

Abstract
Metformin is a widely used oral antihyperglycemic drug for the treatment of type II diabetes mellitus. The intestinal absorption of metformin is dose-dependent and involves an active, saturable uptake process. Metformin has been shown to be transported by the human organic cation transporters 1 and 2 (hOCT1–2). We recently cloned and characterized a novel proton-activated organic cation transporter, plasma membrane monoamine transporter (PMAT). We previously showed that PMAT transports many classic organic cations (e.g., monoamine neurotransmitters, 1-methyl-4-phenylpyridinium) in a pH-dependent manner and its mRNA is expressed in multiple human tissues. The goal of this study is to investigate whether metformin is a substrate of PMAT and whether PMAT plays a role in the intestinal uptake of metformin. Using Madin-Darby canine kidney cells stably expressing human PMAT, we showed that metformin is avidly transported by PMAT, with an apparent affinity (Km = 1.32 mM) comparable to those reported for hOCT1–2. Interestingly, the concentration-velocity profile of PMAT-mediated metformin uptake is sigmoidal, with a Hill coefficient of 2.64. PMAT-mediated metformin transport is greatly stimulated by acidic pH, with the uptake rate being ~4-fold higher at pH 6.6 than at pH 7.4. Using a polyclonal antibody against PMAT, we showed that the PMAT protein (58 kDa) was expressed in human small intestine and concentrated on the tips of the mucosal epithelial layer. Taken together, our results suggest that PMAT transports metformin, is expressed in human intestine, and may play a role in the intestinal absorption of metformin and possibly other cationic drugs.
The biguanide drug, metformin (Glucophage), is widely used for the treatment of hyperglycemia in patients with type II diabetes mellitus. It lowers blood glucose concentration without causing hypoglycemia (Caspary and Creutzfeldt, 1971; Hundal et al., 2000; Borst and Snellen, 2001). Although the exact mechanism leading to its blood glucose-lowering effect has not been fully understood, metformin seems to ameliorate hyperglycemia by improving peripheral sensitivity to insulin, reducing gastrointestinal glucose absorption, and decreasing hepatic glucose production (Caspary and Creutzfeldt, 1971; Hundal et al., 2000; Borst and Snellen, 2001). After oral administration, metformin is slowly absorbed from the proximal small intestine (e.g., duodenum) and mainly excreted from urine (>90%) without undergoing significant biotransformation (Scheen, 1996; Bell and Hadden, 1997). Because the rate of metformin absorption is slower than its rate of plasma elimination, intestinal absorption is the rate-limiting step of metformin disposition (Scheen, 1996; Bell and Hadden, 1997). The absorption of metformin is incomplete and dose-dependent (Scheen, 1996; Bell and Hadden, 1997). More metformin was absorbed after a lower dose than after a higher dose (Tucker et al., 1981), and an inverse relationship was observed between the amount of metformin ingested (from 0.25 to 2.0 g) and its bioavailability (from 86 to 42%) (Scheen, 1996; Bell and Hadden, 1997). Based on the observation of dose-dependent absorption kinetics, it was suggested that metformin absorption is mediated by an active, saturable absorption process (Scheen, 1996; Bell and Hadden, 1997; Klepser and Kelly, 1997). However, the transporters underlying the active uptake process of metformin in the intestine have not been clearly defined.
Organic cation transporters (OCTs) in the solute carrier 22 (SLC22) family have been implicated in the disposition of metformin. The OCTs are polyspecific transporters most prominently expressed in the liver and kidney, where they play a key role in the elimination of organic cations from systemic circulation (Koepsell, 1998; Dresser et al., 2001; Wright, 2005). Metformin has been shown to be transported by both OCT1 and OCT2 (Dresser et al., 2002; Wang et al., 2002; Kimura et al., 2005a,b). Using gene-knockout mice, Oct1 was demonstrated to play an important role in hepatic metformin uptake, which is an important site for its therapeutic effect as well as some side effects (e.g., lactic acidosis) (Wang et al., 2002). On the other hand, hOCT2, which is expressed on the basolateral membrane of kidney tubular cells, was implicated in the renal excretion of metformin (Kimura et al., 2005b; Masuda et al., 2006). However, compared with the liver and kidney, little is known about the transporter(s) involved in the intestinal absorption of metformin. Because of their basolateral locations and low expression levels in the human enterocytes (Koepsell, 1998; Muller et al., 2005; Wright, 2005), OCT1 and OCT2 may play a limited role in intestinal absorption of metformin. OCT3, the third member of the OCT family, was reported to have a relatively higher expression level in the intestine and appeared to be localized on the apical membrane of human enterocytes (Wu et al., 1998; Muller et al., 2005). However, OCT3 tends to exhibit relatively narrower substrate specificity toward xenobiotics (Wright, 2005), and the contribution of OCT3 to metformin absorption is still unknown because metformin has not been shown to be transported by OCT3. In addition, the transport activity of the OCTs, including OCT3, is generally significantly decreased at lower pH (Urakami et al., 1998; Wu et al., 1998; Sweet and Pritchard, 1999), and the acidic environment in the gut lumen (pH 4.0–7.0) may limit the ability of OCT3 in drug absorption.
