

Abstract
Excessive activation of G-protein coupled receptor (GPCR) and receptor tyrosine kinase (RTK) pathways has been linked to prostate cancer metastasis. Rac activation by guanine-nucleotide exchange factors (GEFs) plays an important role in directional cell migration, a critical step of tumor metastasis cascades. We found that upregulation of P-Rex1, a Rac-selective GEF synergistically activated by Gβγ freed during GPCR signaling and PIP3 generated during either RTK or GPCR signaling, strongly correlates with metastatic phenotypes in both prostate cancer cell lines and human prostate cancer specimens. Silencing endogenous P-Rex1 in metastatic prostate cancer PC-3 cells selectively inhibited Rac activity and reduced cell migration and invasion in response to ligands of both epidermal growth factor receptor and G-protein coupled CXC chemokine receptor 4. Conversely, expression of recombinant P-Rex1, but not its “GEF-dead” mutant, in non-metastatic prostate cancer CWR22Rv1 cells increased cell migration and invasion via Rac-dependent lamellipodia formation. More importantly, using a mouse xenograft model, we demonstrated that expression of P-Rex1, but not its mutant, induced lymph node metastasis of CWR22Rv1 cells without an effect on primary tumor growth. Thus, by functioning as a coincidence detector of chemotactic signals from both GPCRs and RTKs, P-Rex1-dependent activation of Rac promotes prostate cancer metastasis.
Keywords: Prostate cancer metastasis, P-Rex1, Rac-selective guanine-nucleotide exchange factor, G-protein coupled receptors, receptor tyrosine kinases
Introduction
Prostate cancer is the most commonly diagnosed noncutaneous cancer in American men and the second leading cause of cancer mortality (Jemal et al. 2007). Metastasis, the major cause of prostate cancer death, begins with the migration and invasion of the cancer cells into the surrounding tissues and lymphatics in response to chemotactic and structural signals (Steeg and Theodorescu 2008). Chemoattractants, including chemokines, growth factors and matrix metalloproteases, bind to specific membrane receptors on cancer cells to direct their migration and invasion (Kedrin et al. 2007). Earlier research focused on chemoattractant stimulation of receptor tyrosine kinases (RTKs), but recent evidence from both preclinical and clinical studies has shown that G-protein coupled receptor (GPCR) systems are excessively activated in prostate cancer cells (Daaka 2004; Dorsam and Gutkind 2007), and contribute to the migratory potential of cancer cells during metastasis (Dorsam and Gutkind 2007).
Cell migration towards chemoattractants is dependent on actin cytoskeleton reorganization regulated by the Rho family of small GTPases including Rac, Cdc42 and Rho (Yamazaki et al. 2005). Aberrant Rac signaling is found in some human cancers and mediates cancer metastasis (Sun et al. 2006; Yamaguchi and Condeelis 2007) in part by generating the F-actin-rich thinned-out lamellipodia protrusions at the leading edge of migrating cancer cells. Hyperactivation of Rac has been found in metastatic prostate cancer cells (Knight-Krajewski et al. 2004) and inhibition of Rac activation blocked prostate cancer cell migration and invasion (Gao et al. 2004), suggesting an important role of Rac activation in prostate cancer metastasis.
Rac acts as a molecular switch and cycles between an inactive GDP-bound form and an active GTP-bound form catalyzed by guanine nucleotide exchange factors (GEFs) (Servitja et al. 2003). Both GPCRs and RTKs can activate Rac via Rac-selective GEFs (RacGEFs) (Ma et al. 1998; Schiller 2006). Tiam1, a RacGEF, that contributes to the metastasis of colorectal and breasts cancers (Minard et al. 2004), is also increased in prostate cancer (Engers et al. 2006). However, Tiam1 is already elevated in pre-cancerous high-grade prostatic intraepithelial neoplasia (Engers et al. 2006), suggesting that Tiam1 upregulation may not directly trigger prostate cancer metastasis.
During a recent screen of cancer cell lines, we found a selective elevation of P-Rex1 expression in metastatic prostate cancer cells. P-Rex1 is a GEF that selectively activates Rac through its catalytic Dbl-homology (DH) domain (Welch et al. 2002; Rosenfeldt et al. 2004; Hill et al. 2005). P-Rex1 helps regulate the function of neutrophils (Welch et al. 2005), macrophages (Wang et al. 2008) and neuronal migration in the developing nervous system (Yoshizawa et al. 2005). P-Rex1 is among the few RacGEFs that is known to be directly activated by free Gβγ subunits released from G-proteins during GPCR signaling (Welch et al. 2002). In the developing nervous system, P-Rex1 was also stimulated by agonists such as nerve growth factor which act through RTKs to activate phosphoinositide-3 (PI3) kinases and their downstream effectors (Yoshizawa et al. 2005). Interestingly, mammalian target of rapamycin (mTOR), which integrates nutrient and growth factor signals (Fingar and Blenis 2004), can also activate P-Rex1 via mTOR complex 2 formation to activate Rac and promote cell migration (Hernandez-Negrete et al. 2007; Dada et al. 2008). Thus, P-Rex1 could function as a molecule that integrates signals from several input pathways simultaneously activated by the cell microenvironment and could be a novel therapeutic target in many pathological conditions, including human cancers (Hernandez-Negrete et al. 2007).
The purpose of this study was to determine the potential roles of upregulated P-Rex1 in human prostate cancer metastasis. Our studies revealed that immunohistochemical staining for P-Rex1 was more intense and uniform in human lymph node metastases of prostate cancer than in the corresponding primary prostate tumors or adjacent benign tissues. Moreover, silencing endogenous P-Rex1 protein in metastatic prostate cancer PC-3 cells suppresses Rac-dependent cell migration and invasion while exogenous expression of P-Rex1 in nonmetastatic CWR22Rv1 increases cell migration and invasion via Rac activation in vitro and leads to the development of lymph node metastases following orthotopic transplantation in SCID mice. These data collectively identify P-Rex1 as a novel promoter of prostate cancer metastasis.
