholar]
- 21.Lesser G J, Rose G D. Proteins Struct Funct Genet. 1990;8:6–13. doi: 10.1002/prot.340080104. [DOI] [PubMed] [Google Scholar]
- 22.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 23.Lee C, Levitt M. Nature (London) 1991;352:448–451. doi: 10.1038/352448a0. [DOI] [PubMed] [Google Scholar]
- 24.Varadarajan R, Connelly P R, Sturtevant J M, Richards F M. Biochemistry. 1992;31:1421–1426. doi: 10.1021/bi00120a019. [DOI] [PubMed] [Google Scholar]
- 25.Rohl C A, Chakrabartty A, Baldwin R L. Protein Sci. 1996;5:2623–2637. doi: 10.1002/pro.5560051225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stapley B J, Rohl C A, Doig A J. Protein Sci. 1995;4:2383–2391. doi: 10.1002/pro.5560041117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Armstrong K M, Fairman R, Baldwin R L. J Mol Biol. 1993;230:284–291. doi: 10.1006/jmbi.1993.1142. [DOI] [PubMed] [Google Scholar]
- 28.Matouschek A, Kellis J T, Jr, Serrano L, Fersht A R. Nature (London) 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 29.Oliveberg M, Tan Y J, Silow M, Fersht A R. J Mol Biol. 1998;277:933–943. doi: 10.1006/jmbi.1997.1612. [DOI] [PubMed] [Google Scholar]
- 30.Wyckoff H W, Tsernoglou D, Hanson A W, Knox J R, Lee B, Richards F M. J Biol Chem. 1970;245:305–328. [PubMed] [Google Scholar]
- 31.Fersht A R, Itzhaki L S, elMasry N F, Matthews J M, Otzen D E. Proc Natl Acad Sci USA. 1994;91:10426–10429. doi: 10.1073/pnas.91.22.10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D E, Yi Q, Gladwin S T, Goldberg J M, Baker D. J Mol Biol. 1998;284:807–815. doi: 10.1006/jmbi.1998.2200. [DOI] [PubMed] [Google Scholar]
- 33.Eigen M, Kruse W, Maass G, De Maeyer L. In: Progress In Reaction Kinetics. Porter G, editor. Vol. 2. New York: Macmillan; 1964. pp. 285–318. [Google Scholar]
- 34.Rohl C A, Baldwin R L. Biochemistry. 1994;33:7760–7767. doi: 10.1021/bi00191a003. [DOI] [PubMed] [Google Scholar]
- 35.Chakrabartty A, Schellman J A, Baldwin R L. Nature (London) 1991;351:586–588. doi: 10.1038/351586a0. [DOI] [PubMed] [Google Scholar]
- 36.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 37.Grantcharova V P, Riddle D S, Santiago J V, Baker D. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 38.Martinez J C, Pisabarro M T, Serrano L. Nat Struct Biol. 1998;5:721–729. doi: 10.1038/1418. [DOI] [PubMed] [Google Scholar]
- 39.Sosnick T R, Jackson S, Wilk R R, Englander S W, De Grado W F. Proteins Struct Funct Genet. 1996;24:427–432. doi: 10.1002/(SICI)1097-0134(199604)24:4<427::AID-PROT2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]