We recently reported the cloning and characterization of a novel organic cation transporter, plasma membrane monoamine transporter (PMAT), which shows low sequence homology to equilibrative nucleoside transporters (SLC29). Unlike the equilibrative nucleoside transporters, PMAT mainly accepts organic cations as substrates, such as 1-methyl-4-phenylpyridinium (MPP+), tetraethylammonium, and monoamine neurotransmitters (Engel et al., 2004; Engel and Wang, 2005). The substrate and inhibitor specificity of PMAT largely overlaps with that of the OCTs (Engel and Wang, 2005). However, unlike the OCTs, the activity of PMAT is greatly stimulated by acidic pH, presumably due to transport-coupling with an inwardly directed proton gradient (Xia et al., 2007). Our previous Northern analysis showed that PMAT mRNA is widely expressed in a number of human tissues, including brain, skeletal muscle, kidney, liver, and heart (Engel et al., 2004). Recent probing of a multi-tissue RNA array by Barnes et al. (2006) revealed a high level of PMAT mRNA expression in various segments of the human intestinal tract. More recently, our immunofluorescence studies showed that the PMAT protein is primarily targeted to the apical membrane of polarized epithelial cells (Xia et al., 2007). On the basis of these observations, we hypothesized that PMAT protein is expressed in the apical membrane of the enterocytes and may use luminal proton to drive the absorption of certain organic cation drugs. The goal of this study is to investigate whether metformin is a substrate of PMAT and whether PMAT is likely to play a role in the intestinal absorption of metformin.
Materials and Methods
Chemicals
[14C]Metformin (26 mCi/mmol) was obtained from Moravek Biochemicals, Inc. (Brea, CA). [3H]MPP+ (39.3 Ci/mmol) was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO). MPP+, metformin, and phenformin were purchased from Sigma-Aldrich (St. Louis, MO). Buformin was obtained from Wako Pure Chemicals Inc. (Osaka, Japan).
Transport Assay in MDCK Cells
Madin-Darby canine kidney (MDCK) cell lines stably transfected with PMAT cDNA or the pcDNA3 vector were used in these studies (Engel et al., 2004). Cells were maintained in minimal essential medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (Invitrogen) and G418 (500 μg/ml). For uptake studies, cells were plated (105 cells/well) in 24-well plates and allowed to grow at 37°C for 2 to 3 days until confluent. Growth medium was aspirated and each well was rinsed with Krebs-Ringer-Henseleit (KRH) buffer (5.6 mM glucose, 125 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM CaCl2, 1.2 mM MgSO4, 25 mM HEPES, pH 7.4 or 6.6). Cells were then incubated in KRH buffer for 15 min at 37°C. Transport assays were performed at 37°C by incubating cells in KRH buffer containing a radiolabeled substrate. For inhibition studies, cells were incubated with a radiolabeled substrate in the absence (control) or presence of an inhibitor at desired concentrations. Uptake was terminated by aspirating the reaction mixture and washing the cells three times with ice-cold KRH buffer. Cells were then solubilized and the radioactivity was quantified by liquid scintillation counting. Protein content in each well was measured using a BCA protein assay kit (Pierce, Rockford, IL) and the uptake in each well was normalized to the corresponding protein content.
Reverse Transcription-Polymerase Chain Reaction
Human small intestine cDNA (a kind gift from Dr. Kenneth Thummel, University of Washington) was synthesized as described before (Xu et al., 2006a,b). Fifty nanograms of cDNA was used as the template to amplify PMAT and glyceraldehyde-3-phosphate dehydrogenase GAPDH genes. PCR amplification for PMAT was performed using the following pair of primers: 5′-CGGGCGTGAT-GATCTCTCTGAGCCGCATC-3′ and 5′-GGTTGAACACAGCCATGATGAGGATGGGCA-3′ (564-base pair amplicon). PCR amplification for GAPDH was performed using primers 5′-CGTATTGGGCGCCTGGTCACCAGGGCTGCT-3′ and 5′-TTGAGGGCAATGCCAGCCCCAGCGTCGAAG-3′ (875-base pair amplicon). Amplification conditions were 94°C for 1 min, 61°C for 1 min, 72°C for 1 min for 35 cycles, and finally 72°C for 10 min.
Production and Purification of PMAT Peptide Antibody
A polyclonal peptide antibody (P469) was generated toward amino acid residues 469 to 482 (ILAAGKVSPKQREL) in human PMAT. The generation and validation of this anti-PMAT peptide antibody was described previously (Dahlin et al., 2007). In brief, the peptide was chemically synthesized and conjugated to keyhole limpet hemocyanin. The synthetic peptide-keyhole limpet hemocyanin conjugates were purified to >95% homogeneity and immunized into rabbits. The polyclonal antisera were commercially prepared using standard protocols by Sigma-Genosys (The Woodlands, TX). The antibody was further affinity-purified by chromatography on a column prepared by crosslinking of peptide to cyanogen bromide-activated Sepharose 4B (ProSci Incorporated, Poway, CA).
Western Blotting
To detect PMAT expression in human small intestine, commercial blots containing human small intestine whole lysates (20 μg/lane) were purchased from Imgenex (San Diego, CA). Immunodetection was performed with the PMAT antibody (1:500) as described above. Control blots included detection with prebleed rabbit serum or antibody preabsorbed with the purified peptide antigen. To detect GAPDH, an internal control, the same blot used for PMAT detection was stripped using Restore Western Blot Stripping Buffer from Pierce and incubated with mouse monoclonal anti-GAPDH antibody (1:1000) (Imgenex), followed by horseradish peroxidase-conjugated goat anti-mouse IgG (1:20,000 dilution).
Immunohistochemistry and Confocal Microscopy
To detect cellular localization of PMAT in human small intestine, commercial slides containing human small intestine tissue were purchased from BioChain Institute Inc. (Hayward, CA). Slides were rinsed twice with PBS and fixed for 30 min at room temperature with 4% (v/v) paraformaldehyde. Slides were then rinsed three times with PBS and incubated in 50 mM NH4Cl in PBS for 15 min to quench the fixative. Slides were then permeabilized with 0.2% Triton X-100 in PBS for 10 min. Potential sites for nonspecific antibody binding were blocked by a 90-min incubation with a blocking buffer (10% fetal bovine serum, 0.1% Triton X-100 in PBS). The slides were incubated with the primary rabbit anti-PMAT antibody (1:500 dilution in blocking buffer) for 1 h, washed three times with PBS containing 0.05% Tween 20, and incubated 30 min with a secondary Alexa Fluor 488 conjugated goat anti-rabbit IgG (1:1000 in blocking buffer) (Invitrogen) in the dark. The slides were then washed three times with PBS containing 0.05% Tween 20. The slides were observed and photographed by a Leica SP confocal microscope (Leica Microsystems, Inc., Bannockburn, IL) with inverted lenses.