Results
P-Rex1 is upregulated in metastatic human prostate cancer cell lines
Expression levels of P-Rex1 mRNA were very low in human normal primary prostate epithelial cells (PrEC) and in nonmetastatic prostate cancer LNCaP and CWR22Rv1 cells, but were remarkably higher in metastatic prostate cancer PC3-LN4 and PC-3 cells (Figure 1a). Transwell chamber assays were used to measure the directional migration and invasion abilities of those prostate cancer cells in response to NIH3T3 fibroblast conditioned medium (CM), which contains many cytokines and growth factors, and is commonly used for in vitro metastasis assays (Albini and Benelli 2007; Mensing et al. 1983). As shown in Figure 1b, there is a direct correlation between the P-Rex1 mRNA expression levels in these cells and their ability to migrate and invade through the Matrigel. Western blot analysis and immunofluorescent staining using P-Rex1-specific antibody 6F12 (Welch et al. 2005) and 4A3 (Yoshizawa et al. 2005), respectively, confirmed that P-Rex1 protein was significantly higher in PC-3 cells than in LNCaP and CWR22Rv1 cells (Figure 1c). Most P-Rex1 in PC-3 cells is cytosolic, with a small fraction found near the plasma membrane (Figure 1c, right).
Figure 1.

Upregulation of P-Rex1 in human metastatic prostate cancer cell lines and in human prostate cancer specimens. (a) Analysis of P-Rex1 mRNA expression in human prostate cell lines by quantitative real-time RT-PCR. Bars represent the mean ± S.E. of P-Rex1 mRNA levels (n=5). Inset: Conventional RT-PCR for P-Rex1 and β-actin. (b) Transwell migration and invasion assays of prostate cancer cells toward NIH3T3 CM. Results are represented as mean ± S.E. per 10,000 loaded cells (n=3). (c) Left: Western blot analysis of P-Rex1 protein using the anti-P-Rex1 antibody 6F12. Right: Representative immunofluorescence staining of prostate cancer cells for P-Rex1 protein with the anti-P-Rex1 antibody 4A3, followed with AlexaF488 (green)-linked secondary antibody. DAPI staining (blue) indicates the nuclei. (d) Immunohistochemical staining of human specimens for P-Rex1 protein using the antibody 4A3 as indicated by brown color. The left panel shows representative results from primary tumors in two cases (d1 and d2) as well as their paired prostate tumor metastases in lymph nodes (d3 and d4, respectively). Green and black arrows point to noncancerous prostate epithelial cells and localized prostate cancer, respectively, while red arrows indicate prostate cancer metastasis to lymph nodes. The average H-score was used to grade P-Rex1 expression levels. The right panel shows the mean ± S.E of eight cases. *p=0.016, **p=0.0055, ***p=0.0004.
P-Rex1 protein is upregulated in human prostate adenocarcinoma and lymph node metastases
The P-Rex1 protein level in cells was measured immunohistochemically in primary tumors and a paired lymph node metastasis in archived samples from 8 prostate cancer patients. Figure 1d shows representative results from primary tumors (d1 and d2) alongside the paired lymph node metastases (d3 and d4, respectively) for two cases. Compared with the noncancerous adjacent prostate tissue in primary tumors (green arrows), P-Rex1 staining intensity is only slightly and heterogeneously increased in the localized prostate carcinoma (black arrows), but higher and more homogeneous in lymph node metastases (red arrows). An Automated Cellular Imaging System (Gao et al. 2005) was used to get the histo-score (H-score) indices of P-Rex1 protein levels shown in Figure 1d (right panel). The H-score was increased an average of 3.7-fold in lymph node metastases and 1.6-fold in localized prostate cancer relative to nearby noncancerous prostate tissues, differences that were statistically significant. Furthermore, the H-score was significantly higher in lymph node metastases than in the localized prostate cancer from the same patient, raising the possibility that the heterogeneous upregulation of P-Rex1 in the primary prostate tumor is associated with progression to the metastatic phenotype.
Silencing endogenous P-Rex1 suppresses metastatic prostate cancer cell migration and invasion by reducing Rac activity
To directly demonstrate a role for P-Rex1 in metastatic prostate cancer cells, endogenous P-Rex1 expression in PC-3 cells was silenced with a P-Rex1-specific siRNA (Yoshizawa et al. 2005). Both western blot assay (Figure 2a, upper panel) and immunostaining analysis (Figure 2a, lower panel) indicated that this P-Rex1 siRNA reduced the P-Rex1 protein level in PC-3 cells approximately 80% compared to P-Rex1 levels with mock or scramble siRNA transfection. Silencing P-Rex1 reduced PC-3 cell migration and invasion in response to NIH3T3 CM by approximately 50% when compared to the scramble siRNA (Figure 2b). In addition, silencing P-Rex1 caused a large decrease in the level of activated GTP-bound Rac1 with minimal effect on activated GTP-bound RhoA (Figure 2c, upper panel), confirming that endogenous P-Rex1 functions as an activator of Rac1 but not RhoA in PC-3 cells. These results were further supported by the data shown in Figure 2c, lower panel, where silencing P-Rex1 significantly attenuated active Rac-dependent lamellipodia formation in PC-3 cells. To understand the relationship between the decreased Rac activity and reduced cell migration ability, we verified that NSC23766 (NSC), which selectively blocks Rac activation by Tiam1 (Gao et al. 2004), also blocked the P-Rex1/Rac1 interaction (see Supplement). NSC reduced PC-3 cell migration in a dose-dependent manner, causing about a 70% inhibition at 100 μM (Figure 2d). In contrast, Rho inhibition with 2 μg/ml of cell permeable C3 transferase (C3), a Rho inhibitor (recommended concentration is 0.5 – 1 μg/ml) or selective inhibition of downstream effector Rho kinase with 15 μM Y-27632 (Y, Ki = 140 nM) only caused a 35% reduction. Altogether, our data show that silencing endogenous P-Rex1 impairs Rac activation, resulting in decreased lamellipodia formation and an inhibition of metastatic prostate cancer cell migration and invasion.
Figure 2.