Data Analysis
All experiments were performed in triplicate and repeated three times. Data were expressed as mean ± S.D. Statistical significance was determined by Student’s t test. IC50 or Ki values were determined by nonlinear least-squares regression fitting as described previously (Engel and Wang, 2005; Zhou et al., 2006a,b). The Vmax and Km were determined by nonlinear least-squares regression fitting data into the following sigmoidal equation: V = (Vmax · Sγ)/(Sγ + Kmγ) using WinNonlin 5.0 software (Pharsight, Mountain View, CA).
Results
Inhibition of PMAT-Mediated Organic Cation Uptake by Biguanides
As the first step toward characterizing the interaction of metformin with PMAT, we examined the inhibitory effect of metformin on the uptake of a prototype PMAT substrate, MPP+, in MDCK cells stably expressing PMAT protein. MPP+ was chosen as the probe substrate because it is not metabolized in cells, has a low background uptake, and is the best substrate identified for PMAT to date. One-minute uptake of [3H]MPP+ (1 μM) was measured in the presence of metformin at various concentrations. As shown in Fig. 1b, metformin exhibited dose-dependent inhibition on PMAT-mediated MPP+ uptake. The apparent inhibition potency of metformin toward PMAT (Ki) was determined as 2.27 mM. Two structurally related biguanides, buformin and phenformin, were also examined (see structures in Fig. 1a). Buformin and phenformin both inhibited PMAT-mediated MPP+ uptake in a dose-dependent manner (Fig. 1b). The calculated Ki values are 1.51 mM for buformin and 0.25 mM for phenformin. These data demonstrated that biguanides inhibit PMAT. The order of inhibition potency is phenformin > buformin ≥ metformin.
Fig. 1.

Structures of biguanides (a) and inhibition of PMAT-specific MPP+ uptake by biguanides (b). Transport was measured in PMAT-expressing cells and vector-transfected cells (control) at 1 min with 1 μM [3H]MPP+. The PMAT-specific uptake was calculated by subtracting the transport activity in control cells. Each value represents mean ± S.D. (n = 3).
pH-Dependent Metformin Transport by PMAT
To determine whether PMAT accepts metformin as a substrate, direct uptake of [14C]metformin was performed using radiotracer flux studies in MDCK cells stably expressing human PMAT. Figure 2 shows 1-min uptake of [14C]metformin (1 μM) in vector- and PMAT-transfected MDCK cells at two different pH conditions. At pH 7.4, metformin uptake was about 20- to 30-fold higher in PMAT-transfected cells than in vector-transfected cells. At pH 6.6, uptake of [14C]metformin (1 μM) in PMAT-transfected cells was 80-fold higher in vector-transfected cells (Fig. 2). PMAT-mediated metformin uptake at pH 6.6 was ~4-fold higher than that at pH 7.4. Together, our data unequivocally demonstrated that metformin is transported by PMAT, and PMAT-mediated metformin uptake is stimulated by lower extra-cellular pH.
Fig. 2.
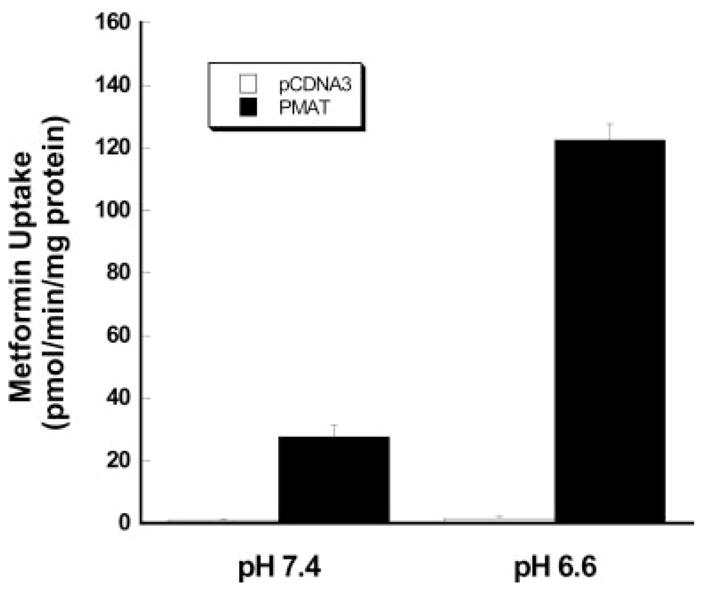
Influence of pH on PMAT-mediated [14C]metformin (1 μM). Vector-transfected cells and PMAT-transfected cells were incubated for 1 min at 37°C. Each value represents mean ± S.D. (n = 3).
Effect of Membrane Potential
We previously showed that PMAT-mediated transport of MPP+ uptake is sensitive to changes in membrane potential (Engel et al., 2004). To examine whether PMAT-mediated metformin transport is also dependent on membrane potential, uptake was measured under various depolarization conditions at pH 7.4. Depolarization of cells with increased extracellular K+ strongly reduced PMAT-mediated metformin uptake (Fig. 3). Barium (Ba2+), an effective potassium channel blocker, also substantially reduced PMAT-mediated metformin uptake in normal physiological buffer. These data suggest that PMAT-mediated metformin transport is electrogenic and facilitated by the physiologic inside-negative membrane potential in mammalian cells.
Fig. 3.
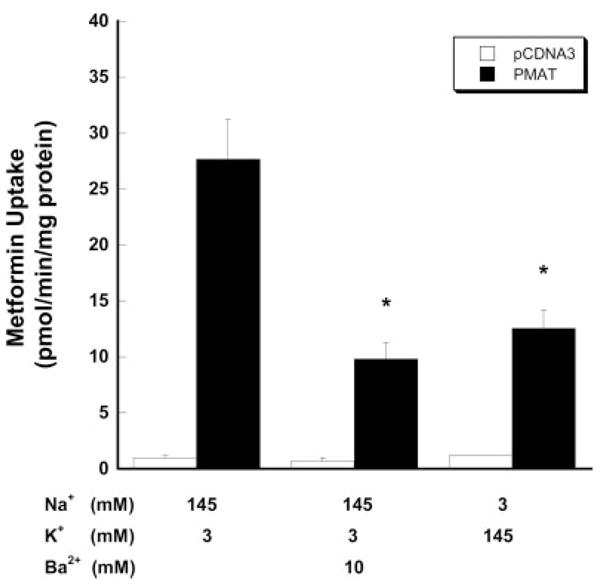
Influence of membrane potential on PMAT-mediated [14C]metformin (1 μM). Vector-transfected cells and PMAT-transfected cells were incubated for 1 min at 37°C. Ba2+ was added to block the potassium channels. To avoid the precipitation of barium by sulfate and phosphate in the KRH buffer, a chloride salt-based buffer (5 mM glucose, 145 mM NaCl, 3 mM KCl, 1 mM CaCl2, 0.5 mM MgCl2, and 5 mM HEPES, pH 7.4) with different compositions of potassium and sodium was used. Each value represents mean ± S.D. (n = 3).
Kinetics of PMAT-Mediated Metformin Transport
To define the initial rate phase of PMAT-mediated metformin transport, time-dependent uptake of [14C]metformin was examined at a low (1 μM) and a high (4 mM) substrate concentration using radiotracer flux studies. As shown in Fig. 4, accumulation of metformin in PMAT-expressing cells increased with time. At both substrate concentrations, PMAT-specific uptake was linear up to 1 to 5 min. Based on these data, kinetic studies were first carried out using 1 min as the incubation time. As shown in Fig. 5a, PMAT-mediated metformin uptake was saturable. Interestingly, the concentration-velocity profile was sigmoidal, which is in contrast to the typical hyperbolic curves observed for other PMAT substrates such as MPP+ and serotonin (Engel et al., 2004). This nontypical kinetic behavior is particularly evident after Eadie-Hofstee transformation, which revealed a homotropic but not a linear fitting (Fig. 5a, inset). Because of the nonclassic kinetics, the data were fitted to the sigmoidal kinetics equation shown under Materials and Methods by nonlinear least-squares regression to obtain the kinetic parameters. The calculated apparent affinity (Km) is 1.32 ± 0.11 mM and maximal velocity (Vmax) is 16.86 ± 0.94 nmol/min/mg protein. The Vmax/Km ratio is 12.77 μl/min/mg protein and the fitted Hill coefficient (γ) is 2.64. To ensure that the sigmoidal kinetics of metformin uptake are not caused by pre-steady-state kinetics, especially at low substrate concentrations, the transport kinetics were further examined with a longer incubation time (3 min) (Fig. 5b). Similar to the 1-min uptake, PMAT-mediated metformin uptake at 3 min was sigmoidal with Km of 1.68 ± 0.28 mM, Vmax of 15.28 ± 1.88 nmol/min/mg protein, and γ of 2.21. None of those parameters obtained from 3-min incubation is significantly different form those determined at 1-min incubation. In addition, it is also unlikely that the atypical kinetics were due to substrate depletion or limitation of detection at low substrate concentrations, since the consumption of extracellular metformin at the lowest substrate concentration is less than 1% of total metformin in the incubation buffer and the absolute metformin uptake radioactivity is well above the measurement limit of liquid scintillation counting.
Fig. 4.
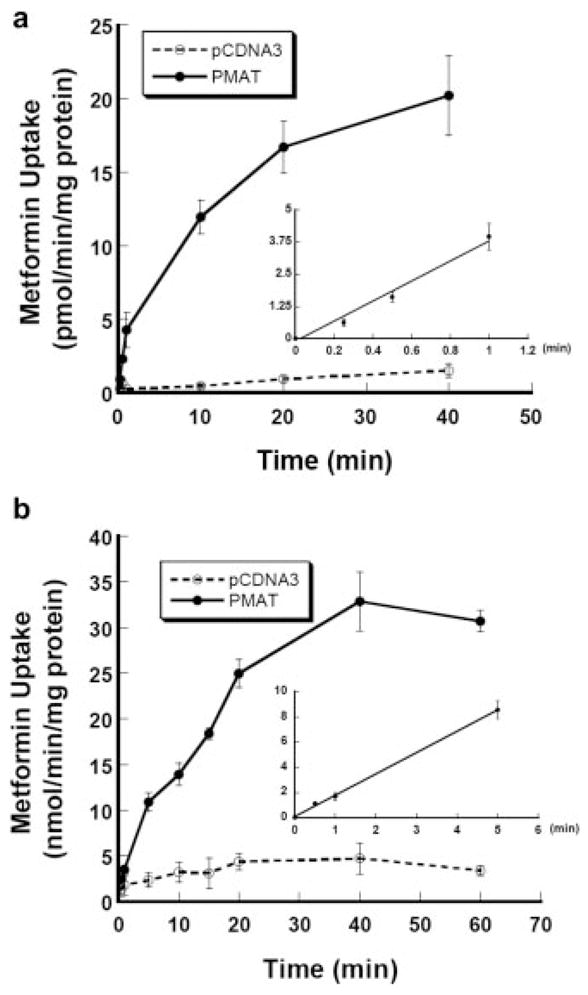
Time courses of [14C]metformin uptake at 1 μM (a) and 4 mM (b) mediated by PMAT. Vector-transfected cells (open circles) and PMAT-transfected cells (solid circles) were incubated at 37°C. Inset, a plot of initial phase with linear fitting of PMAT-specific metformin uptake calculated by subtracting the transport activity in the control cells. Each value represents mean ± S.D. (n = 3).