Silencing endogenous P-Rex1 suppressed prostate cancer PC-3 cell migration and invasion by reducing Rac activation. (a) Silencing P-Rex1 by siRNA as demonstrated by western blot analysis (upper panel) and immunofluorescence staining (lower panel) of P-Rex1 protein in PC-3 cells. A mock transfection and scramble siRNA were used as controls. The western blot image is representative of 3 separate experiments. (b) Silencing P-Rex1 reduced PC-3 cell migration and invasion in response to NIH3T3 CM. Bars show mean ± S.E. with *p<0.05 compared to control (n=3). (c) Silencing P-Rex1 in PC-3 cells reduced Rac1 activity (upper left panel) without affecting RhoA activity (upper right panel). Immunofluorescence staining of F-actin (lower panel) shows a corresponding loss of lamellipodia formation in P-Rex1 silenced PC-3 cells. Graphed data are mean ± S.E (n=3). At least 50 cells were counted in each experiment with *p<0.05 when compared to scramble siRNA; inset images are representative cells. (d) Migration of PC-3 cells without (CN) or with indicated concentrations of Rac activation inhibitor NSC, Rho inhibitor C3 transferase (C3) or Rho-kinase inhibitor Y27632 (Y). Bars show the mean ± S.E. (n=3) with *p<0.05.
Endogenous P-Rex1 contributes to directional prostate cancer cell migration in response to both GPCR and RTK ligands
Because NIH3T3 CM contains a range of known and unknown factors capable of acting on both GPCRs and RTKs, we next examined the effects of silencing endogenous P-Rex1 on GPCR- or RTK-dependent prostate cancer cell migration. CXCL12 is a selective agonist for the G-protein coupled CXC chemokine receptor 4 (CXCR4). Epidermal growth factor (EGF) activates RTK pathways. Both CXCR4 and EGF receptor have been linked to prostate cancer metastasis (Arya et al. 2004; Ching et al. 1993). In the absence of CXCL12 or EGF, PC-3 cells displayed less spreading and less lamellipodia formation when transfected with P-Rex1 siRNA (Figure 3a). Silencing endogenous P-Rex1 caused a noticeable decrease in lamellipodia formation in response to CXCL12 (200 ng/ml). When PC-3 cells were treated with EGF (50 ng/ml), they were spread out with lamellipodia formation irrespective of whether they were transfected with scramble siRNA or P-Rex1 siRNA, with a tendency for lamellipodia formation to be decreased after P-Rex1 siRNA transfection. Figure 3b shows complementary data from Transwell migration assays. When cells were transfected with scramble siRNA (Figure 3b), CXCL12 and EGF increased cell migration by about 1.7 fold and 2.1 fold, respectively, from a basal level of 272 ± 28 migrated cells/10,000 cells loaded in the Transwell chamber. However, after transfecting cells with P-Rex1 siRNA, CXCL12 had little stimulatory effect (1.1 fold) while EGF still increased cell migration about 1.7-fold from a basal level of 217 ± 25 migrated cells/10,000 cells loaded. Thus, transient transfection with P-Rex1 siRNA, which was capable of decreasing P-Rex1 protein levels about 80% (see Figure 2a) also caused about an 80% reduction in the relative ability of the PC-3 cells to migrate in response to CXCL12 but only reduced EGF-induced cell migration by 40%.
Figure 3.

Endogenous P-Rex1 contributes to directional prostate cancer cell migration in response to both GPCR and RTK ligands. (a) PC3 cells transiently transfected with scramble or P-Rex1 siRNA were plated on coverslips. The cells were starved in RPMI 1640 medium for 14 h, stimulated without (none) or with CXCL12 (200 ng/ml) or EGF (50 ng/ml) for 30 min. Cells were stained for F-actin (red) and with DAPI to visualize the nuclei (blue). (b) Silencing P-Rex1 reduced PC-3 cell migration in response to CXCL12 (200 ng/ml) or EGF (50 ng/ml). Bars show mean ± S.E. with *p<0.05 compared to respective control (n=4). The basal migratory abilities (none) of cells transfected with scramble siRNA and P-Rex1 siRNA were 272 ± 28 and 217 ± 25 per 10,000 cells loaded in the Transwell chamber, respectively. Arrows point to lamellipodia.
Exogenous P-Rex1 promotes prostate cancer cell migration and invasion via Rac activation
We next used nonmetastatic prostate cancer CWR22Rv1 cells as an in vitro model to investigate the effect of exogenously expressed P-Rex1 on prostate cancer metastasis. As shown in Figure 4a (left panel), with about 50% transfection efficiency, expression of P-Rex1 increased CWR22Rv1 cell migration by about two-fold. Deletion of the DH domain of P-Rex1 completely abolished its ability to enhance the CWR22Rv1 cell migration. A similar dependence on the DH domain was seen in PC3-LN4 cells when green fluorescent protein (GFP)-tagged P-Rex1 or its P-Rex1 (-DH) mutant was transiently expressed at similar levels (Figure 4a, right panel). Thus, increasing P-Rex1 expression in prostate cancer cells increases their RacGEF-dependent migration ability.
Figure 4.

P-Rex1 promotes prostate cancer cell migration and invasion via Rac activation. (a) CWR22Rv1 cells (left panel) or PC3-LN4 cells (right panel) were transfected with control GFP, GFP-tagged wild-type P-Rex1 or P-Rex1(-DH) mutant for 48 h, and then subjected to Transwell migration assay for 12 h (CWR22Rv1 cells) or 5 h (PC3-LN4) toward NIH3T3 CM. Transfected PC3-LN4 cells were selected by flow cytometry cell sorting based on GFP fluorescence prior to migration assays due to a low transfection efficiency (~25%). Bars show mean ± S.E. with *p<0.05 compared to control set at 100% (n=3). Inset: Expression of GFP-tagged P-Rex1 and P-Rex1(-DH) protein detected by western blot analysis using anti-GFP antibody. (b) CWR22Rv1 cells stably expressing wild-type P-Rex1, P-Rex1 “GEF-dead” mutant or control vector were subjected to migration and invasion assays for 12 h. Cells stably expressing wild-type P-Rex1 were pretreated without (none) or with 50 μM NSC for 15 min. Bars show mean ± S.E. with *p<0.01 compared to control (n=3). Inset: Western blot analysis of expression levels of P-Rex1 and its mutant. (c) P-Rex1 activates Rac1 in prostate cancer cells. The amount of activated GTP-bound Rac1 was measured by a Rac activation assay. The image shown is representative of 3 separate experiments. (d) P-Rex1 increases lamellipodia formation. Serum-starved CWR22Rv1 cells or PC-3 cells (positive control) were incubated with NIH3T3 CM for 5 h and then stained with rhodamine-conjugated phalloidin (red) for detection of F-actin.