Fig. 5.
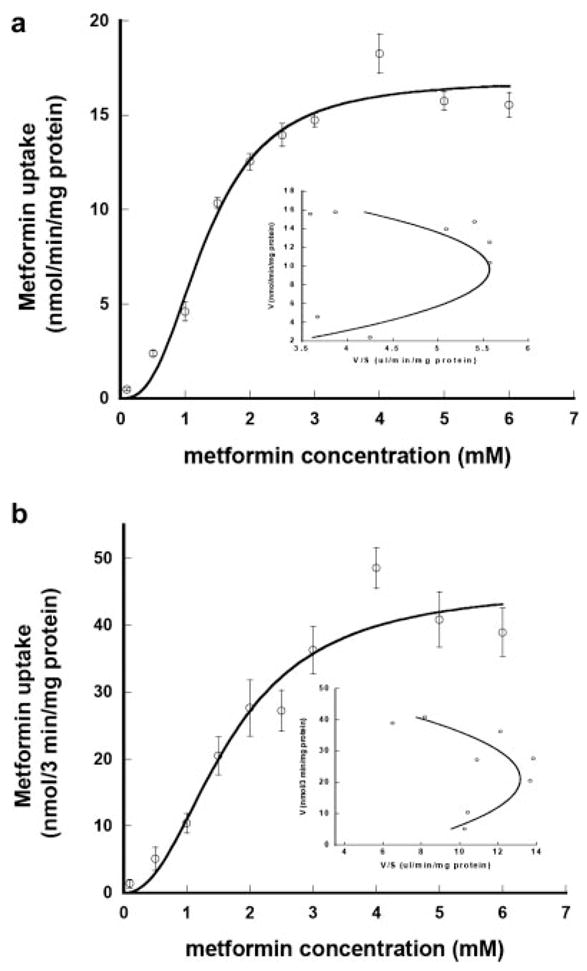
Concentration-dependent transport of [14C]metformin by PMAT at 1 min (a) or 3 min (b) of incubation. The PMAT-specific uptake was measured at 37°C at pH 7.4 and calculated by subtracting the transport activity in control cells. Inset, Eadie-Hofstee plots. Each value represents mean ± S.D. (n = 3).
Expression of PMAT mRNA and Protein in the Human Intestine
To determine whether PMAT transcripts and protein are present in human small intestine, semiquantitative RT-PCR and immunoblotting were used using cDNA and tissue lysates prepared from human small intestine. The primers used in RT-PCR were validated in a previous study (Engel et al., 2004). Figure 6a showed that PMAT transcripts can be specifically amplified from human small intestine cDNA. To determine PMAT protein expression in human small intestine, Western blotting was carried out with a polyclonal affinity-purified peptide antibody against PMAT recently developed in this laboratory (Dahlin et al., 2007). A single, strong band consistent with the expected molecular mass of PMAT (58 kDa) was observed (Fig. 6b). This band disappeared when immunoblotting was carried out with prebleed antiserum (data not shown) or with the anti-PMAT antibody preabsorbed with the synthetic peptide antigen (Fig. 6b). These data clearly demonstrated that the PMAT protein is expressed in human small intestine. To further determine the intestinal expression of PMAT protein at the cellular level, immunofluorescence staining was performed using cryosections of normal human small intestine tissue. Strong PMAT-specific immunolabeling was observed in the mucosal layer (Fig. 6c). Very weak or no staining was seen in submucosal and muscularis externa layers. Within the mucosa, the highest immunofluorescence signal was found at the tips of villi and was primarily concentrated in the thin layer of absorptive cells (enterocytes) (Fig. 6c). Although punctate staining was observed throughout these cells, it appeared to be most concentrated on the apical surface of the enterocytes. The specificity of staining for PMAT was confirmed by the lack of discernible fluorescence when prebleed antisera were used. Together, our data showed that PMAT mRNA and protein are expressed in human small intestine and are present on the apical surface of the enterocytes.
Fig. 6.

Determination of PMAT expression in human small intestine by RT-PCR (a), Western blot (b), and immunohistochemistry (c). a, human small intestine was analyzed for PMAT mRNA expression by semiquantitative RT-PCR. GAPDH primers were used as an internal control. b, immunoblotting was carried out with the anti-PMAT antibody at 1:1000 dilution. The same blots were stripped and blotted with an antibody against an internal standard, GAPDH. c, immunoblotting was carried out with the anti-PMAT antibody at 1:500 dilution. Slides containing human small intestine tissue were permeabilized and stained with either anti-PMAT antibody or prebleed serum (control).
Discussion
In this study, we characterized metformin transport by a novel organic cation transporter, PMAT, and investigated the expression and cellular localization of PMAT in human small intestine. We first showed that metformin is avidly transported by PMAT. We then demonstrated that PMAT-mediated metformin transport is greatly stimulated by acidic pH and exhibits sigmoidal kinetics. Finally, we showed that PMAT protein is expressed in human small intestine and concentrates on the tips of the mucosal epithelial cells. These findings suggest that PMAT may represent an important organic cation transporter in the human intestine and may serve as an absorptive route for metformin and other orally administered organic cation drugs.
Metformin is widely used for the treatment of type II diabetes mellitus. With a pKa value of 12.4, the biguanide drug primarily exists in the protonated cation form at physiological pH. A number of studies have suggested that organic cation transporters play important roles in the disposition and pharmacological action of metformin (Dresser et al., 2002; Wang et al., 2002; Kimura et al., 2005a,b). In particular, metformin was shown to be a substrate of human OCT1 and OCT2, which have been, respectively, implicated in the hepatic uptake and renal excretion of this drug (Wang et al., 2002; Kimura et al., 2005b). In this study, we demonstrated that metformin is an excellent substrate for PMAT, a novel organic cation transporter that transports many structurally diverse type I cations and shares a large substrate overlap with the OCTs (Engel et al., 2004; Engel and Wang, 2005). In Table 1, we compiled literature data of the apparent affinities of OCTs toward biguanides and compared them with those of PMAT determined in this study. The apparent affinity (Km or IC50) of PMAT toward metformin is comparable to that of OCT1 and OCT2, which are all in the low millimolar range. PMAT also interacted with buformin and phenformin, which share the biguanide moiety but are more hydrophobic than metformin (Fig. 1a; Table 1). With increasing hydrophobicity, the apparent affinity of PMAT toward the biguanides increases. A similar trend was also observed for OCT2 (Table 1). However, although the addition of aliphatic hydrophobicity in buformin greatly increased its affinity toward OCT2, its effect on PMAT is rather minor. In contrast, the addition of aromatic hydrophobicity in phenformin significantly increased the affinity toward PMAT. These results are in concert with our previous observation that PMAT favors aromatic over aliphatic hydrophobicity for high-affinity substrate/inhibitor interaction (Engel and Wang, 2005).
TABLE 1.
Apparent affinities of human PMAT and OCT toward biguanides
| Transporter |
Km or IC50 |
||
|---|---|---|---|
| Metformin | Buformin | Phenformin | |
| μM | |||
| PMAT | 1320 | 1500 | 245 |
| hOCT1 | 1470a | N.D. | 10b |
| hOCT2 | 1380c | 203c | 65b |
N.D., not determined.
Values were taken from Kimura et al. (2005a);
Kimura, et al. (2005b). Italic values indicate IC50.
Interestingly, kinetic analysis revealed that PMAT-mediated metformin uptake exhibits sigmoidal kinetics (Fig. 5), which were in contrast to the typical hyperbolic curves observed for other PMAT substrates such as MPP+ and serotonin (Engel et al., 2004). Eadie-Hofstee transformation revealed a “hook”-shaped V/S – V profile, which simulates the homotropic allosteric effect observed in several cytochrome P450 drug-metabolizing enzymes (Shou et al., 1994; Kenworthy et al., 2001; Galetin et al., 2003; Atkins, 2005) and drug efflux transporters (Shapiro and Ling, 1997; Shapiro et al., 1999). For example, such positive cooperativity has been reported for the multidrug resistance protein MDR1 in its interaction with rhodamine 123 and Hoechst 33342 (Shapiro and Ling, 1997; Shapiro et al., 1999). It was proposed, based on kinetic observations, that many of the atypical kinetic profiles might result from multiple substrate bindings within the active site, and sigmoidal kinetics often indicate a positive cooperativity among these sites (Shapiro and Ling, 1997; Shapiro et al., 1999). Thus, it is likely that the sigmoidal kinetics observed in our study may indicate multiple allosteric metformin binding sites with positive cooperativity in PMAT. Multiple allosteric substrate binding sites have been observed in a number of drug-metabolizing enzymes (e.g., CYP3A) and transporters (e.g., MDR1) that exhibit diverse substrate specificity. Positive cooperativity will occur when substrate (or effector) binding to the allosteric site enhances the binding affinity of the substrate to the primary active site (Ueng et al., 1997). Similar to MDR1 or CYP3A, the substrate specificity of PMAT is broad and includes a variety of compounds with diverse structures, such as monoamine neurotransmitters, various aliphatic and aromatic organic cations, and certain nucleosides. Thus, it is likely that PMAT protein may contain a large substrate binding pocket, which allows different substrates to interact at different sites or more than one molecule of the same substrate to be recognized simultaneously. Interaction between these sites may lead to nonhyperbolic kinetic behaviors for some substrates. However, we have no direct evidence of multiple metformin binding sites, and further experiments are needed to elucidate the mechanisms underlining the atypical kinetic behavior of PMAT.
We previously reported that PMAT-mediated transport of the prototype organic cation MPP+ is Na+-independent and sensitive to changes in membrane potential and extracellular pH (Engel et al., 2004; Xia et al., 2006). Consistent with these results, we observed that PMAT-mediated metformin uptake was significantly decreased under depolarization conditions (Fig. 3), indicating that metformin transport is an electrogenic process and that it is the protonated form (i.e., cation) that is being transported into the cells. On the other hand, lowering extracellular pH greatly enhanced the initial rates of PMAT-mediated metformin uptake (Fig. 2). These observations are in contrast to the OCTs, the activities of which are generally compromised under acidic pH (Urakami et al., 1998; Wu et al., 1998; Sweet and Pritchard, 1999; Fujita et al., 2006). With a pKa of 12.4, metformin is almost completely charged at pH 6.6 and 7.4. Thus, the stimulatory effect of acidic pH on PMAT-mediated metformin uptake is unlikely to be caused by a pH effect on substrate fractional protonation. Whereas our recent study indicates that the stimulatory effect of proton may be due to transporter coupling with the inwardly directed proton gradient, the exact molecular mechanism underlying the observed pH effect on PMAT activity awaits further investigation. Regardless, the observation that PMAT favors acidic conditions suggests that the pH of the microenvironment in tissues expressing this transporter may play an important role in regulating the transport activity of PMAT.