Furthermore we generated stable CWR22Rv1 cells expressing either wild-type P-Rex1 or a P-Rex1(E56A/N238A) “GEF-dead” mutant (Hill et al. 2005). Although P-Rex1 mutant expression was similar to wild-type P-Rex1 (Figure 4b, inset), cells expressing wild-type P-Rex1, but not its mutant, showed a 3-fold increase in cell migration and invasion (Figure 4b) when compared to control cells transfected with vector. Rac activation inhibitor NSC (50 μM) abolished the increases in migration and invasion observed in cells stably expressing wild-type P-Rex1 (Figure 4b), suggesting that stably expressed P-Rex1 in CWR22Rv1 cells functioned in a Rac-dependent manner similar to that seen in PC-3 cells. Indeed, CWR22Rv1 cells expressing wild-type P-Rex1 had significantly higher Rac activity, similar to PC-3 cells, when compared to CWR22Rv1 control cells or cells expressing the P-Rex1 mutant (Figure 4c). In addition, cell spreading was seen in PC-3 cells and CWR22Rv1 cells expressing wild-type P-Rex1 in association with active Rac-dependent lamellipodia (Figure 4d). In contrast, CWR22Rv1 control cells or cells expressing the P-Rex1 mutant had fusiform shapes and lacked lamellipodia. These findings are consistent with the notion that P-Rex1 stimulates Rac activity to induce the lamellipodia formation needed for effective migration and invasion of prostate cancer cells.
P-Rex1 does not affect prostate tumor growth but induces spontaneous lymph node metastasis in mouse models
Metastatic PC3-LN4 cells, CWR22Rv1 control cells, and cells stably expressing wild-type P-Rex1 or its “GEF dead” mutant were subcutaneously injected into nude mice (6 mice per group), and tumor volumes were measured once per week. There was no statistical difference in the subcutaneous tumor growth rate among these groups (Figure 5a) and tumor incidence was 100% in all four groups, suggesting that P-Rex1 does not have a direct impact on prostate tumor growth.
Figure 5.
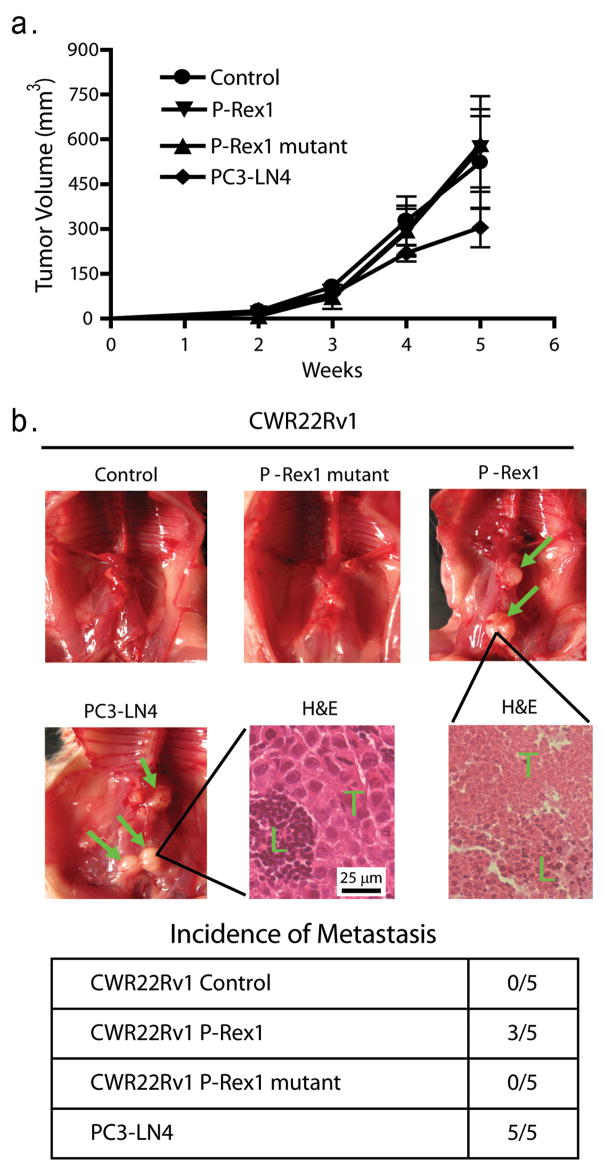
In vivo prostate tumor growth and spontaneous metastasis to mouse lymph nodes (a) 1 × 106 PC3-LN4 cells or CWR22Rv1 cells stably expressing either wild-type P-Rex1, P-Rex1 “GEF-dead” mutant or vector were injected subcutaneously into Nu/Nu mice (6 mice per group). Tumor growth was monitored once per week for 4 weeks. One way ANOVA indicates no statistical differences in tumor growth among the four groups. (b) Intraprostatic injection of CWR22Rv1 cells or PC3-LN4 cells into male NOD/SCID mice (50,000 cells in 15 μl culture medium/mouse). Nine weeks post-injection, mice were sacrificed to examine spontaneous metastases (green arrow) of prostate tumor into lymph nodes. H&E staining of mouse lymph nodes from the PC3-LN4 group and the CWR22Rv1 expressing P-Rex1 group show infiltration of tumor cells (T) into the lymph node tissue (L). The incidence of spontaneous metastasis in each group is shown in the table.
We next used an orthotopic model (Kovar et al. 2006) to determine the role of P-Rex1 in the spontaneous metastasis of prostate cancer to lymph nodes. The CWR22Rv1 stable transfectants were implanted into an anterior prostate lobe of immunodeficient male NOD/SCID mice. The metastatic PC3-LN4 cell line was used as a positive control. All mice bore large primary prostate tumors of similar size (data not shown). All five mice in the PC3-LN4 group had macroscopic lymph node metastases (green arrows, Figure 5b). There was no visible metastasis detected in any mice bearing tumors derived from CWR22Rv1 control cells or cells expressing the P-Rex1 mutant. In contrast, expression of wild-type P-Rex1 in CWR22Rv1 cells gave rise to lymph node metastases (3 out of 5 mice). H&E staining of the mouse lymph nodes showed widespread infiltration of tumor cells (T) into the lymph node tissue (L) for both the PC3-LN4 group with the larger tumor cells (bottom middle panel) and the CWR22Rv1/P-Rex1 group with smaller tumor cells (bottom right panel). These results demonstrate that P-Rex1 promotes spontaneous prostate cancer metastasis in vivo.