In the intestine, luminal pH could be as acidic as pH 6. If PMAT is expressed in the apical membranes of the intestinal epithelial cells, the naturally acidic environment in the lumen can serve as a driving force to promote PMAT-mediated metformin uptake. Recently, Barnes et al. (2006) reported that PMAT mRNA is highly expressed in various segments of the human intestinal tract. Our RT-PCR results further confirmed the presence of PMAT mRNA in the human small intestine (Fig. 6a). Using a polyclonal anti-peptide antibody recently developed in our laboratory (Dahlin et al., 2007), we further demonstrated that PMAT protein (calculated molecular mass of 58 kDa) was expressed in human small intestine (Fig. 6b). Immunofluorescence studies in human intestinal tissue sections revealed strong labeling of the thin layer of mucosal absorptive cells (enterocytes) (Fig. 6c). The immunolabeling appeared to be most concentrated on the apical surface of the enterocytes, but punctate intracellular staining was observed (Fig. 6c). In a different study, Barnes et al. (2006) also reported both intracellular and cell surface staining in various human heart tissue sections using a different antibody. Thus it appears that some PMAT protein may also be expressed in intracellular organelles. Nevertheless, the strong PMAT immunoreactivity observed in the tips of villous epithelium indicate that PMAT protein is expressed at the apical membrane of the enterocytes, where it may play a role in the intestinal absorption of metformin and possibly other cationic drugs.
In summary, our results suggest that PMAT transports metformin, is expressed in human intestine, and may play a role in oral absorption of metformin. Because many drugs and dietary substances are also organic cations, PMAT may also play a role in intestinal absorption of these compounds if they are substrates of PMAT. However, it should be pointed out that the in vitro data present in the current study are circumstantial. Further in vivo/ex vivo studies that directly demonstrate PMAT-mediated metformin transport into intestinal tissues are needed to define the in vivo role of PMAT in intestinal absorption of organic cations.
Acknowledgments
We thank Dr. Karen Engel for her work in setting up the PMAT-MDCK cell line. We also thank Dr. Kelly Hudkins for her insights into the distribution of PMAT in human small intestine.
This work was supported by a grant (GM66233) from the National Institutes of Health. M.Z. is partially supported by a predoctoral fellowship from Eli Lilly Foundation.
ABBREVIATIONS
- OCT
organic cation transporter
- MPP+
1-methyl-4-phenylpyridinium
- MDCK
Madin-Darby canine kidney
- PMAT
plasma membrane monoamine transporter
- KRH
Krebs-Ringer-Henseleit
- RT-PCR
reverse-transcription polymerase chain reaction
- PBS
phosphate-buffered saline
References
- Atkins WM. Non-Michaelis-Menten kinetics in cytochrome P450-catalyzed reactions. Annu Rev Pharmacol Toxicol. 2005;45:291–310. doi: 10.1146/annurev.pharmtox.45.120403.100004. [DOI] [PubMed] [Google Scholar]
- Barnes K, Dobrzynski H, Foppolo S, Beal PR, Ismat F, Scullion ER, Sun L, Tellez J, Ritzel MW, Claycomb WC, et al. Distribution and functional characterization of equilibrative nucleoside transporter-4, a novel cardiac adenosine transporter activated at acidic pH. Circ Res. 2006;99:510–519. doi: 10.1161/01.RES.0000238359.18495.42. [DOI] [PubMed] [Google Scholar]
- Bell PM, Hadden DR. Metformin. Endocrinol Metab Clin North Am. 1997;199(26):523–537. doi: 10.1016/s0889-8529(05)70265-6. [DOI] [PubMed] [Google Scholar]
- Borst SE, Snellen HG. Metformin, but not exercise training, increases insulin responsiveness in skeletal muscle of Sprague-Dawley rats. Life Sci. 2001;69:1497–1507. doi: 10.1016/s0024-3205(01)01225-5. [DOI] [PubMed] [Google Scholar]
- Caspary WF, Creutzfeldt W. Analysis of the inhibitory effect of biguanides on glucose absorption: inhibition of active sugar transport. Diabetologia. 1971;7:379–385. doi: 10.1007/BF01219474. [DOI] [PubMed] [Google Scholar]
- Dahlin A, Xia L, Kong W, Hevner R, Wang J. Expression and immunolocalization of the plasma membrane monoamine transporter (PMAT) in the brain. Neuroscience. 2007;146:1193–1211. doi: 10.1016/j.neuroscience.2007.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresser MJ, Leabman MK, Giacomini KM. Transporters involved in the elimination of drugs in the kidney: organic anion transporters and organic cation transporters. J Pharm Sci. 2001;90:397–421. doi: 10.1002/1520-6017(200104)90:4<397::aid-jps1000>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Dresser MJ, Xiao G, Leabman MK, Gray AT, Giacomini KM. Interactions of n-tetraalkylammonium compounds and biguanides with a human renal organic cation transporter (hOCT2) Pharm Res. 2002;19:1244–1247. doi: 10.1023/a:1019870831174. [DOI] [PubMed] [Google Scholar]
- Engel K, Wang J. Interaction of organic cations with a newly identified plasma membrane monoamine transporter. Mol Pharmacol. 2005;68:1397–1407. doi: 10.1124/mol.105.016832. [DOI] [PubMed] [Google Scholar]
- Engel K, Zhou M, Wang J. Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem. 2004;279:50042–50049. doi: 10.1074/jbc.M407913200. [DOI] [PubMed] [Google Scholar]
- Fujita T, Urban TJ, Leabman MK, Fujita K, Giacomini KM. Transport of drugs in the kidney by the human organic cation transporter, OCT2 and its genetic variants. J Pharm Sci. 2006;95:25–36. doi: 10.1002/jps.20536. [DOI] [PubMed] [Google Scholar]