Discussion
Current evidence suggests that Rac activity is crucial for migration and invasion of cancer cells, and aberrant Rac signaling is often seen in cancer metastasis (Sun et al. 2006). Since Rac mutations are rarely detected in cancer, research is focused on the factors that govern Rac activation. RTKs and GPCRs are among the plasma membrane proteins capable of initiating responses that lead to Rac activation through RacGEFs. Here, we report that P-Rex1, a RacGEF that functions downstream of both GPCR and RTK signaling, is markedly upregulated in metastatic prostate cancer and may function as one of the important molecules in metastatic signaling pathways in prostate cancer cells.
P-Rex1 is a relatively abundant protein normally expressed in cells of a hematopoietic or neuronal lineage (Welch et al. 2002). We found that endogenous P-Rex1 expression levels are very low in normal prostate epithelial cells and nonmetastatic prostate cancer cells, but remarkably elevated in the metastatic prostate cancer cells. In addition, our studies using human prostate cancer specimens showed a progressive increase of P-Rex1 protein in cells from non-cancerous prostate tissues, localized prostate carcinoma and prostate cancer that has metastasized to a lymph node. Using a mouse xenograft model, we demonstrated that P-Rex1 expression induced lymph node metastasis of nonmetastatic prostate cancer CWR22Rv1 cells without an effect on primary tumor development and growth. These results suggest that upregulated P-Rex1 may contribute to prostate cancer metastasis.
P-Rex1 is characterized by a DH domain possessing its catalytic activity in tandem with a pleckstrin-homology (PH) domain, two adjacent DEP (disheveled, EGL-10 and pleckstrin homology) domains, two adjacent PDZ (postsynaptic density disc-large zo-1) domains and a tail with polyphosphate 4-phosphatase homology (Welch et al. 2002). The PH domain of RacGEFs binds to PIP3 in the cell membrane, which can either directly activate the RacGEF or anchor the molecule to facilitate its activation by other proteins. P-Rex1 is among the few RacGEFs also known to be selectively activated by Gβγ subunits (Welch et al. 2002; Rosenfeldt et al. 2004; Hill et al. 2005). Moreover, the binding of cytosolic P-Rex1 to both Gβγ subunits and PIP3 leads to a synergistic activation of this RacGEF (Welch et al. 2002; Barber et al. 2007). Interestingly, P-Rex1 has recently been shown to interact with the cellular nutrient sensor mTOR via a DEP domain and functions as an effector mediating Rac activation through the rapamycin-insensitive mTORC2 pathway (Hernandez-Negrete et al. 2007). Thus, P-Rex1 appears to be positioned as a coincidence detector, something that could be crucial for a cancer cell navigating through a range of chemotactic signal gradients reaching it from different directions.
Indeed, our data indicate endogenous P-Rex1 can regulate metastatic prostate cancer cell migration and invasion. For instance, silencing endogenous P-Rex1 in metastatic PC-3 cells reduced their migration and invasion abilities toward chemoattractants in NIH3T3 CM by approximately 50%. As expected, silencing endogenous P-Rex1 also largely decreased Rac1 activity without a significant effect on RhoA activity. The loss of Rac1 activity is important for the reduction of the PC-3 cell migration since NSC, which blocks the activation of Rac1 by P-Rex1, also significantly reduced PC-3 cell migration. Those results indicate a significant contribution of endogenous P-Rex1 to the maintenance of Rac activity that is important for PC-3 cells to migrate in response to chemoattractants in NIH3T3 CM. Furthermore, silencing P-Rex1 attenuated the cell migratory response to CXCL12, an agonist of G-protein coupled CXCR4, by over 80% while RTK agonist EGF-stimulated cell migration was only reduced about 40%, suggesting that P-Rex1 is especially important for chemotactic factors that stimulate GPCRs. The relative insensitivity of EGF-stimulated responses to P-Rex1 silencing may be due to activation of a greater range of RacGEFs following stimulation of RTK pathways.
A wide range of evidence has appeared over the past several years supporting a role for activation of G-proteins by GPCRs during cancer growth and metastasis (Daaka 2004; Dorsam and Gutkind 2007). The Gα subunit of the G12 subfamily of G-proteins is directly linked to intracellular pathways that promote prostate cancer invasion (Kelly et al. 2006). Pertussis toxin (PTx), which selectively inactivates Gi-proteins, also suppresses prostate cancer metastasis in a mouse model (Bex et al. 1999), demonstrating the importance of Gi-proteins. Several prostate cancer metastasis-associated GPCRs including CXCR4 (Arya et al. 2004) and CC chemokine receptor 2 (Lu et al. 2007) are coupled to Gi-proteins (Moepps et al. 1997; Jimenez-Sainz et al. 2003). Gαi is not required for neutrophil chemotaxis mediated by Gi-coupled receptors (Neptune et al. 1999). Instead, disruption of Gβγ signaling inhibits this neutrophil chemotaxis (Lehmann et al. 2008). Inhibition of Gβγ-mediated signaling has been shown to inhibit prostate cancer cell growth in vitro and prostate tumor formation in vivo (Bookout et al. 2003). In addition, we found (Qin, unpublished observations) that the migration and invasion of P-Rex1-expressing metastatic prostate cancer cells are significantly attenuated by PTx and M119, a Gβγ inhibitor (Bonacci et al. 2006). It therefore appears that chemotactic factors that stimulate GPCRs promote prostate cancer metastasis via a Gβγ-dependent P-Rex1/Rac activation pathway. Further investigation is in progress.
Characteristically, the DH domain accounts for the GEF activity leading to Rac activation (Hoffman and Cerione 2002). We found that transient expression of recombinant P-Rex1, but not a DH-deleted P-Rex1 mutant, increases cell migration in both CWR22Rv1 cells and PC3-LN4 cells normally having undetectable and mid-range levels of endogenous P-Rex1, respectively. These results prompted us to establish stable CWR22Rv1 cells expressing P-Rex1 or its “GEF-dead” mutant. Compared to control cells, cells stably expressing P-Rex1 have 3-fold higher migration and invasion abilities that could be largely abolished by NSC, the inhibitor of Rac activation. The failure of the P-Rex1 “GEF-dead” mutant to activate Rac caused a corresponding loss of its ability to induce lamellipodia formation in prostate cancer cells. Consequently, the P-Rex1 “GEF-dead” mutant failed to stimulate prostate cancer cell migration and invasion in vitro and spontaneous metastasis in an orthotopic xenograft model. Altogether, our studies demonstrated the importance of the classical action of P-Rex1 as a RacGEF in prostate cancer metastasis.
Other chemotactic factors acting through RTKs can increase PIP3 production through the PI3K signaling pathway (Cantley 2002) and contribute to prostate cancer metastasis (Mimeault and Batra 2006). In fact, the silencing of P-Rex1 reduced EGF stimulated migration by 40%, suggesting the importance of PIP3-dependent P-Rex1 activation in RTK-promoted prostate cancer metastasis. Furthermore, mTOR, the nutrient sensor in cells (Fingar and Blenis 2004), also uses P-Rex1 to stimulate cell migration (Hernandez-Negrete et al. 2007). Thus, by functioning as a molecule that integrates the simultaneous but separate inputs from GPCRs, RTKs, and mTOR nutrient sensing in its local microenvironment, the synergistic activation of P-Rex1 could function as a coincidence detector that helps regulate the direction of prostate cancer cell movement. Unlike Rac1, P-Rex1 has limited tissue expression and its knockout is not embryonically lethal (Welch et al. 2005). P-Rex1 contributes to the regulation of neutrophil function, but is not essential for either chemotaxis or degranulation (Welch et al. 2005). Thus, P-Rex1 could be an attractive drug target since blockade of its expression or actions may impair metastatic prostate cancer cell navigation with less destructive side effects than traditional chemotherapy, thereby making the cancer a better target for surgery, radiation and immunotherapy.
Materials and Methods
Cells and reagents
PC-3, LNCaP and CWR22Rv1 cell lines (ATCC) were grown in RPMI-1640 medium with 10% fetal bovine serum (FBS). The PC3-LN4 cell line (Melanie Simpson, University of Nebraska at Lincoln) was cultured in DMEM with 10% FBS, non-essential amino acids and 1.0 mM sodium pyruvate. Human prostate epithelial cell line PrEC (Zafar Nawaz, University of Miami) was cultured in PrEGM (Cambrex).
Additional reagents were NSC23766, Y27632 and anti-GFP antibody (EMD Biosciences); cell permeable C3 transferase and F-actin visualization kit (Cytoskeleton); Rac activation assay kit, Rho assay reagent, anti-Rac1 antibody (clone 23A8), and Immobilon-FL (Millipore); mouse anti-RhoA (26C4), and rabbit anti-HA and anti-β-actin antibodies (Santa Cruz); FBS (Hyclone); Lipofectamine™ 2000, AlexaF488-labeled secondary antibody, RPMI-1640 and DMEM (Invitrogen); EGF, Matrigel and fibronectin (BD Biosciences); CXCL12 (R&D Systems); and IRDye-labeled secondary antibodies (LI-COR Biosciences). Descriptions of rat monoclonal anti-P-Rex1 antibody 4A3, untagged and GFP-tagged wild-type P-Rex1 and DH-domain deleted mutant have been published (Yoshizawa et al. 2005). P-Rex1 monoclonal antibody 6F12 was a gift from Marcus Thelen, Institute for Research in Biomedicine, Bellinzona, Switzerland. P-Rex1 “GEF-dead” mutant was generated as described previously (Hill et al. 2005).
Cell invasion and migration assays
Matrigel invasion assays were performed at 37 °C using 24-well Transwell inserts (Corning) coated with 30 μg of Matrigel. Cells (50,000) suspended in 200 μl of serum-free medium were seeded into the upper chamber with 600 μl of medium containing chemoattractants in the lower chamber. Cells that invaded through the membrane were stained, counted, and normalized relative to 10,000 seeded cells. Transwell cell migration assays were performed similarly, but without Matrigel. NIH3T3 CM (Albini and Benelli 2007) was the chemoattractant in routine experiments. To study the effect of silencing endogenous P-Rex1 on the EGF receptor- or CXCR4-dependent cell migration, the insert membrane was pre-coated with fibronectin (2 μg/ml) and EGF or CXCL12 was used as the chemoattractant.
Conventional RT-PCR and quantitative real-time RT-PCR
Conventional and real-time RT-PCR analyses were done as described previously (Cao et al. 2006). P-Rex1 primers resulting in a 163-bp product are, forward 5′-CCTTCTTCCTCTTC GACAAC-3′ and reverse 5′-CCATCTTCCACATTCTCCAC-3′. Conventional PCR products were separated by 4% agarose gel electrophoresis and confirmed by DNA sequencing.
Fluorescence microscopy
P-Rex1 was visualized in methanol-fixed prostate cancer cells with antibody 4A3 and AlexaF488-conjugated secondary antibody. F-actin was stained with rhodamine-labeled phalloidin. Nikon Ti-80 microscope images were captured by a CoolSNAP-CF camera and processed with Image-Pro® Plus software (v6.1). Lamellipodia were identified as smooth convex stretches of perpendicular actin stain along cell edges. Images are representative of at least 50 cells.
Immunohistochemistry analysis of human prostate tissues
Formalin-fixed, paraffin-embedded blocks of prostate cancer were archived with informed consent for research purposes by the Creighton University Department of Pathology, and this use was approved by our Institutional Review Board. Immunohistochemistry was performed using standard techniques. Tissue sections were stained with the 4A3 anti-P-Rex1 antibody, counterstained with hematoxylin, and analyzed by the Automated Cellular Imaging System (ChromaVision Medical Systems) as described previously (Gao et al. 2005). Negative controls were performed without the primary antibody. A “histo-score” (H score) was calculated from 10 randomly chosen 200X fields by multiplying the percentage (P) of cells staining positive for P-Rex1 with the average intensity (I), and averaged for each sample. The maximum H-score of 255 would be obtained if all the cells (P=1.0) in the lesion stained with maximal intensity (I=255).
RNA interference and stable cell lines
PC-3 cells in suspension were transfected with 50 nM scramble siRNA (negative control #1 siRNA, Ambion) or synthesized P-Rex1 siRNA (target sequence 5′-GCAACGACTTCAA GCTGGTGGAGAA-3′) using Lipofectamine™ 2000. After 5 h of incubation, 90% of the transfection medium was replaced with fresh culture medium. The following day, adherent PC-3 cells were re-transfected with the same siRNA. Two days later, cells were harvested for western blot analysis of Rex1 protein expression or used in Transwell assays. Stable CWR22Rv1 cell lines expressing wild-type P-Rex1, P-Rex1 mutant or vector were established with standard methods. Cells were selected with G-418 (400 μg/ml) for three weeks. Positive clones were amplified and verified by western blotting for P-Rex1 expression.