- Galetin A, Clarke SE, Houston JB. Multisite kinetic analysis of interactions between prototypical CYP3A4 subgroup substrates: midazolam, testosterone, and nifedipine. Drug Metab Dispos. 2003;31:1108–1116. doi: 10.1124/dmd.31.9.1108. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF, Landau BR, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy KE, Clarke SE, Andrews J, Houston JB. Multisite kinetic models for CYP3A4: simultaneous activation and inhibition of diazepam and testosterone metabolism. Drug Metab Dispos. 2001;29:1644–1651. [PubMed] [Google Scholar]
- Kimura N, Masuda S, Tanihara Y, Ueo H, Okuda M, Katsura T, Inui K. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet. 2005a;20:379–386. doi: 10.2133/dmpk.20.379. [DOI] [PubMed] [Google Scholar]
- Kimura N, Okuda M, Inui K. Metformin transport by renal basolateral organic cation transporter hOCT2. Pharm Res. 2005b;22:255–259. doi: 10.1007/s11095-004-1193-3. [DOI] [PubMed] [Google Scholar]
- Klepser TB, Kelly MW. Metformin hydrochloride: an antihyperglycemic agent. Am J Health Syst Pharm. 1997;54:893–903. doi: 10.1093/ajhp/54.8.893. [DOI] [PubMed] [Google Scholar]
- Koepsell H. Organic cation transporters in intestine, kidney, liver, and brain. Annu Rev Physiol. 1998;60:243–266. doi: 10.1146/annurev.physiol.60.1.243. [DOI] [PubMed] [Google Scholar]
- Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, Ogawa O, Inui K. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol. 2006;17:2127–2135. doi: 10.1681/ASN.2006030205. [DOI] [PubMed] [Google Scholar]
- Muller J, Lips KS, Metzner L, Neubert RH, Koepsell H, Brandsch M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT) Biochem Pharmacol. 2005;70:1851–1860. doi: 10.1016/j.bcp.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Scheen AJ. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 1996;30:359–371. doi: 10.2165/00003088-199630050-00003. [DOI] [PubMed] [Google Scholar]
- Shapiro AB, Fox K, Lam P, Ling V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur J Biochem. 1999;259:841–850. doi: 10.1046/j.1432-1327.1999.00098.x. [DOI] [PubMed] [Google Scholar]
- Shapiro AB, Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur J Biochem. 1997;250:130–137. doi: 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- Shou M, Grogan J, Mancewicz JA, Krausz KW, Gonzalez FJ, Gelboin HV, Korzekwa KR. Activation of CYP3A4: evidence for the simultaneous binding of two substrates in a cytochrome P450 active site. Biochemistry. 1994;33:6450–6455. doi: 10.1021/bi00187a009. [DOI] [PubMed] [Google Scholar]
- Sweet DH, Pritchard JB. rOCT2 is a basolateral potential-driven carrier, not an organic cation/proton exchanger. Am J Physiol. 1999;277:F890–F898. doi: 10.1152/ajprenal.1999.277.6.F890. [DOI] [PubMed] [Google Scholar]
- Tucker GT, Casey C, Phillips PJ, Connor H, Ward JD, Woods HF. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol. 1981;12:235–246. doi: 10.1111/j.1365-2125.1981.tb01206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueng YF, Kuwabara T, Chun YJ, Guengerich FP. Cooperativity in oxidations catalyzed by cytochrome P450 3A4. Biochemistry. 1997;36:370–381. doi: 10.1021/bi962359z. [DOI] [PubMed] [Google Scholar]
- Urakami Y, Okuda M, Masuda S, Saito H, Inui KI. Functional characteristics and membrane localization of rat multispecific organic cation transporters, OCT1 and OCT2, mediating tubular secretion of cationic drugs. J Pharmacol Exp Ther. 1998;287:800–805. [PubMed] [Google Scholar]
- Wang DS, Jonker JW, Kato Y, Kusuhara H, Schinkel AH, Sugiyama Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther. 2002;302:510–515. doi: 10.1124/jpet.102.034140. [DOI] [PubMed] [Google Scholar]
- Wright SH. Role of organic cation transporters in the renal handling of therapeutic agents and xenobiotics. Toxicol Appl Pharmacol. 2005;204:309–319. doi: 10.1016/j.taap.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Wu X, Kekuda R, Huang W, Fei YJ, Leibach FH, Chen J, Conway SJ, Ganapathy V. Identity of the organic cation transporter OCT3 as the extraneuronal monoamine transporter (uptake2) and evidence for the expression of the transporter in the brain. J Biol Chem. 1998;273:32776–32786. doi: 10.1074/jbc.273.49.32776. [DOI] [PubMed] [Google Scholar]
- Xia L, Engel K, Zhou M, Wang J. Membrane Localization and pH-dependent Transport of a Newly Cloned Organic Cation Transporter (PMAT) in Kidney Cells. Am J Physiol Renal Physiol. 2007;292:F682–F690. doi: 10.1152/ajprenal.00302.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Hashizume T, Shuhart MC, Davis CL, Nelson WL, Sakaki T, Kalhorn TF, Watkins PB, Schuetz EG, Thummel KE. Intestinal and hepatic CYP3A4 catalyze hydroxylation of 1alpha,25-dihydroxyvitamin D(3): implications for drug-induced osteomalacia. Mol Pharmacol. 2006a;69:56–65. doi: 10.1124/mol.105.017392. [DOI] [PubMed] [Google Scholar]
- Xu Y, Iwanaga K, Zhou C, Cheesman MJ, Farin F, Thummel KE. Selective induction of intestinal CYP3A23 by 1alpha,25-dihydroxyvitamin D3 in rats. Biochem Pharmacol. 2006b;72:385–392. doi: 10.1016/j.bcp.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Zhou M, Engel K, Wang J. Evidence for significant contribution of a newly identified monoamine transporter (PMAT) to serotonin uptake in the human brain. Biochem Pharmacol. 2007a;73:147–154. doi: 10.1016/j.bcp.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Xia L, Engel K, Wang J. Molecular determinants of substrate selectivity of a novel organic cation transporter (PMAT) in the SLC29 family. J Biol Chem. 2007b;282:3188–3195. doi: 10.1074/jbc.M609421200. [DOI] [PMC free article] [PubMed] [Google Scholar]