Protein extraction, electrophoresis and western blot analysis
Protein was extracted from cells using 1xRIPA complete lysis buffer (Santa Cruz). Prestained protein standards and protein samples (40 μg) were subjected to SDS-PAGE before transfer to Immobilon-FL. Primary antibodies were used to identify the relevant protein and loading control (β-actin). IRDye-labeled secondary antibodies were used for band detection with an Odyssey infrared imaging system (LI-COR Biosciences).
Rac and Rho activation assays
Cell lysates were assayed according to the Rac Activation Assay Kit or Rhotekin RBD Rho Assay Reagent instructions. Lysates and activated-Rac1 or -RhoA were subjected to western blot analysis using anti-Rac1 or anti-RhoA antibody, respectively.
Subcutaneous and orthotopic injections of prostate cancer cells
These experiments were approved by the Creighton University Institutional Animal Care and Use Committee. A prostate cancer cell suspension (106 cells in 200 μl of 1:1 culture medium and Matrigel) was injected subcutaneously in the flank region of male athymic Nu/Nu mice (Charles River, 6 mice per group). Tumor growth was monitored once per week and tumor volume was estimated using the ellipsoid formula. Orthotopic injections of tumor cells were performed as described elsewhere (Kovar et al. 2006). Briefly, the left anterior prostate of male NOD/SCID mice (Charles River) was exposed through a small suprapubic incision for injection of 50,000 CWR22Rv1 or PC3-LN4 cells in 15 μl of growth media. Mice were monitored for 9 weeks. At the endpoint, primary prostate tumors were removed and the carcasses were dissected to examine para-aortic lymph nodes for visible tumor metastases.
Statistical analysis
Results are means ± SE of at least three determinations and statistical comparisons used Student’s t-test. A probability (P) value of < 0.05 was considered significant.
Supplementary Material
Acknowledgments
This work was supported in part by Nebraska State Grant LB595, NIH 1R011CA125661 and Department of Defense Prostate Cancer Research Program W81XWH-07-1-0189 (Y.T). We thank Dr. Laura Hansen for helpful comments and discussions; Dr. Haihong Jiang, Lyudmila Batalkina, Dr. Greg Perry and Lisa Linder-Stephenson for their technical assistance. The project described was also supported by Grant Number G20RR024001 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
References
- Albini A, Benelli R. The chemoinvasion assay: a method to assess tumor and endothelial cell invasion and its modulation. Nat Protoc. 2007;2:504–511. doi: 10.1038/nprot.2006.466. [DOI] [PubMed] [Google Scholar]
- Arya M, Patel HR, McGurk C, Tatoud R, Klocker H, Masters J, et al. The importance of the CXCL12-CXCR4 chemokine ligand-receptor interaction in prostate cancer metastasis. J Exp Ther Oncol. 2004;4:291–303. [PubMed] [Google Scholar]
- Barber MA, Donald S, Thelen S, Anderson KE, Thelen M, Welch HC. Membrane translocation of P-Rex1 is mediated by G protein betagamma subunits and phosphoinositide 3-kinase. J Biol Chem. 2007;282:29967–29976. doi: 10.1074/jbc.M701877200. [DOI] [PubMed] [Google Scholar]
- Bex A, Lummen G, Rembrink K, Otto T, Metz K, Rubben H. Influence of pertussis toxine on local progression and metastasis after orthotopic implantation of the human prostate cancer cell line PC3 in nude mice. Prostate Cancer Prostatic Dis. 1999;2:36–40. doi: 10.1038/sj.pcan.4500286. [DOI] [PubMed] [Google Scholar]
- Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, et al. Differential targeting of Gbetagamma-subunit signaling with small molecules. Science. 2006;312:443–446. doi: 10.1126/science.1120378. [DOI] [PubMed] [Google Scholar]
- Bookout AL, Finney AE, Guo R, Peppel K, Koch WJ, Daaka Y. Targeting Gbetagamma signaling to inhibit prostate tumor formation and growth. J Biol Chem. 2003;278:37569–37573. doi: 10.1074/jbc.M306276200. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Cao X, Qin J, Xie Y, Khan O, Dowd F, Scofield M, et al. Regulator of G-protein signaling 2 (RGS2) inhibits androgen-independent activation of androgen receptor in prostate cancer cells. Oncogene. 2006;25:3719–3734. doi: 10.1038/sj.onc.1209408. [DOI] [PubMed] [Google Scholar]
- Ching KZ, Ramsey E, Pettigrew N, D’Cunha R, Jason M, Dodd JG. Expression of mRNA for epidermal growth factor, transforming growth factor-alpha and their receptor in human prostate tissue and cell lines. Mol Cell Biochem. 1993;126:151–158. doi: 10.1007/BF00925693. [DOI] [PubMed] [Google Scholar]
- Daaka Y. G proteins in cancer: the prostate cancer paradigm. Sci STKE. 2004;2004:re2. doi: 10.1126/stke.2162004re2. [DOI] [PubMed] [Google Scholar]
- Dada S, Demartines N, Dormond O. mTORC2 regulates PGE2-mediated endothelial cell survival and migration. Biochem Biophys Res Commun. 2008;372:875–879. doi: 10.1016/j.bbrc.2008.05.154. [DOI] [PubMed] [Google Scholar]
- Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- Engers R, Mueller M, Walter A, Collard JG, Willers R, Gabbert HE. Prognostic relevance of Tiam1 protein expression in prostate carcinomas. Br J Cancer. 2006;95:1081–1086. doi: 10.1038/sj.bjc.6603385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–3171. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- Gao X, Mohsin SK, Gatalica Z, Fu G, Sharma P, Nawaz Z. Decreased expression of e6-associated protein in breast and prostate carcinomas. Endocrinology. 2005;146:1707–1712. doi: 10.1210/en.2004-1198. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Negrete I, Carretero-Ortega J, Rosenfeldt H, Hernandez-Garcia R, Calderon-Salinas JV, Reyes-Cruz G, et al. P-Rex1 links mammalian target of rapamycin signaling to Rac activation and cell migration. J Biol Chem. 2007;282:23708–23715. doi: 10.1074/jbc.M703771200. [DOI] [PubMed] [Google Scholar]
- Hill K, Krugmann S, Andrews SR, Coadwell WJ, Finan P, Welch HC, et al. Regulation of P-Rex1 by phosphatidylinositol (3,4,5)-trisphosphate and Gbetagamma subunits. J Biol Chem. 2005;280:4166–4173. doi: 10.1074/jbc.M411262200. [DOI] [PubMed] [Google Scholar]
- Hoffman GR, Cerione RA. Signaling to the Rho GTPases: networking with the DH domain. FEBS Lett. 2002;513:85–91. doi: 10.1016/s0014-5793(01)03310-5. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sainz MC, Fast B, Mayor F, Jr, Aragay AM. Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Mol Pharmacol. 2003;64:773–782. doi: 10.1124/mol.64.3.773. [DOI] [PubMed] [Google Scholar]
- Kedrin D, van RJ, Hernandez L, Condeelis J, Segall JE. Cell motility and cytoskeletal regulation in invasion and metastasis. J Mammary Gland Biol Neoplasia. 2007;12:143–152. doi: 10.1007/s10911-007-9046-4. [DOI] [PubMed] [Google Scholar]
- Kelly P, Stemmle LN, Madden JF, Fields TA, Daaka Y, Casey PJ. A role for the G12 family of heterotrimeric G proteins in prostate cancer invasion. J Biol Chem. 2006;281:26483–26490. doi: 10.1074/jbc.M604376200. [DOI] [PubMed] [Google Scholar]
- Knight-Krajewski S, Welsh CF, Liu Y, Lyons LS, Faysal JM, Yang ES, et al. Deregulation of the Rho GTPase, Rac1, suppresses cyclin-dependent kinase inhibitor p21(CIP1) levels in androgen-independent human prostate cancer cells. Oncogene. 2004;23:5513–5522. doi: 10.1038/sj.onc.1207708. [DOI] [PubMed] [Google Scholar]
- Kovar JL, Johnson MA, Volcheck WM, Chen J, Simpson MA. Hyaluronidase expression induces prostate tumor metastasis in an orthotopic mouse model. Am J Pathol. 2006;169:1415–1426. doi: 10.2353/ajpath.2006.060324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann DM, Seneviratne AM, Smrcka AV. Small molecule disruption of G protein beta gamma subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol. 2008;73:410–418. doi: 10.1124/mol.107.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Cai Z, Xiao G, Liu Y, Keller ET, Yao Z, et al. CCR2 expression correlates with prostate cancer progression. J Cell Biochem. 2007;101:676–685. doi: 10.1002/jcb.21220. [DOI] [PubMed] [Google Scholar]
- Ma AD, Metjian A, Bagrodia S, Taylor S, Abrams CS. Cytoskeletal reorganization by G protein-coupled receptors is dependent on phosphoinositide 3-kinase gamma, a Rac guanosine exchange factor, and Rac. Mol Cell Biol. 1998;18:4744–4751. doi: 10.1128/mcb.18.8.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensing H, Pontz BF, Muller PK, Gauss-Muller V. A study on fibroblast chemotaxis using fibronectin and conditioned medium as chemoattractants. Eur J Cell Biol. 1983;29:268–273. [PubMed] [Google Scholar]
- Mimeault M, Batra SK. Recent advances on multiple tumorigenic cascades involved in prostatic cancer progression and targeting therapies. Carcinogenesis. 2006;27:1–22. doi: 10.1093/carcin/bgi229. [DOI] [PubMed] [Google Scholar]
- Minard ME, Kim LS, Price JE, Gallick GE. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res Treat. 2004;84:21–32. doi: 10.1023/B:BREA.0000018421.31632.e6. [DOI] [PubMed] [Google Scholar]
- Moepps B, Frodl R, Rodewald HR, Baggiolini M, Gierschik P. Two murine homologues of the human chemokine receptor CXCR4 mediating stromal cell-derived factor 1alpha activation of Gi2 are differentially expressed in vivo. Eur J Immunol. 1997;27:2102–2112. doi: 10.1002/eji.1830270839. [DOI] [PubMed] [Google Scholar]
- Neptune ER, Iiri T, Bourne HR. Galphai is not required for chemotaxis mediated by Gi-coupled receptors. J Biol Chem. 1999;274:2824–2828. doi: 10.1074/jbc.274.5.2824. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt H, Vazquez-Prado J, Gutkind JS. P-REX2, a novel PI-3-kinase sensitive Rac exchange factor. FEBS Lett. 2004;572:167–171. doi: 10.1016/j.febslet.2004.06.097. [DOI] [PubMed] [Google Scholar]
- Schiller MR. Coupling receptor tyrosine kinases to Rho GTPases--GEFs what’s the link. Cell Signal. 2006;18:1834–1843. doi: 10.1016/j.cellsig.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Servitja JM, Marinissen MJ, Sodhi A, Bustelo XR, Gutkind JS. Rac1 function is required for Src-induced transformation. Evidence of a role for Tiam1 and Vav2 in Rac activation by Src. J Biol Chem. 2003;278:34339–34346. doi: 10.1074/jbc.M302960200. [DOI] [PubMed] [Google Scholar]
- Steeg PS, Theodorescu D. Metastasis: a therapeutic target for cancer. Nat Clin Pract Oncol. 2008;5:206–219. doi: 10.1038/ncponc1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Xu D, Zhang B. Rac signaling in tumorigenesis and as target for anticancer drug development. Drug Resist Updat. 2006;9:274–287. doi: 10.1016/j.drup.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Wang Z, Dong X, Li Z, Smith JD, Wu D. Lack of a significant role of P-Rex1, a major regulator of macrophage Rac1 activation and chemotaxis, in atherogenesis. Prostaglandins Other Lipid Mediat. 2008;87:9–13. doi: 10.1016/j.prostaglandins.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, et al. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, et al. P-Rex1 regulates neutrophil function. Curr Biol. 2005;15:1867–1873. doi: 10.1016/j.cub.2005.09.050. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642–652. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005;96:379–386. doi: 10.1111/j.1349-7006.2005.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa M, Kawauchi T, Sone M, Nishimura YV, Terao M, Chihama K, et al. Involvement of a Rac activator, P-Rex1, in neurotrophin-derived signaling and neuronal migration. J Neurosci. 2005;25:4406–4419. doi: 10.1523/JNEUROSCI.4955-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.